Impact of Nutrition on Pulmonary Arterial Hypertension


 and
and 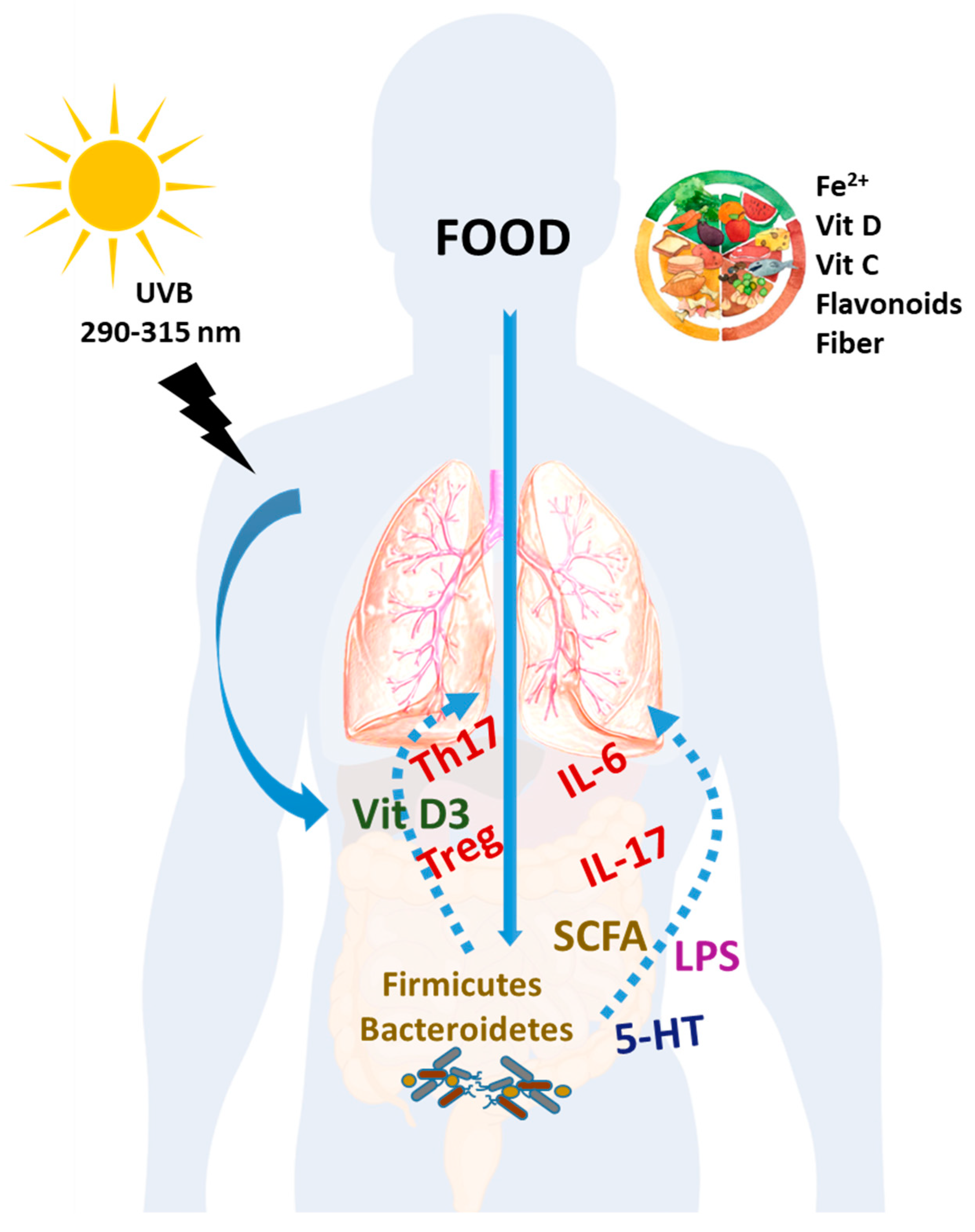{kind=link}
Abstract
1. Pulmonary Hypertension
1.1. Etiology
1.2. Pathophysiology
1.3. Current Pharmacological Therapies
1.4. Non-Pharmacological Therapies
2. Dietary Components with an Impact on PAH
2.1. Vitamin C
2.2. Vitamin D
2.3. Iron
2.4. Flavonoids and Other Polyphenols
2.5. Microbiota
3. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Simonneau, G.; Montani, D.; Celermajer, D.S.; Denton, C.P.; Gatzoulis, M.A.; Krowka, M.; Williams, P.G.; Souza, R. Haemodynamic definitions and updated clinical classification of pulmonary hypertension. Eur. Respir. J. 2019, 53. [Google Scholar] [CrossRef] [PubMed]
- Humbert, M.; Guignabert, C.; Bonnet, S.; Dorfmuller, P.; Klinger, J.R.; Nicolls, M.R.; Olschewski, A.J.; Pullamsetti, S.S.; Schermuly, R.T.; Stenmark, K.R.; et al. Pathology and pathobiology of pulmonary hypertension: State of the art and research perspectives. Eur. Respir. J. 2019, 53. [Google Scholar] [CrossRef] [PubMed]
- Galie, N.; Humbert, M.; Vachiery, J.L.; Gibbs, S.; Lang, I.; Torbicki, A.; Simonneau, G.; Peacock, A.; Vonk Noordegraaf, A.; Beghetti, M.; et al. 2015 ESC/ERS Guidelines for the diagnosis and treatment of pulmonary hypertension: The Joint Task Force for the Diagnosis and Treatment of Pulmonary Hypertension of the European Society of Cardiology (ESC) and the European Respiratory Society (ERS): Endorsed by: Association for European Paediatric and Congenital Cardiology (AEPC), International Society for Heart and Lung Transplantation (ISHLT). Eur. Respir. J. 2015, 46, 903–975. [Google Scholar] [CrossRef] [PubMed]
- Simonneau, G.; Robbins, I.M.; Beghetti, M.; Channick, R.N.; Delcroix, M.; Denton, C.P.; Elliott, C.G.; Gaine, S.P.; Gladwin, M.T.; Jing, Z.C.; et al. Updated clinical classification of pulmonary hypertension. J. Am. Coll. Cardiol. 2009, 54, S43–S54. [Google Scholar] [CrossRef] [PubMed]
- Lau, E.M.T.; Giannoulatou, E.; Celermajer, D.S.; Humbert, M. Epidemiology and treatment of pulmonary arterial hypertension. Nat. Rev. Cardiol. 2017, 14, 603–614. [Google Scholar] [CrossRef]
- Badesch, D.B.; Raskob, G.E.; Elliott, C.G.; Krichman, A.M.; Farber, H.W.; Frost, A.E.; Barst, R.J.; Benza, R.L.; Liou, T.G.; Turner, M.; et al. Pulmonary arterial hypertension: Baseline characteristics from the REVEAL Registry. Chest 2010, 137, 376–387. [Google Scholar] [CrossRef]
- Benza, R.L.; Miller, D.P.; Barst, R.J.; Badesch, D.B.; Frost, A.E.; McGoon, M.D. An evaluation of long-term survival from time of diagnosis in pulmonary arterial hypertension from the REVEAL Registry. Chest 2012, 142, 448–456. [Google Scholar] [CrossRef]
- Humbert, M.; Sitbon, O.; Yaici, A.; Montani, D.; O’Callaghan, D.S.; Jais, X.; Parent, F.; Savale, L.; Natali, D.; Gunther, S.; et al. Survival in incident and prevalent cohorts of patients with pulmonary arterial hypertension. Eur. Respir. J. 2010, 36, 549–555. [Google Scholar] [CrossRef]
- Morrell, N.W.; Aldred, M.A.; Chung, W.K.; Elliott, C.G.; Nichols, W.C.; Soubrier, F.; Trembath, R.C.; Loyd, J.E. Genetics and genomics of pulmonary arterial hypertension. Eur. Respir. J. 2019, 53. [Google Scholar] [CrossRef]
- Evans, J.D.; Girerd, B.; Montani, D.; Wang, X.J.; Galie, N.; Austin, E.D.; Elliott, G.; Asano, K.; Grunig, E.; Yan, Y.; et al. BMPR2 mutations and survival in pulmonary arterial hypertension: An individual participant data meta-analysis. Lancet Respir. Med. 2016, 4, 129–137. [Google Scholar] [CrossRef]
- Austin, E.D.; Loyd, J.E. The genetics of pulmonary arterial hypertension. Circ. Res. 2014, 115, 189–202. [Google Scholar] [CrossRef] [PubMed]
- Ma, L.; Roman-Campos, D.; Austin, E.D.; Eyries, M.; Sampson, K.S.; Soubrier, F.; Germain, M.; Tregouet, D.A.; Borczuk, A.; Rosenzweig, E.B.; et al. A novel channelopathy in pulmonary arterial hypertension. N. Engl. J. Med. 2013, 369, 351–361. [Google Scholar] [CrossRef] [PubMed]
- Remillard, C.V.; Tigno, D.D.; Platoshyn, O.; Burg, E.D.; Brevnova, E.E.; Conger, D.; Nicholson, A.; Rana, B.K.; Channick, R.N.; Rubin, L.J.; et al. Function of Kv1.5 channels and genetic variations of KCNA5 in patients with idiopathic pulmonary arterial hypertension. Am. J. Physiol. Cell Physiol. 2007, 292, C1837–C1853. [Google Scholar] [CrossRef] [PubMed]
- Orcholski, M.E.; Yuan, K.; Rajasingh, C.; Tsai, H.; Shamskhou, E.A.; Dhillon, N.K.; Voelkel, N.F.; Zamanian, R.T.; de Jesus Perez, V.A. Drug-induced pulmonary arterial hypertension: A primer for clinicians and scientists. Am. J. Physiol. Lung Cell Mol. Physiol. 2018, 314, L967–L983. [Google Scholar] [CrossRef] [PubMed]
- Southgate, L.; Machado, R.D.; Graf, S.; Morrell, N.W. Molecular genetic framework underlying pulmonary arterial hypertension. Nat. Rev. Cardiol. 2019. [Google Scholar] [CrossRef] [PubMed]
- Jaafar, S.; Visovatti, S.; Young, A.; Huang, S.; Cronin, P.; Vummidi, D.; McLaughlin, V.; Khanna, D. Impact of the revised haemodynamic definition on the diagnosis of pulmonary hypertension in patients with systemic sclerosis. Eur. Respir. J. 2019, 54. [Google Scholar] [CrossRef]
- Jarrett, H.; Barnett, C. HIV-associated pulmonary hypertension. Curr. Opin. HIV AIDS 2017, 12, 566–571. [Google Scholar] [CrossRef]
- Rabinovitch, M. Molecular pathogenesis of pulmonary arterial hypertension. J. Clin. Investig. 2012, 122, 4306–4313. [Google Scholar] [CrossRef]
- Guignabert, C.; Dorfmuller, P. Pathology and pathobiology of pulmonary hypertension. Semin. Respir. Crit. Care Med. 2013, 34, 551–559. [Google Scholar] [CrossRef]
- Mondejar-Parreno, G.; Callejo, M.; Barreira, B.; Morales-Cano, D.; Esquivel-Ruiz, S.; Moreno, L.; Cogolludo, A.; Perez-Vizcaino, F. miR-1 is increased in pulmonary hypertension and downregulates Kv1.5 channels in rat pulmonary arteries. J. Physiol. 2019, 597, 1185–1197. [Google Scholar] [CrossRef]
- Cogolludo, A.; Moreno, L.; Lodi, F.; Frazziano, G.; Cobeno, L.; Tamargo, J.; Perez-Vizcaino, F. Serotonin inhibits voltage-gated K+ currents in pulmonary artery smooth muscle cells: Role of 5-HT2A receptors, caveolin-1, and KV1.5 channel internalization. Circ. Res. 2006, 98, 931–938. [Google Scholar] [CrossRef] [PubMed]
- Antigny, F.; Hautefort, A.; Meloche, J.; Belacel-Ouari, M.; Manoury, B.; Rucker-Martin, C.; Pechoux, C.; Potus, F.; Nadeau, V.; Tremblay, E.; et al. Potassium Channel Subfamily K Member 3 (KCNK3) Contributes to the Development of Pulmonary Arterial Hypertension. Circulation 2016, 133, 1371–1385. [Google Scholar] [CrossRef] [PubMed]
- Olschewski, A.; Veale, E.L.; Nagy, B.M.; Nagaraj, C.; Kwapiszewska, G.; Antigny, F.; Lambert, M.; Humbert, M.; Czirjak, G.; Enyedi, P.; et al. TASK-1 (KCNK3) channels in the lung: From cell biology to clinical implications. Eur. Respir. J. 2017, 50. [Google Scholar] [CrossRef] [PubMed]
- Guignabert, C.; Tu, L.; Girerd, B.; Ricard, N.; Huertas, A.; Montani, D.; Humbert, M. New molecular targets of pulmonary vascular remodeling in pulmonary arterial hypertension: Importance of endothelial communication. Chest 2015, 147, 529–537. [Google Scholar] [CrossRef]
- Budhiraja, R.; Tuder, R.M.; Hassoun, P.M. Endothelial dysfunction in pulmonary hypertension. Circulation 2004, 109, 159–165. [Google Scholar] [CrossRef]
- Rabinovitch, M.; Guignabert, C.; Humbert, M.; Nicolls, M.R. Inflammation and immunity in the pathogenesis of pulmonary arterial hypertension. Circ. Res. 2014, 115, 165–175. [Google Scholar] [CrossRef]
- Montani, D.; Chaumais, M.C.; Guignabert, C.; Gunther, S.; Girerd, B.; Jais, X.; Algalarrondo, V.; Price, L.C.; Savale, L.; Sitbon, O.; et al. Targeted therapies in pulmonary arterial hypertension. Pharmacol. Ther. 2014, 141, 172–191. [Google Scholar] [CrossRef]
- O’Callaghan, D.S.; Savale, L.; Montani, D.; Jais, X.; Sitbon, O.; Simonneau, G.; Humbert, M. Treatment of pulmonary arterial hypertension with targeted therapies. Nat. Rev. Cardiol. 2011, 8, 526–538. [Google Scholar] [CrossRef]
- Kemp, K.; Savale, L.; O’Callaghan, D.S.; Jais, X.; Montani, D.; Humbert, M.; Simonneau, G.; Sitbon, O. Usefulness of first-line combination therapy with epoprostenol and bosentan in pulmonary arterial hypertension: An observational study. J. Heart Lung Transplant. 2012, 31, 150–158. [Google Scholar] [CrossRef]
- Richter, M.J.; Grimminger, J.; Kruger, B.; Ghofrani, H.A.; Mooren, F.C.; Gall, H.; Pilat, C.; Kruger, K. Effects of exercise training on pulmonary hemodynamics, functional capacity and inflammation in pulmonary hypertension. Pulm. Circ. 2017, 7, 20–37. [Google Scholar] [CrossRef]
- Galie, N.; Corris, P.A.; Frost, A.; Girgis, R.E.; Granton, J.; Jing, Z.C.; Klepetko, W.; McGoon, M.D.; McLaughlin, V.V.; Preston, I.R.; et al. Updated treatment algorithm of pulmonary arterial hypertension. J. Am. Coll. Cardiol. 2013, 62, D60–D72. [Google Scholar] [CrossRef] [PubMed]
- Cuspidi, C.; Tadic, M.; Grassi, G.; Mancia, G. Treatment of hypertension: The ESH/ESC guidelines recommendations. Pharmacol. Res. 2018, 128, 315–321. [Google Scholar] [CrossRef] [PubMed]
- Vinke, P.; Bowen, T.S.; Boekschoten, M.V.; Witkamp, R.F.; Adams, V.; van Norren, K. Anti-inflammatory nutrition with high protein attenuates cardiac and skeletal muscle alterations in a pulmonary arterial hypertension model. Sci. Rep. 2019, 9, 10160. [Google Scholar] [CrossRef] [PubMed]
- Carpenter, K.J. The discovery of vitamin C. Ann. Nutr. Metab. 2012, 61, 259–264. [Google Scholar] [CrossRef] [PubMed]
- Granger, M.; Eck, P. Dietary Vitamin C in Human Health. Adv. Food Nutr. Res. 2018, 83, 281–310. [Google Scholar] [CrossRef] [PubMed]
- Kattoor, A.J.; Pothineni, N.V.K.; Palagiri, D.; Mehta, J.L. Oxidative Stress in Atherosclerosis. Curr. Atheroscler. Rep. 2017, 19, 42. [Google Scholar] [CrossRef]
- Incalza, M.A.; D’Oria, R.; Natalicchio, A.; Perrini, S.; Laviola, L.; Giorgino, F. Oxidative stress and reactive oxygen species in endothelial dysfunction associated with cardiovascular and metabolic diseases. Vasc. Pharmacol. 2018, 100, 1–19. [Google Scholar] [CrossRef]
- Ye, Y.; Li, J.; Yuan, Z. Effect of antioxidant vitamin supplementation on cardiovascular outcomes: A meta-analysis of randomized controlled trials. PLoS ONE 2013, 8, e56803. [Google Scholar] [CrossRef]
- Frank, B.S.; Runciman, M.; Manning, W.A.; Ivy, D.D.; Abman, S.H.; Howley, L. Pulmonary Hypertension Secondary to Scurvy in a Developmentally Typical Child. J. Pediatr. 2019, 208, 291. [Google Scholar] [CrossRef]
- Dean, T.; Kaushik, N.; Williams, S.; Zinter, M.; Kim, P. Cardiac arrest and pulmonary hypertension in scurvy: A case report. Pulm. Circ. 2019, 9. [Google Scholar] [CrossRef]
- Kupari, M.; Rapola, J. Reversible pulmonary hypertension associated with vitamin C deficiency. Chest 2012, 142, 225–227. [Google Scholar] [CrossRef] [PubMed]
- Ghulam Ali, S.; Pepi, M. A Very Uncommon Case of Pulmonary Hypertension. CASE 2018, 2, 279–281. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Taddei, S.; Virdis, A.; Ghiadoni, L.; Magagna, A.; Salvetti, A. Vitamin C improves endothelium-dependent vasodilation by restoring nitric oxide activity in essential hypertension. Circulation 1998, 97, 2222–2229. [Google Scholar] [CrossRef] [PubMed]
- Knowles, H.J.; Raval, R.R.; Harris, A.L.; Ratcliffe, P.J. Effect of ascorbate on the activity of hypoxia-inducible factor in cancer cells. Can. Res. 2003, 63, 1764–1768. [Google Scholar]
- Urrutia, A.A.; Aragones, J. HIF Oxygen Sensing Pathways in Lung Biology. Biomedicines 2018, 6. [Google Scholar] [CrossRef]
- Xiang, R.P.; Sun, W.D.; Wang, J.Y.; Wang, X.L. Effect of vitamin C on pulmonary hypertension and muscularisation of pulmonary arterioles in broilers. Br. Poult. Sci. 2002, 43, 705–712. [Google Scholar] [CrossRef]
- Jones, G. Extrarenal vitamin D activation and interactions between vitamin D(2), vitamin D(3), and vitamin D analogs. Annu. Rev. Nutr. 2013, 33, 23–44. [Google Scholar] [CrossRef]
- Dusso, A.S.; Brown, A.J.; Slatopolsky, E. Vitamin D. Am. J. Physiol. Ren. Physiol. 2005, 289, F8–F28. [Google Scholar] [CrossRef]
- Moore, D.D.; Kato, S.; Xie, W.; Mangelsdorf, D.J.; Schmidt, D.R.; Xiao, R.; Kliewer, S.A. International Union of Pharmacology. LXII. The NR1H and NR1I receptors: Constitutive androstane receptor, pregnene X receptor, farnesoid X receptor alpha, farnesoid X receptor beta, liver X receptor alpha, liver X receptor beta, and vitamin D receptor. Pharmacol. Rev. 2006, 58, 742–759. [Google Scholar] [CrossRef]
- Carlberg, C.; Campbell, M.J. Vitamin D receptor signaling mechanisms: Integrated actions of a well-defined transcription factor. Steroids 2013, 78, 127–136. [Google Scholar] [CrossRef]
- Giangreco, A.A.; Nonn, L. The sum of many small changes: microRNAs are specifically and potentially globally altered by vitamin D3 metabolites. J. Steroid Biochem. Mol. Biol. 2013, 136, 86–93. [Google Scholar] [CrossRef] [PubMed]
- Battault, S.; Whiting, S.J.; Peltier, S.L.; Sadrin, S.; Gerber, G.; Maixent, J.M. Vitamin D metabolism, functions and needs: From science to health claims. Eur. J. Nutr. 2013, 52, 429–441. [Google Scholar] [CrossRef] [PubMed]
- Norman, P.E.; Powell, J.T. Vitamin D and cardiovascular disease. Circ. Res. 2014, 114, 379–393. [Google Scholar] [CrossRef] [PubMed]
- Roth, D.E.; Abrams, S.A.; Aloia, J.; Bergeron, G.; Bourassa, M.W.; Brown, K.H.; Calvo, M.S.; Cashman, K.D.; Combs, G.; De-Regil, L.M.; et al. Global prevalence and disease burden of vitamin D deficiency: A roadmap for action in low- and middle-income countries. Ann. N. Y. Acad. Sci. 2018, 1430, 44–79. [Google Scholar] [CrossRef] [PubMed]
- Demer, L.L.; Hsu, J.J.; Tintut, Y. Steroid Hormone Vitamin D: Implications for Cardiovascular Disease. Circ. Res. 2018, 122, 1576–1585. [Google Scholar] [CrossRef]
- Bivona, G.; Agnello, L.; Ciaccio, M. The immunological implication of the new vitamin D metabolism. Cent. Eur. J. Immunol. 2018, 43, 331–334. [Google Scholar] [CrossRef]
- Wang, T.J.; Pencina, M.J.; Booth, S.L.; Jacques, P.F.; Ingelsson, E.; Lanier, K.; Benjamin, E.J.; D’Agostino, R.B.; Wolf, M.; Vasan, R.S. Vitamin D deficiency and risk of cardiovascular disease. Circulation 2008, 117, 503–511. [Google Scholar] [CrossRef]
- Schottker, B.; Jorde, R.; Peasey, A.; Thorand, B.; Jansen, E.H.; Groot, L.; Streppel, M.; Gardiner, J.; Ordonez-Mena, J.M.; Perna, L.; et al. Vitamin D and mortality: Meta-analysis of individual participant data from a large consortium of cohort studies from Europe and the United States. BMJ 2014, 348, g3656. [Google Scholar] [CrossRef]
- Tanaka, H.; Kataoka, M.; Isobe, S.; Yamamoto, T.; Shirakawa, K.; Endo, J.; Satoh, T.; Hakamata, Y.; Kobayashi, E.; Sano, M.; et al. Therapeutic impact of dietary vitamin D supplementation for preventing right ventricular remodeling and improving survival in pulmonary hypertension. PLoS ONE 2017, 12, e0180615. [Google Scholar] [CrossRef]
- Yu, H.; Xu, M.; Dong, Y.; Liu, J.; Li, Y.; Mao, W.; Wang, J.; Wang, L. 1,25(OH)2D3 attenuates pulmonary arterial hypertension via microRNA-204 mediated Tgfbr2/Smad signaling. Exp. Cell Res. 2018, 362, 311–323. [Google Scholar] [CrossRef]
- Ulrich, S.; Hersberger, M.; Fischler, M.; Huber, L.C.; Senn, O.; Treder, U.; Speich, R.; Schmid, C. Bone mineral density and secondary hyperparathyroidism in pulmonary hypertension. Open Respir. Med. J. 2009, 3, 53–60. [Google Scholar] [CrossRef]
- Demir, M.; Uyan, U.; Keceoclu, S.; Demir, C. The relationship between vitamin D deficiency and pulmonary hypertension. Prague Med. Rep. 2013, 114, 154–161. [Google Scholar] [CrossRef] [PubMed]
- Mirdamadi, A.; Moshkdar, P. Benefits from the correction of vitamin D deficiency in patients with pulmonary hypertension. Casp. J. Intern. Med. 2016, 7, 253–259. [Google Scholar]
- Jolliffe, D.A.; Greenberg, L.; Hooper, R.L.; Griffiths, C.J.; Camargo, C.A., Jr.; Kerley, C.P.; Jensen, M.E.; Mauger, D.; Stelmach, I.; Urashima, M.; et al. Vitamin D supplementation to prevent asthma exacerbations: A systematic review and meta-analysis of individual participant data. Lancet Respir. Med. 2017, 5, 881–890. [Google Scholar] [CrossRef]
- Jolliffe, D.A.; Greenberg, L.; Hooper, R.L.; Mathyssen, C.; Rafiq, R.; de Jongh, R.T.; Camargo, C.A.; Griffiths, C.J.; Janssens, W.; Martineau, A.R. Vitamin D to prevent exacerbations of COPD: Systematic review and meta-analysis of individual participant data from randomised controlled trials. Thorax 2019, 74, 337–345. [Google Scholar] [CrossRef]
- Manson, J.E.; Cook, N.R.; Lee, I.M.; Christen, W.; Bassuk, S.S.; Mora, S.; Gibson, H.; Gordon, D.; Copeland, T.; D’Agostino, D.; et al. Vitamin D Supplements and Prevention of Cancer and Cardiovascular Disease. N. Engl. J. Med. 2019, 380, 33–44. [Google Scholar] [CrossRef]
- Bolland, M.J.; Grey, A.; Gamble, G.D.; Reid, I.R. The effect of vitamin D supplementation on skeletal, vascular, or cancer outcomes: A trial sequential meta-analysis. Lancet Diabetes Endocrinol. 2014, 2, 307–320. [Google Scholar] [CrossRef]
- Beveridge, L.A.; Khan, F.; Struthers, A.D.; Armitage, J.; Barchetta, I.; Bressendorff, I.; Cavallo, M.G.; Clarke, R.; Dalan, R.; Dreyer, G.; et al. Effect of Vitamin D Supplementation on Markers of Vascular Function: A Systematic Review and Individual Participant Meta-Analysis. J. Am. Heart Assoc. 2018, 7. [Google Scholar] [CrossRef]
- Camaschella, C. Iron-deficiency anemia. N. Engl. J. Med. 2015, 372, 1832–1843. [Google Scholar] [CrossRef]
- Munoz, M.; Gomez-Ramirez, S.; Besser, M.; Pavia, J.; Gomollon, F.; Liumbruno, G.M.; Bhandari, S.; Cladellas, M.; Shander, A.; Auerbach, M. Current misconceptions in diagnosis and management of iron deficiency. Blood Transfus. 2017, 15, 422–437. [Google Scholar] [CrossRef]
- Anand, I.S.; Gupta, P. Anemia and Iron Deficiency in Heart Failure: Current Concepts and Emerging Therapies. Circulation 2018, 138, 80–98. [Google Scholar] [CrossRef] [PubMed]
- Rhodes, C.J.; Howard, L.S.; Busbridge, M.; Ashby, D.; Kondili, E.; Gibbs, J.S.; Wharton, J.; Wilkins, M.R. Iron deficiency and raised hepcidin in idiopathic pulmonary arterial hypertension: Clinical prevalence, outcomes, and mechanistic insights. J. Am. Coll. Cardiol. 2011, 58, 300–309. [Google Scholar] [CrossRef] [PubMed]
- Ruiter, G.; Lankhorst, S.; Boonstra, A.; Postmus, P.E.; Zweegman, S.; Westerhof, N.; van der Laarse, W.J.; Vonk-Noordegraaf, A. Iron deficiency is common in idiopathic pulmonary arterial hypertension. Eur. Respir. J. 2011, 37, 1386–1391. [Google Scholar] [CrossRef] [PubMed]
- Ramakrishnan, L.; Pedersen, S.L.; Toe, Q.K.; Quinlan, G.J.; Wort, S.J. Pulmonary Arterial Hypertension: Iron Matters. Front. Physiol. 2018, 9, 641. [Google Scholar] [CrossRef] [PubMed]
- Hampole, C.V.; Mehrotra, A.K.; Thenappan, T.; Gomberg-Maitland, M.; Shah, S.J. Usefulness of red cell distribution width as a prognostic marker in pulmonary hypertension. Am. J. Cardiol. 2009, 104, 868–872. [Google Scholar] [CrossRef] [PubMed]
- Krasuski, R.A.; Hart, S.A.; Smith, B.; Wang, A.; Harrison, J.K.; Bashore, T.M. Association of anemia and long-term survival in patients with pulmonary hypertension. Int. J. Cardiol. 2011, 150, 291–295. [Google Scholar] [CrossRef]
- Yu, X.; Zhang, Y.; Luo, Q.; Liu, Z.; Zhao, Z.; Zhao, Q.; Gao, L.; Jin, Q.; Yan, L. Iron deficiency in pulmonary arterial hypertension associated with congenital heart disease. Scand. Cardiovasc. J. 2018, 52, 378–382. [Google Scholar] [CrossRef]
- Anker, S.D.; Comin Colet, J.; Filippatos, G.; Willenheimer, R.; Dickstein, K.; Drexler, H.; Luscher, T.F.; Bart, B.; Banasiak, W.; Niegowska, J.; et al. Ferric carboxymaltose in patients with heart failure and iron deficiency. N. Engl. J. Med. 2009, 361, 2436–2448. [Google Scholar] [CrossRef]
- Bolger, A.P.; Bartlett, F.R.; Penston, H.S.; O’Leary, J.; Pollock, N.; Kaprielian, R.; Chapman, C.M. Intravenous iron alone for the treatment of anemia in patients with chronic heart failure. J. Am. Coll. Cardiol. 2006, 48, 1225–1227. [Google Scholar] [CrossRef]
- Cotroneo, E.; Ashek, A.; Wang, L.; Wharton, J.; Dubois, O.; Bozorgi, S.; Busbridge, M.; Alavian, K.N.; Wilkins, M.R.; Zhao, L. Iron homeostasis and pulmonary hypertension: Iron deficiency leads to pulmonary vascular remodeling in the rat. Circ. Res. 2015, 116, 1680–1690. [Google Scholar] [CrossRef]
- Wolin, M.S.; Patel, D.; Alhawaj, R.; Gupte, S.A.; Sun, D. Iron Metabolism and Vascular Remodeling: Novel Insights Provided by Transferrin-1 Receptor Depletion in Mice With Pulmonary Hypertension. Am. J. Hypertens. 2016, 29, 676–678. [Google Scholar] [CrossRef] [PubMed]
- Robinson, J.C.; Graham, B.B.; Rouault, T.C.; Tuder, R.M. The crossroads of iron with hypoxia and cellular metabolism. Implications in the pathobiology of pulmonary hypertension. Am. J. Respir. Cell Mol. Biol. 2014, 51, 721–729. [Google Scholar] [CrossRef] [PubMed]
- Smith, T.G.; Balanos, G.M.; Croft, Q.P.; Talbot, N.P.; Dorrington, K.L.; Ratcliffe, P.J.; Robbins, P.A. The increase in pulmonary arterial pressure caused by hypoxia depends on iron status. J. Physiol. 2008, 586, 5999–6005. [Google Scholar] [CrossRef] [PubMed]
- Smith, T.G.; Talbot, N.P.; Privat, C.; Rivera-Ch, M.; Nickol, A.H.; Ratcliffe, P.J.; Dorrington, K.L.; Leon-Velarde, F.; Robbins, P.A. Effects of iron supplementation and depletion on hypoxic pulmonary hypertension: Two randomized controlled trials. JAMA 2009, 302, 1444–1450. [Google Scholar] [CrossRef]
- Naito, Y.; Hosokawa, M.; Sawada, H.; Oboshi, M.; Iwasaku, T.; Okuhara, Y.; Eguchi, A.; Nishimura, K.; Soyama, Y.; Hirotani, S.; et al. Iron is associated with the development of hypoxia-induced pulmonary vascular remodeling in mice. Heart Vessel. 2016, 31, 2074–2079. [Google Scholar] [CrossRef]
- Lakhal-Littleton, S.; Crosby, A.; Frise, M.C.; Mohammad, G.; Carr, C.A.; Loick, P.A.M.; Robbins, P.A. Intracellular iron deficiency in pulmonary arterial smooth muscle cells induces pulmonary arterial hypertension in mice. Proc. Natl. Acad. Sci. USA 2019, 116, 13122–13130. [Google Scholar] [CrossRef]
- Yu, X.; Luo, Q.; Liu, Z.; Zhao, Z.; Zhao, Q.; An, C.; Huang, Z.; Jin, Q.; Gao, L.; Yan, L. Prevalence of iron deficiency in different subtypes of pulmonary hypertension. Heart Lung 2018, 47, 308–313. [Google Scholar] [CrossRef]
- Soon, E.; Treacy, C.M.; Toshner, M.R.; MacKenzie-Ross, R.; Manglam, V.; Busbridge, M.; Sinclair-McGarvie, M.; Arnold, J.; Sheares, K.K.; Morrell, N.W.; et al. Unexplained iron deficiency in idiopathic and heritable pulmonary arterial hypertension. Thorax 2011, 66, 326–332. [Google Scholar] [CrossRef]
- Rhodes, C.J.; Wharton, J.; Howard, L.; Gibbs, J.S.; Vonk-Noordegraaf, A.; Wilkins, M.R. Iron deficiency in pulmonary arterial hypertension: A potential therapeutic target. Eur. Respir. J. 2011, 38, 1453–1460. [Google Scholar] [CrossRef]
- Soon, E.; Holmes, A.M.; Treacy, C.M.; Doughty, N.J.; Southgate, L.; Machado, R.D.; Trembath, R.C.; Jennings, S.; Barker, L.; Nicklin, P.; et al. Elevated levels of inflammatory cytokines predict survival in idiopathic and familial pulmonary arterial hypertension. Circulation 2010, 122, 920–927. [Google Scholar] [CrossRef]
- Howard, L.S.; Watson, G.M.; Wharton, J.; Rhodes, C.J.; Chan, K.; Khengar, R.; Robbins, P.A.; Kiely, D.G.; Condliffe, R.; Elliott, C.A.; et al. Supplementation of iron in pulmonary hypertension: Rationale and design of a phase II clinical trial in idiopathic pulmonary arterial hypertension. Pulm. Circ. 2013, 3, 100–107. [Google Scholar] [CrossRef] [PubMed]
- Ruiter, G.; Manders, E.; Happe, C.M.; Schalij, I.; Groepenhoff, H.; Howard, L.S.; Wilkins, M.R.; Bogaard, H.J.; Westerhof, N.; van der Laarse, W.J.; et al. Intravenous iron therapy in patients with idiopathic pulmonary arterial hypertension and iron deficiency. Pulm. Circ. 2015, 5, 466–472. [Google Scholar] [CrossRef] [PubMed]
- Sanchez, M.; Romero, M.; Gomez-Guzman, M.; Tamargo, J.; Perez-Vizcaino, F.; Duarte, J. Cardiovascular effects of flavonoids. Curr. Med. Chem. 2018. [Google Scholar] [CrossRef] [PubMed]
- Perez-Vizcaino, F.; Fraga, C.G. Research trends in flavonoids and health. Arch. Biochem. Biophys. 2018, 646, 107–112. [Google Scholar] [CrossRef]
- Perez-Vizcaino, F.; Duarte, J. Flavonols and cardiovascular disease. Mol. Asp. Med. 2010, 31, 478–494. [Google Scholar] [CrossRef]
- Quinones, M.; Miguel, M.; Aleixandre, A. Beneficial effects of polyphenols on cardiovascular disease. Pharmacol. Res. 2013, 68, 125–131. [Google Scholar] [CrossRef]
- Csiszar, A.; Labinskyy, N.; Olson, S.; Pinto, J.T.; Gupte, S.; Wu, J.M.; Hu, F.; Ballabh, P.; Podlutsky, A.; Losonczy, G.; et al. Resveratrol prevents monocrotaline-induced pulmonary hypertension in rats. Hypertension 2009, 54, 668–675. [Google Scholar] [CrossRef]
- Chicoine, L.G.; Stewart, J.A., Jr.; Lucchesi, P.A. Is resveratrol the magic bullet for pulmonary hypertension? Hypertension 2009, 54, 473–474. [Google Scholar] [CrossRef]
- Chen, B.; Xue, J.; Meng, X.; Slutzky, J.L.; Calvert, A.E.; Chicoine, L.G. Resveratrol prevents hypoxia-induced arginase II expression and proliferation of human pulmonary artery smooth muscle cells via Akt-dependent signaling. Am. J. Physiol. Lung Cell Mol. Physiol. 2014, 307, L317–L325. [Google Scholar] [CrossRef] [PubMed]
- Guan, Z.; Shen, L.; Liang, H.; Yu, H.; Hei, B.; Meng, X.; Yang, L. Resveratrol inhibits hypoxia-induced proliferation and migration of pulmonary artery vascular smooth muscle cells by inhibiting the phosphoinositide 3-kinase/protein kinase B signaling pathway. Mol. Med. Rep. 2017, 16, 1653–1660. [Google Scholar] [CrossRef]
- Xu, D.; Li, Y.; Zhang, B.; Wang, Y.; Liu, Y.; Luo, Y.; Niu, W.; Dong, M.; Liu, M.; Dong, H.; et al. Resveratrol alleviate hypoxic pulmonary hypertension via anti-inflammation and anti-oxidant pathways in rats. Int. J. Med. Sci. 2016, 13, 942–954. [Google Scholar] [CrossRef] [PubMed]
- Shi, W.; Zhai, C.; Feng, W.; Wang, J.; Zhu, Y.; Li, S.; Wang, Q.; Zhang, Q.; Yan, X.; Chai, L.; et al. Resveratrol inhibits monocrotaline-induced pulmonary arterial remodeling by suppression of SphK1-mediated NF-kappaB activation. Life Sci. 2018, 210, 140–149. [Google Scholar] [CrossRef] [PubMed]
- Perez-Vizcaino, F.; Duarte, J.; Jimenez, R.; Santos-Buelga, C.; Osuna, A. Antihypertensive effects of the flavonoid quercetin. Pharmacol. Rep. 2009, 61, 67–75. [Google Scholar] [CrossRef]
- Duarte, J.; Perez-Palencia, R.; Vargas, F.; Ocete, M.A.; Perez-Vizcaino, F.; Zarzuelo, A.; Tamargo, J. Antihypertensive effects of the flavonoid quercetin in spontaneously hypertensive rats. Br. J. Pharmacol. 2001, 133, 117–124. [Google Scholar] [CrossRef]
- Gao, H.; Chen, C.; Huang, S.; Li, B. Quercetin attenuates the progression of monocrotaline-induced pulmonary hypertension in rats. J. Biomed. Res. 2012, 26, 98–102. [Google Scholar] [CrossRef][Green Version]
- Morales-Cano, D.; Menendez, C.; Moreno, E.; Moral-Sanz, J.; Barreira, B.; Galindo, P.; Pandolfi, R.; Jimenez, R.; Moreno, L.; Cogolludo, A.; et al. The flavonoid quercetin reverses pulmonary hypertension in rats. PLoS ONE 2014, 9, e114492. [Google Scholar] [CrossRef]
- Jimenez, R.; Lopez-Sepulveda, R.; Romero, M.; Toral, M.; Cogolludo, A.; Perez-Vizcaino, F.; Duarte, J. Quercetin and its metabolites inhibit the membrane NADPH oxidase activity in vascular smooth muscle cells from normotensive and spontaneously hypertensive rats. Food Funct. 2015, 6, 409–414. [Google Scholar] [CrossRef]
- He, Y.; Cao, X.; Liu, X.; Li, X.; Xu, Y.; Liu, J.; Shi, J. Quercetin reverses experimental pulmonary arterial hypertension by modulating the TrkA pathway. Exp. Cell Res. 2015, 339, 122–134. [Google Scholar] [CrossRef]
- Huang, S.; Zhu, X.; Huang, W.; He, Y.; Pang, L.; Lan, X.; Shui, X.; Chen, Y.; Chen, C.; Lei, W. Quercetin Inhibits Pulmonary Arterial Endothelial Cell Transdifferentiation Possibly by Akt and Erk1/2 Pathways. Biomed. Res. Int. 2017, 2017. [Google Scholar] [CrossRef]
- He, Y.; Cao, X.; Guo, P.; Li, X.; Shang, H.; Liu, J.; Xie, M.; Xu, Y.; Liu, X. Quercetin induces autophagy via FOXO1-dependent pathways and autophagy suppression enhances quercetin-induced apoptosis in PASMCs in hypoxia. Free Radic. Biol. Med. 2017, 103, 165–176. [Google Scholar] [CrossRef]
- Cao, X.; He, Y.; Li, X.; Xu, Y.; Liu, X. The IRE1alpha-XBP1 pathway function in hypoxia-induced pulmonary vascular remodeling, is upregulated by quercetin, inhibits apoptosis and partially reverses the effect of quercetin in PASMCs. Am. J. Transl. Res. 2019, 11, 641–654. [Google Scholar] [PubMed]
- Evans, M.; Elliott, J.G.; Sharma, P.; Berman, R.; Guthrie, N. The effect of synthetic genistein on menopause symptom management in healthy postmenopausal women: A multi-center, randomized, placebo-controlled study. Maturitas 2011, 68, 189–196. [Google Scholar] [CrossRef] [PubMed]
- Zaheer, K.; Humayoun Akhtar, M. An updated review of dietary isoflavones: Nutrition, processing, bioavailability and impacts on human health. Crit. Rev. Food Sci. Nutr. 2017, 57, 1280–1293. [Google Scholar] [CrossRef] [PubMed]
- Vera, R.; Jimenez, R.; Lodi, F.; Sanchez, M.; Galisteo, M.; Zarzuelo, A.; Perez-Vizcaino, F.; Duarte, J. Genistein restores caveolin-1 and AT-1 receptor expression and vascular function in large vessels of ovariectomized hypertensive rats. Menopause 2007, 14, 933–940. [Google Scholar] [CrossRef]
- Karamsetty, M.R.; Klinger, J.R.; Hill, N.S. Phytoestrogens restore nitric oxide-mediated relaxation in isolated pulmonary arteries from chronically hypoxic rats. J. Pharmacol. Exp. Ther. 2001, 297, 968–974. [Google Scholar]
- Matori, H.; Umar, S.; Nadadur, R.D.; Sharma, S.; Partow-Navid, R.; Afkhami, M.; Amjedi, M.; Eghbali, M. Genistein, a soy phytoestrogen, reverses severe pulmonary hypertension and prevents right heart failure in rats. Hypertension 2012, 60, 425–430. [Google Scholar] [CrossRef]
- Weigand, L.; Sylvester, J.T.; Shimoda, L.A. Mechanisms of endothelin-1-induced contraction in pulmonary arteries from chronically hypoxic rats. Am. J. Physiol. Lung Cell. Mol. Physiol. 2006, 290, L284–L290. [Google Scholar] [CrossRef]
- Homma, N.; Morio, Y.; Takahashi, H.; Yamamoto, A.; Suzuki, T.; Sato, K.; Muramatsu, M.; Fukuchi, Y. Genistein, a phytoestrogen, attenuates monocrotaline-induced pulmonary hypertension. Respir. Int. Rev. Thorac. Dis. 2006, 73, 105–112. [Google Scholar] [CrossRef]
- Kuriyama, S.; Morio, Y.; Toba, M.; Nagaoka, T.; Takahashi, F.; Iwakami, S.; Seyama, K.; Takahashi, K. Genistein attenuates hypoxic pulmonary hypertension via enhanced nitric oxide signaling and the erythropoietin system. Am. J. Physiol. Lung Cell. Mol. Physiol. 2014, 306, L996–L1005. [Google Scholar] [CrossRef]
- Zheng, Z.; Yu, S.; Zhang, W.; Peng, Y.; Pu, M.; Kang, T.; Zeng, J.; Yu, Y.; Li, G. Genistein attenuates monocrotaline-induced pulmonary arterial hypertension in rats by activating PI3K/Akt/eNOS signaling. Histol. Histopathol. 2017, 32, 35–41. [Google Scholar] [CrossRef]
- Zhang, M.; Wu, Y.; Wang, M.; Wang, Y.; Tausif, R.; Yang, Y. Genistein rescues hypoxia-induced pulmonary arterial hypertension through estrogen receptor and beta-adrenoceptor signaling. J. Nutr. Biochem. 2018, 58, 110–118. [Google Scholar] [CrossRef] [PubMed]
- Levy, M.; Kolodziejczyk, A.A.; Thaiss, C.A.; Elinav, E. Dysbiosis and the immune system. Nat. Rev. Immunol. 2017, 17, 219–232. [Google Scholar] [CrossRef] [PubMed]
- Yang, T.; Santisteban, M.M.; Rodriguez, V.; Li, E.; Ahmari, N.; Carvajal, J.M.; Zadeh, M.; Gong, M.; Qi, Y.; Zubcevic, J.; et al. Gut dysbiosis is linked to hypertension. Hypertension 2015, 65, 1331–1340. [Google Scholar] [CrossRef] [PubMed]
- Marques, F.Z.; Mackay, C.R.; Kaye, D.M. Beyond gut feelings: How the gut microbiota regulates blood pressure. Nat. Rev. Cardiol. 2018, 15, 20–32. [Google Scholar] [CrossRef] [PubMed]
- Robles-Vera, I.; Toral, M.; Romero, M.; Jimenez, R.; Sanchez, M.; Perez-Vizcaino, F.; Duarte, J. Antihypertensive Effects of Probiotics. Curr. Hypertens. Rep. 2017, 19, 26. [Google Scholar] [CrossRef] [PubMed]
- Ley, R.E.; Turnbaugh, P.J.; Klein, S.; Gordon, J.I. Microbial ecology: Human gut microbes associated with obesity. Nature 2006, 444, 1022–1023. [Google Scholar] [CrossRef]
- Mariat, D.; Firmesse, O.; Levenez, F.; Guimaraes, V.; Sokol, H.; Dore, J.; Corthier, G.; Furet, J.P. The Firmicutes/Bacteroidetes ratio of the human microbiota changes with age. BMC Microbiol. 2009, 9, 123. [Google Scholar] [CrossRef]
- Pascale, A.; Marchesi, N.; Govoni, S.; Coppola, A.; Gazzaruso, C. The role of gut microbiota in obesity, diabetes mellitus, and effect of metformin: New insights into old diseases. Curr. Opin. Pharmacol. 2019, 49, 1–5. [Google Scholar] [CrossRef]
- Strati, F.; Cavalieri, D.; Albanese, D.; De Felice, C.; Donati, C.; Hayek, J.; Jousson, O.; Leoncini, S.; Renzi, D.; Calabro, A.; et al. New evidences on the altered gut microbiota in autism spectrum disorders. Microbiome 2017, 5, 24. [Google Scholar] [CrossRef]
- Callejo, M.; Mondejar-Parreno, G.; Barreira, B.; Izquierdo-Garcia, J.L.; Morales-Cano, D.; Esquivel-Ruiz, S.; Moreno, L.; Cogolludo, A.; Duarte, J.; Perez-Vizcaino, F. Pulmonary Arterial Hypertension Affects the Rat Gut Microbiome. Sci. Rep. 2018, 8, 9681. [Google Scholar] [CrossRef]
- Quigley, E.M.M. Nutraceuticals as Modulators of Gut Microbiota: Role in Therapy. Br. J. Pharmacol. 2019. [Google Scholar] [CrossRef] [PubMed]
- Biesalski, H.K. Nutrition meets the microbiome: Micronutrients and the microbiota. Ann. N. Y. Acad. Sci. 2016, 1372, 53–64. [Google Scholar] [CrossRef] [PubMed]
- Skrypnik, K.; Suliburska, J. Association between the gut microbiota and mineral metabolism. J. Sci. Food Agric. 2018, 98, 2449–2460. [Google Scholar] [CrossRef] [PubMed]
- Janeiro, M.H.; Ramirez, M.J.; Milagro, F.I.; Martinez, J.A.; Solas, M. Implication of Trimethylamine N-Oxide (TMAO) in Disease: Potential Biomarker or New Therapeutic Target. Nutrients 2018, 10. [Google Scholar] [CrossRef]
- Robles-Vera, I.; Callejo, M.; Ramos, R.; Duarte, J.; Perez-Vizcaino, F. Impact of Vitamin D Deficit on the Rat Gut Microbiome. Nutrients 2019, 11. [Google Scholar] [CrossRef]
- Yilmaz, B.; Li, H. Gut Microbiota and Iron: The Crucial Actors in Health and Disease. Pharmaceuticals 2018, 11. [Google Scholar] [CrossRef]
- Zhao, L.; Zhang, Q.; Ma, W.; Tian, F.; Shen, H.; Zhou, M. A combination of quercetin and resveratrol reduces obesity in high-fat diet-fed rats by modulation of gut microbiota. Food Funct. 2017, 8, 4644–4656. [Google Scholar] [CrossRef]
- Wedgwood, S.; Warford, C.; Agvatisiri, S.R.; Thai, P.N.; Chiamvimonvat, N.; Kalanetra, K.M.; Lakshminrusimha, S.; Steinhorn, R.H.; Mills, D.A.; Underwood, M.A. The developing gut-lung axis: Postnatal growth restriction, intestinal dysbiosis, and pulmonary hypertension in a rodent model. Pediatr. Res. 2019. [Google Scholar] [CrossRef]
- Thenappan, T.; Khoruts, A.; Chen, Y.; Weir, E.K. Can Intestinal Microbiota and Circulating Microbial Products Contribute to Pulmonary Arterial Hypertension? Am. J. Physiol. Heart Circ. Physiol. 2019. [Google Scholar] [CrossRef]
- Bauer, E.M.; Chanthaphavong, R.S.; Sodhi, C.P.; Hackam, D.J.; Billiar, T.R.; Bauer, P.M. Genetic deletion of toll-like receptor 4 on platelets attenuates experimental pulmonary hypertension. Circ. Res. 2014, 114, 1596–1600. [Google Scholar] [CrossRef]
- Ranchoux, B.; Bigorgne, A.; Hautefort, A.; Girerd, B.; Sitbon, O.; Montani, D.; Humbert, M.; Tcherakian, C.; Perros, F. Gut-Lung Connection in Pulmonary Arterial Hypertension. Am. J. Respir. Cell Mol. Biol. 2017, 56, 402–405. [Google Scholar] [CrossRef] [PubMed]
- Lee, Y.K.; Mazmanian, S.K. Has the microbiota played a critical role in the evolution of the adaptive immune system? Science 2010, 330, 1768–1773. [Google Scholar] [CrossRef] [PubMed]
- Huertas, A.; Phan, C.; Bordenave, J.; Tu, L.; Thuillet, R.; Le Hiress, M.; Avouac, J.; Tamura, Y.; Allanore, Y.; Jovan, R.; et al. Regulatory T Cell Dysfunction in Idiopathic, Heritable and Connective Tissue-Associated Pulmonary Arterial Hypertension. Chest 2016, 149, 1482–1493. [Google Scholar] [CrossRef] [PubMed]
- Hood, K.Y.; Mair, K.M.; Harvey, A.P.; Montezano, A.C.; Touyz, R.M.; MacLean, M.R. Serotonin Signaling Through the 5-HT1B Receptor and NADPH Oxidase 1 in Pulmonary Arterial Hypertension. Arterioscler. Thromb. Vasc. Biol. 2017, 37, 1361–1370. [Google Scholar] [CrossRef]

© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Callejo, M.; Barberá, J.A.; Duarte, J.; Perez-Vizcaino, F. Impact of Nutrition on Pulmonary Arterial Hypertension. Nutrients 2020, 12, 169. https://doi.org/10.3390/nu12010169
Callejo M, Barberá JA, Duarte J, Perez-Vizcaino F. Impact of Nutrition on Pulmonary Arterial Hypertension. Nutrients. 2020; 12(1):169. https://doi.org/10.3390/nu12010169
Chicago/Turabian StyleCallejo, María, Joan Albert Barberá, Juan Duarte, and Francisco Perez-Vizcaino. 2020. "Impact of Nutrition on Pulmonary Arterial Hypertension" Nutrients 12, no. 1: 169. https://doi.org/10.3390/nu12010169
APA StyleCallejo, M., Barberá, J. A., Duarte, J., & Perez-Vizcaino, F. (2020). Impact of Nutrition on Pulmonary Arterial Hypertension. Nutrients, 12(1), 169. https://doi.org/10.3390/nu12010169