The Role of Autophagy in Liver Epithelial Cells and Its Impact on Systemic Homeostasis

 , ,
, ,  , ,
, , 
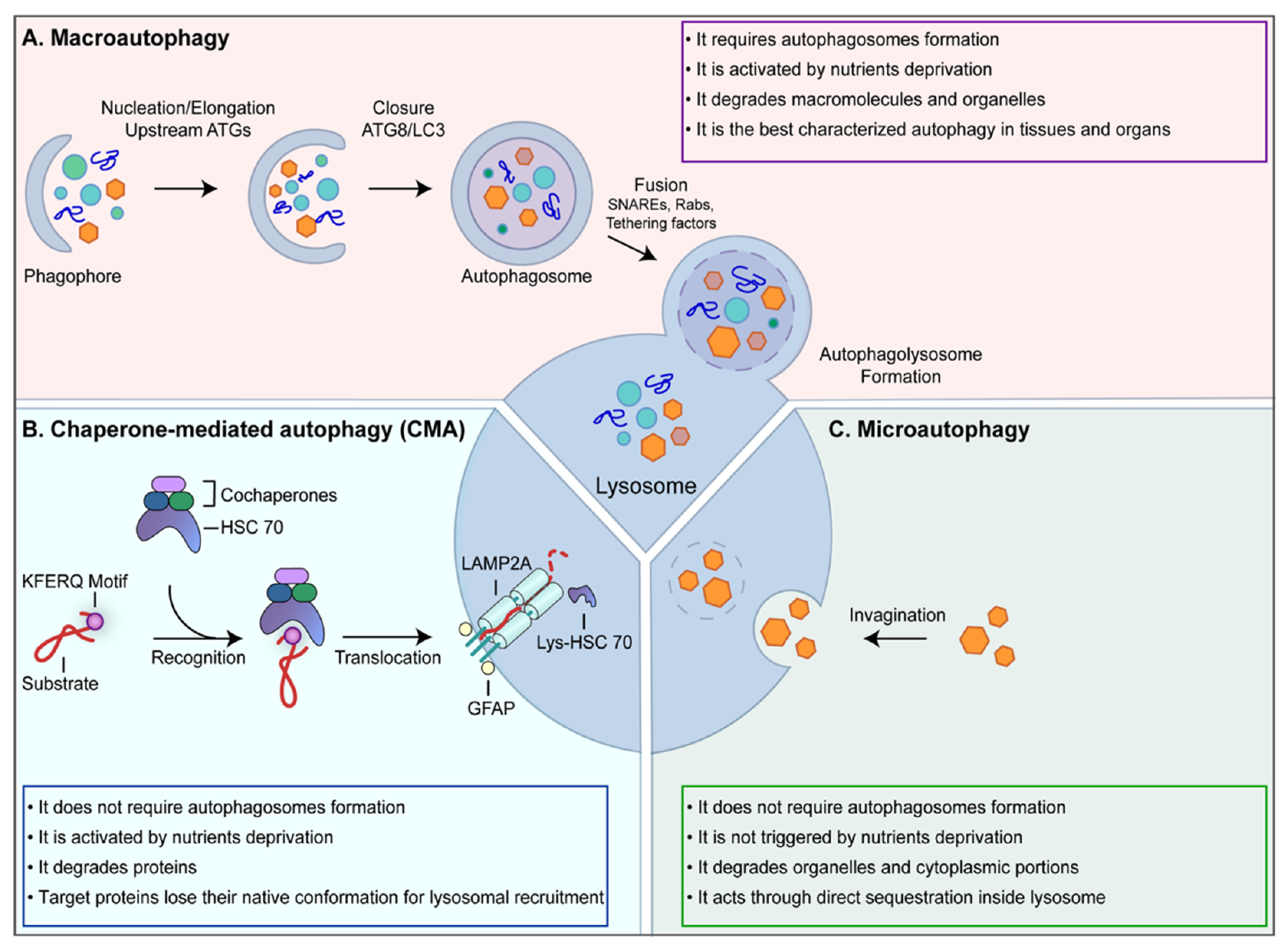{kind=link}
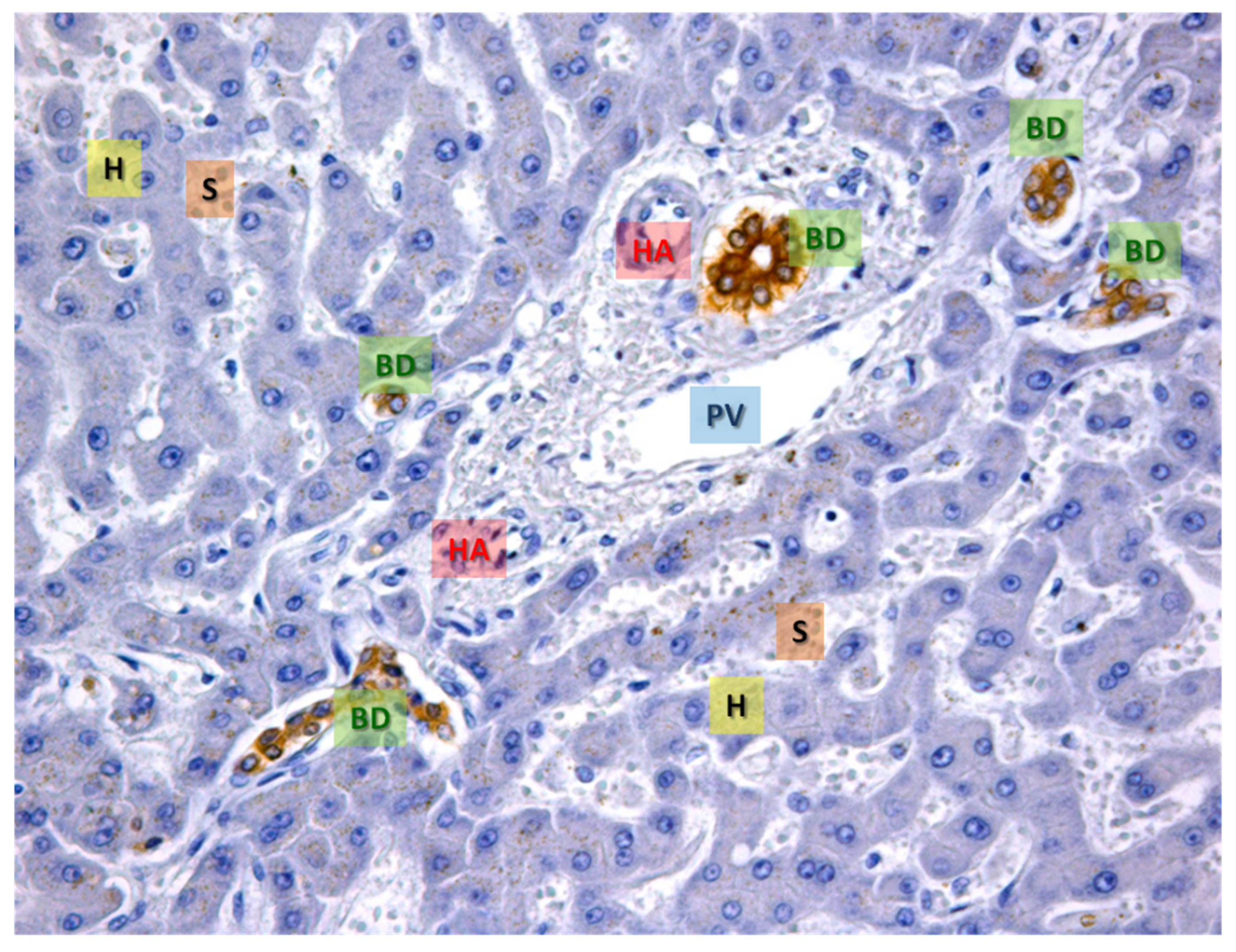{kind=link}
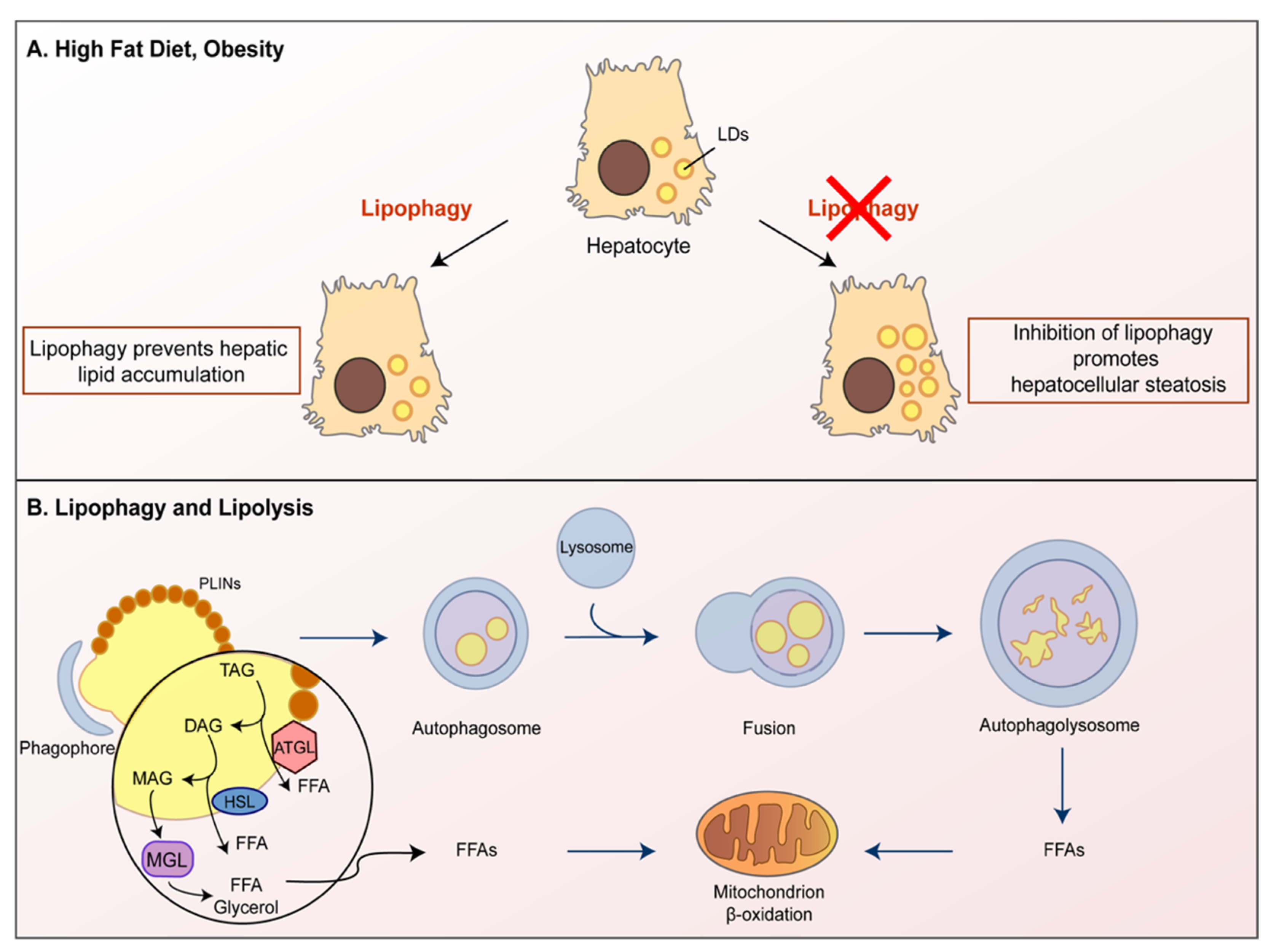{kind=link}
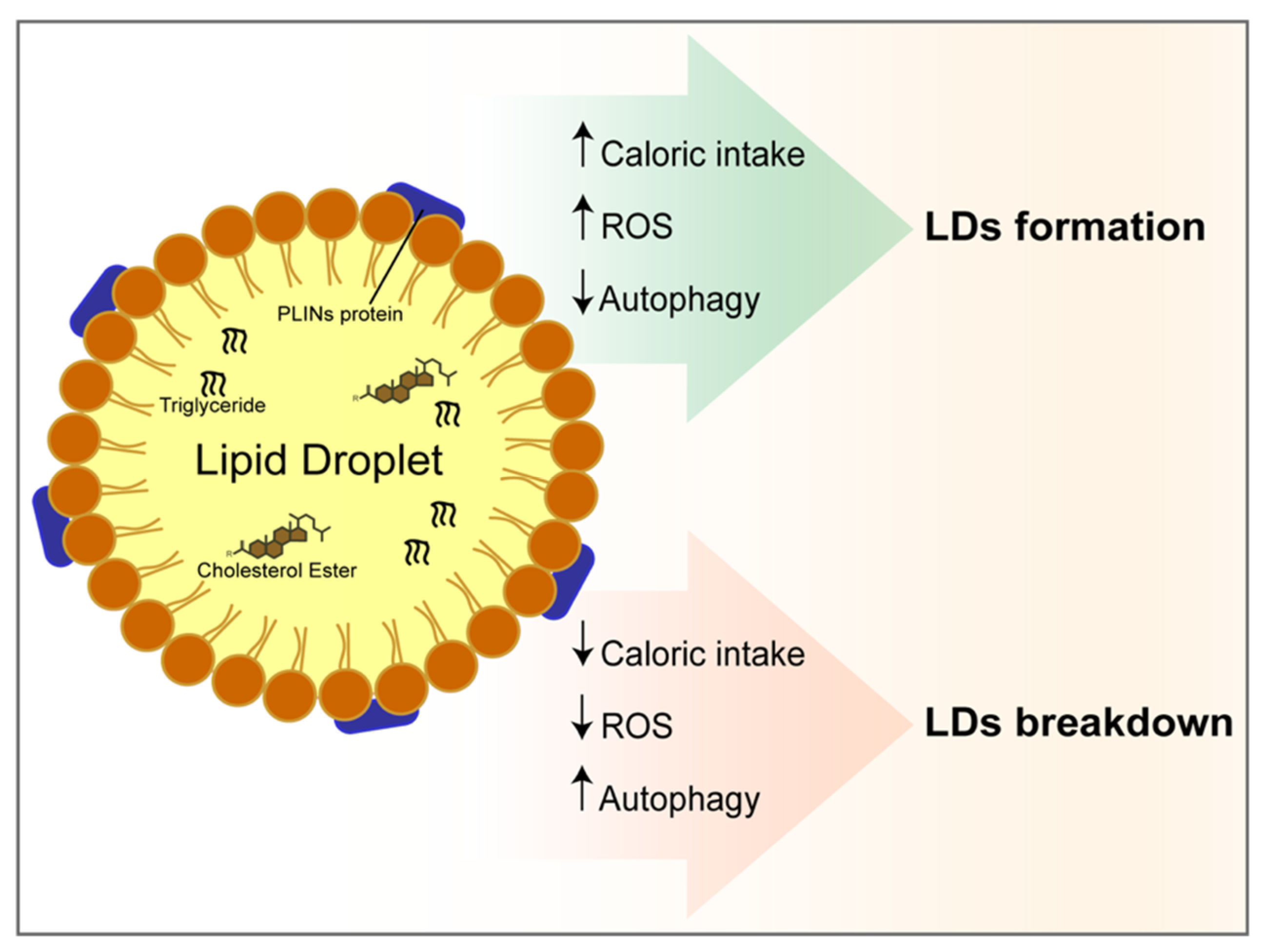{kind=link}
{kind=link}
{kind=link}
Abstract
1. Autophagy Machinery
2. Cell Types within the Liver
3. Autophagy Involvement in Lipid Droplets Turnover
4. The Autophagy at the Crossroad between Oxidative Stress, Lipids Accumulation and Cell Death in a Hypercaloric Diet
5. Liver, Diet Habits and Autophagy
- -
- -
- caffeic acid, found in many vegetables, an autophagy inducer which ameliorates hepatic steatosis [78];
- -
- resveratrol, a non-flavonoid polyphenol found in grapes and red wine, showing proautophagy action and reducing lipid metabolism disorder [79];
- -
- curcumin, a polyphenolic compound present in Curcuma longa, an antioxidant, apoptosis-inhibitor and autophagy-inducer with a protective effect on hepatocellular carcinoma [80] and under evaluation in clinical trials on NAFLD [81].The role diet plays in autophagy control is currently investigated and a large debate is ongoing on the actual role that diets and healthy foods may have on the health [82].
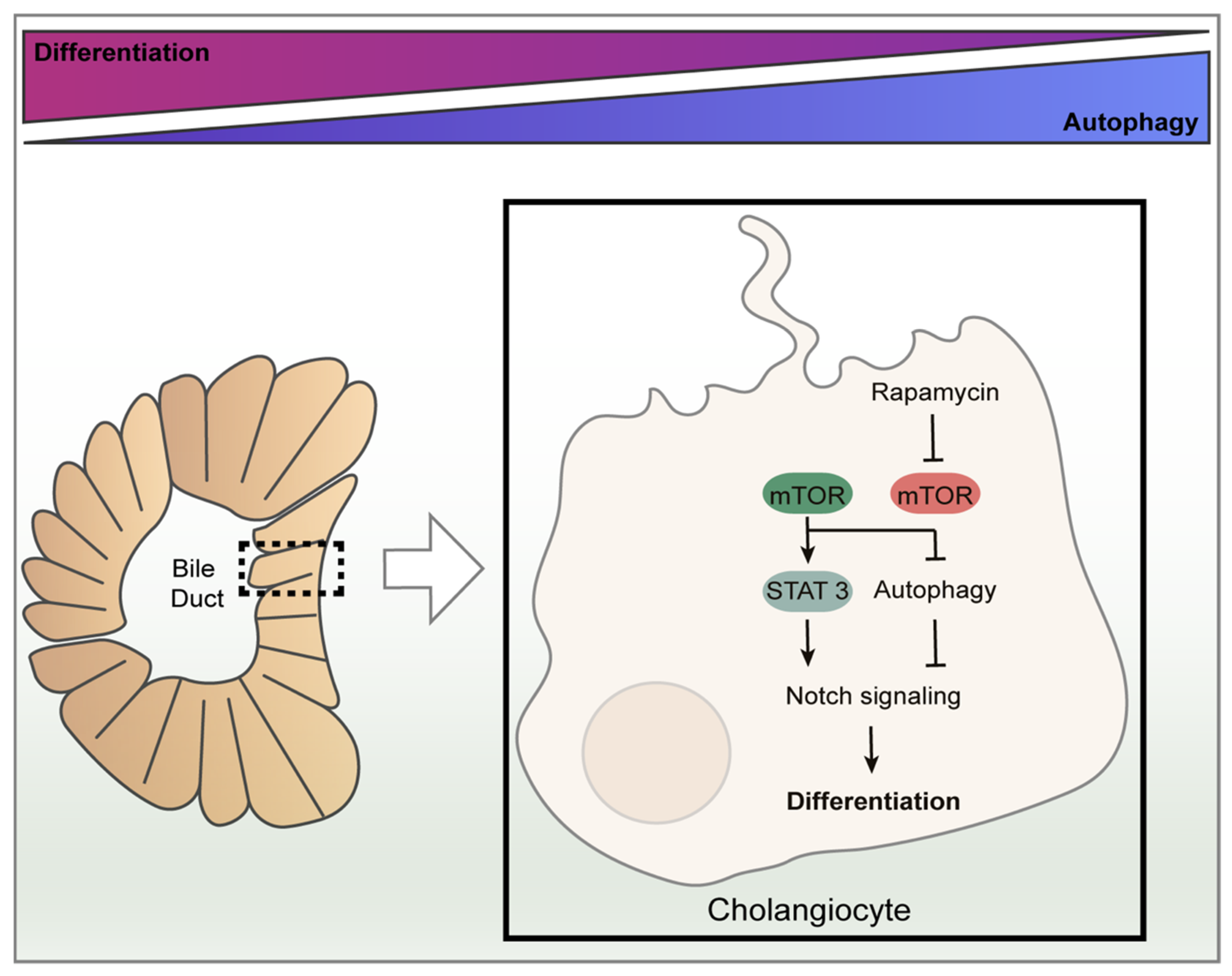
6. The Role of Autophagy on Biliary Epithelium Differentiation and Homeostasis
7. The Role of Autophagy in Endothelial Cells Maintainance
8. The Role of Liver Autophagy in Preserving Systemic Homeostasis Counteracting Cancer Growth
9. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Deter, R.L.; Baudhuin, P.; De Duve, C. Participation of lysosomes in cellular autophagy induced in rat liver by glucagon. J. Cell Biol. 1967, 35, C11–C16. [Google Scholar] [CrossRef] [PubMed]
- Tsukada, M.; Ohsumi, Y. Isolation and characterization of autophagy-defective mutants of Saccharomyces cerevisiae. FEBS Lett. 1993, 333, 169–174. [Google Scholar] [CrossRef]
- Zientara-Rytter, K.; Subramani, S. The Roles of Ubiquitin-Binding Protein Shuttles in the Degradative Fate of Ubiquitinated Proteins in the Ubiquitin-Proteasome System and Autophagy. Cells 2019, 8, 40. [Google Scholar] [CrossRef] [PubMed]
- Giampietri, C.; Petrungaro, S.; Cordella, M.; Tabolacci, C.; Tomaipitinca, L.; Facchiano, A.; Eramo, A.; Filippini, A.; Facchiano, F.; Ziparo, E. Lipid Storage and Autophagy in Melanoma Cancer Cells. Int. J. Mol. Sci. 2017, 18, 1271. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Yao, Z.; Chen, Y.; Qian, L.; Jiang, S.; Zhou, J.; Shao, J.; Chen, A.; Zhang, F.; Zheng, S. Lipophagy and liver disease: New perspectives to better understanding and therapy. Biomed. Pharmacother. 2018, 97, 339–348. [Google Scholar] [CrossRef]
- Hase, K.; Fujiwara, Y.; Kikuchi, H.; Aizawa, S.; Hakuno, F.; Takahashi, S.; Wada, K.; Kabuta, T. RNautophagy/DNautophagy possesses selectivity for RNA/DNA substrates. Nucleic Acids Res. 2015, 43, 6439–6449. [Google Scholar] [CrossRef] [PubMed]
- Sedlackova, L.; Korolchuk, V.I. Mitochondrial quality control as a key determinant of cell survival. Biochim. Biophys. Acta Mol. Cell Res. 2018, 1866, 575–587. [Google Scholar] [CrossRef]
- Beau, I.; Esclatine, A.; Codogno, P. Lost to translation: When autophagy targets mature ribosomes. Trends Cell Biol. 2008, 18, 311–314. [Google Scholar] [CrossRef]
- Grumati, P.; Dikic, I.; Stolz, A. ER-phagy at a glance. J. Cell Sci. 2018, 131. [Google Scholar] [CrossRef] [PubMed]
- Kwon, D.H.; Song, H.K. A Structural View of Xenophagy, a Battle between Host and Microbes. Mol. Cells 2018, 41, 27–34. [Google Scholar] [CrossRef]
- Mizushima, N. A brief history of autophagy from cell biology to physiology and disease. Nat. Cell Biol. 2018, 20, 521–527. [Google Scholar] [CrossRef]
- Mizushima, N. Autophagy: Process and function. Genes Dev. 2007, 21, 2861–2873. [Google Scholar] [CrossRef]
- Kon, M.; Cuervo, A.M. Chaperone-mediated autophagy in health and disease. FEBS Lett. 2010, 584, 1399–1404. [Google Scholar] [CrossRef]
- Klionsky, D.J.; Abdelmohsen, K.; Abe, A.; Abedin, M.J.; Abeliovich, H.; Acevedo Arozena, A.; Adachi, H.; Adams, C.M.; Adams, P.D.; Adeli, K.; et al. Guidelines for the use and interpretation of assays for monitoring autophagy (3rd edition). Autophagy 2016, 12, 1–222. [Google Scholar] [CrossRef]
- Roux, P.P.; Topisirovic, I. Signaling Pathways Involved in the Regulation of mRNA Translation. Mol. Cell. Biol. 2018, 38, e00070-18. [Google Scholar] [CrossRef]
- Hosokawa, N.; Hara, T.; Kaizuka, T.; Kishi, C.; Takamura, A.; Miura, Y.; Iemura, S.; Natsume, T.; Takehana, K.; Yamada, N.; et al. Nutrient-dependent mTORC1 association with the ULK1-Atg13-FIP200 complex required for autophagy. Mol. Biol. Cell 2009, 20, 1981–1991. [Google Scholar] [CrossRef]
- Paquette, M.; El-Houjeiri, L.; Pause, A. mTOR Pathways in Cancer and Autophagy. Cancers 2018, 10, 18. [Google Scholar] [CrossRef]
- Boutouja, F.; Brinkmeier, R.; Mastalski, T.; El Magraoui, F.; Platta, H.W. Regulation of the Tumor-Suppressor BECLIN 1 by Distinct Ubiquitination Cascades. Int. J. Mol. Sci. 2017, 18, 2541. [Google Scholar] [CrossRef]
- D’Arcangelo, D.; Giampietri, C.; Muscio, M.; Scatozza, F.; Facchiano, F.; Facchiano, A. WIPI1, BAG1, and PEX3 Autophagy-Related Genes Are Relevant Melanoma Markers. Oxid. Med. Cell. Longev. 2018, 2018, 1471682. [Google Scholar] [CrossRef]
- Dooley, H.C.; Razi, M.; Polson, H.E.; Girardin, S.E.; Wilson, M.I.; Tooze, S.A. WIPI2 links LC3 conjugation with PI3P, autophagosome formation, and pathogen clearance by recruiting Atg12-5-16L1. Mol. Cell 2014, 55, 238–252. [Google Scholar] [CrossRef]
- Ohsumi, Y. Molecular dissection of autophagy: Two ubiquitin-like systems. Nat. Rev. Mol. Cell Biol. 2001, 2, 211–216. [Google Scholar] [CrossRef]
- Lee, Y.K.; Lee, J.A. Role of the mammalian ATG8/LC3 family in autophagy: Differential and compensatory roles in the spatiotemporal regulation of autophagy. BMB Rep. 2016, 49, 424–430. [Google Scholar] [CrossRef]
- Nakamura, S.; Yoshimori, T. New insights into autophagosome-lysosome fusion. J. Cell Sci. 2017, 130, 1209–1216. [Google Scholar] [CrossRef]
- Chiang, H.L.; Terlecky, S.R.; Plant, C.P.; Dice, J.F. A role for a 70-kilodalton heat shock protein in lysosomal degradation of intracellular proteins. Science 1989, 246, 382–385. [Google Scholar] [CrossRef]
- Rout, A.K.; Strub, M.P.; Piszczek, G.; Tjandra, N. Structure of transmembrane domain of lysosome-associated membrane protein type 2a (LAMP-2A) reveals key features for substrate specificity in chaperone-mediated autophagy. J. Biol. Chem. 2014, 289, 35111–35123. [Google Scholar] [CrossRef]
- Kaushik, S.; Cuervo, A.M. The coming of age of chaperone-mediated autophagy. Nat. Rev. Mol. Cell Biol. 2018, 19, 365–381. [Google Scholar] [CrossRef]
- Todde, V.; Veenhuis, M.; van der Klei, I.J. Autophagy: Principles and significance in health and disease. Biochim. Biophys. Acta 2009, 1792, 3–13. [Google Scholar] [CrossRef]
- Oku, M.; Sakai, Y. Three Distinct Types of Microautophagy Based on Membrane Dynamics and Molecular Machineries. Bioessays 2018, 40, e1800008. [Google Scholar] [CrossRef]
- Takiguchi, M. The C/EBP family of transcription factors in the liver and other organs. Int. J. Exp. Pathol. 1998, 79, 369–391. [Google Scholar] [CrossRef]
- Kmiec, Z. Cooperation of liver cells in health and disease. Adv. Anat. Embryol. Cell Biol. 2001, 161, 1–151. [Google Scholar]
- Fraczek, J.; Bolleyn, J.; Vanhaecke, T.; Rogiers, V.; Vinken, M. Primary hepatocyte cultures for pharmaco-toxicological studies: At the busy crossroad of various anti-dedifferentiation strategies. Arch. Toxicol. 2013, 87, 577–610. [Google Scholar] [CrossRef]
- Gaudio, E.; Onori, P.; Franchitto, A.; Sferra, R.; Riggio, O. Liver metabolic zonation and hepatic microcirculation in carbon tetrachloride-induced experimental cirrhosis. Dig. Dis. Sci. 1997, 42, 167–177. [Google Scholar] [CrossRef]
- Tanimizu, N.; Mitaka, T. Morphogenesis of liver epithelial cells. Hepatol. Res. 2016, 46, 964–976. [Google Scholar] [CrossRef]
- Sato, K.; Meng, F.; Giang, T.; Glaser, S.; Alpini, G. Mechanisms of cholangiocyte responses to injury. Biochim. Biophys. Acta Mol. Basis Dis. 2018, 1864, 1262–1269. [Google Scholar] [CrossRef]
- Bouwens, L.; De Bleser, P.; Vanderkerken, K.; Geerts, B.; Wisse, E. Liver cell heterogeneity: Functions of non-parenchymal cells. Enzyme 1992, 46, 155–168. [Google Scholar] [CrossRef]
- Dixon, L.J.; Barnes, M.; Tang, H.; Pritchard, M.T.; Nagy, L.E. Kupffer cells in the liver. Compr. Physiol. 2013, 3, 785–797. [Google Scholar] [CrossRef]
- Ren, L.; Qi, K.; Zhang, L.; Bai, Z.; Ren, C.; Xu, X.; Zhang, Z.; Li, X. Glutathione Might Attenuate Cadmium-Induced Liver Oxidative Stress and Hepatic Stellate Cell Activation. Biol. Trace Elem. Res. 2019. [Google Scholar] [CrossRef]
- Nakatani, K.; Kaneda, K.; Seki, S.; Nakajima, Y. Pit cells as liver-associated natural killer cells: Morphology and function. Med. Electron Microsc. 2004, 37, 29–36. [Google Scholar] [CrossRef]
- Sorensen, K.K.; Simon-Santamaria, J.; McCuskey, R.S.; Smedsrod, B. Liver Sinusoidal Endothelial Cells. Compr. Physiol. 2015, 5, 1751–1774. [Google Scholar] [CrossRef]
- Gera, S.; Ettel, M.; Acosta-Gonzalez, G.; Xu, R. Clinical features, histology, and histogenesis of combined hepatocellular-cholangiocarcinoma. World J. Hepatol. 2017, 9, 300–309. [Google Scholar] [CrossRef]
- Castorina, S.; Luca, T.; Torrisi, A.; Privitera, G.; Panebianco, M. Isolation of epithelial cells with hepatobiliary phenotype. Ital. J. Anat. Embryol. 2008, 113, 199–207. [Google Scholar]
- Weiskirchen, R.; Tacke, F. Relevance of Autophagy in Parenchymal and Non-Parenchymal Liver Cells for Health and Disease. Cells 2019, 8, 16. [Google Scholar] [CrossRef]
- Allaire, M.; Rautou, P.E.; Codogno, P.; Lotersztajn, S. Autophagy in liver diseases: Time for translation? J. Hepatol. 2019. [Google Scholar] [CrossRef]
- Schulze, R.J.; Rasineni, K.; Weller, S.G.; Schott, M.B.; Schroeder, B.; Casey, C.A.; McNiven, M.A. Ethanol exposure inhibits hepatocyte lipophagy by inactivating the small guanosine triphosphatase Rab7. Hepatol. Commun. 2017, 1, 140–152. [Google Scholar] [CrossRef]
- Sharma, L.; Lone, N.A.; Knott, R.M.; Hassan, A.; Abdullah, T. Trigonelline prevents high cholesterol and high fat diet induced hepatic lipid accumulation and lipo-toxicity in C57BL/6J mice, via restoration of hepatic autophagy. Food Chem. Toxicol. 2018, 121, 283–296. [Google Scholar] [CrossRef]
- Schroeder, B.; Schulze, R.J.; Weller, S.G.; Sletten, A.C.; Casey, C.A.; McNiven, M.A. The small GTPase Rab7 as a central regulator of hepatocellular lipophagy. Hepatology 2015, 61, 1896–1907. [Google Scholar] [CrossRef]
- Brasaemle, D.L. Thematic review series: Adipocyte biology. The perilipin family of structural lipid droplet proteins: Stabilization of lipid droplets and control of lipolysis. J. Lipid Res. 2007, 48, 2547–2559. [Google Scholar] [CrossRef]
- Brasaemle, D.L.; Wolins, N.E. Packaging of fat: An evolving model of lipid droplet assembly and expansion. J. Biol. Chem. 2012, 287, 2273–2279. [Google Scholar] [CrossRef]
- Tsai, T.H.; Chen, E.; Li, L.; Saha, P.; Lee, H.J.; Huang, L.S.; Shelness, G.S.; Chan, L.; Chang, B.H. The constitutive lipid droplet protein PLIN2 regulates autophagy in liver. Autophagy 2017, 13, 1130–1144. [Google Scholar] [CrossRef]
- Zechner, R.; Kienesberger, P.C.; Haemmerle, G.; Zimmermann, R.; Lass, A. Adipose triglyceride lipase and the lipolytic catabolism of cellular fat stores. J. Lipid Res. 2009, 50, 3–21. [Google Scholar] [CrossRef]
- Liu, X.; Liang, Y.; Song, R.; Yang, G.; Han, J.; Lan, Y.; Pan, S.; Zhu, M.; Liu, Y.; Wang, Y.; et al. Long non-coding RNA NEAT1-modulated abnormal lipolysis via ATGL drives hepatocellular carcinoma proliferation. Mol. Cancer 2018, 17, 90. [Google Scholar] [CrossRef]
- Lee, I.H.; Cao, L.; Mostoslavsky, R.; Lombard, D.B.; Liu, J.; Bruns, N.E.; Tsokos, M.; Alt, F.W.; Finkel, T. A role for the NAD-dependent deacetylase Sirt1 in the regulation of autophagy. Proc. Natl. Acad. Sci. USA 2008, 105, 3374–3379. [Google Scholar] [CrossRef]
- Jiang, F. Autophagy in vascular endothelial cells. Clin. Exp. Pharmacol. Physiol. 2016, 43, 1021–1028. [Google Scholar] [CrossRef]
- Pfluger, P.T.; Herranz, D.; Velasco-Miguel, S.; Serrano, M.; Tschop, M.H. Sirt1 protects against high-fat diet-induced metabolic damage. Proc. Natl. Acad. Sci. USA 2008, 105, 9793–9798. [Google Scholar] [CrossRef]
- Di Leo, L.; Vegliante, R.; Ciccarone, F.; Salvatori, I.; Scimeca, M.; Bonanno, E.; Sagnotta, A.; Grazi, G.L.; Aquilano, K.; Ciriolo, M.R. Forcing ATGL expression in hepatocarcinoma cells imposes glycolytic rewiring through PPAR-alpha/p300-mediated acetylation of p53. Oncogene 2018. [Google Scholar] [CrossRef]
- Jiao, M.; Ren, F.; Zhou, L.; Zhang, X.; Zhang, L.; Wen, T.; Wei, L.; Wang, X.; Shi, H.; Bai, L.; et al. Peroxisome proliferator-activated receptor alpha activation attenuates the inflammatory response to protect the liver from acute failure by promoting the autophagy pathway. Cell Death Dis. 2014, 5, e1397. [Google Scholar] [CrossRef]
- Lee, J.M.; Wagner, M.; Xiao, R.; Kim, K.H.; Feng, D.; Lazar, M.A.; Moore, D.D. Nutrient-sensing nuclear receptors coordinate autophagy. Nature 2014, 516, 112–115. [Google Scholar] [CrossRef]
- Khan, S.A.; Sathyanarayan, A.; Mashek, M.T.; Ong, K.T.; Wollaston-Hayden, E.E.; Mashek, D.G. ATGL-catalyzed lipolysis regulates SIRT1 to control PGC-1alpha/PPAR-alpha signaling. Diabetes 2015, 64, 418–426. [Google Scholar] [CrossRef]
- Vegliante, R.; Di Leo, L.; Ciccarone, F.; Ciriolo, M.R. Hints on ATGL implications in cancer: Beyond bioenergetic clues. Cell Death Dis. 2018, 9, 316. [Google Scholar] [CrossRef]
- Sharp, K.P.H.; Schultz, M.; Coppell, K.J. Is non-alcoholic fatty liver disease a reflection of what we eat or simply how much we eat? JGH Open 2018, 2, 59–74. [Google Scholar] [CrossRef]
- Jin, Y.; Tan, Y.; Chen, L.; Liu, Y.; Ren, Z. Reactive Oxygen Species Induces Lipid Droplet Accumulation in HepG2 Cells by Increasing Perilipin 2 Expression. Int. J. Mol. Sci. 2018, 19, 3445. [Google Scholar] [CrossRef]
- Sekiya, M.; Hiraishi, A.; Touyama, M.; Sakamoto, K. Oxidative stress induced lipid accumulation via SREBP1c activation in HepG2 cells. Biochem. Biophys. Res. Commun. 2008, 375, 602–607. [Google Scholar] [CrossRef]
- Filomeni, G.; De Zio, D.; Cecconi, F. Oxidative stress and autophagy: The clash between damage and metabolic needs. Cell Death Differ. 2015, 22, 377–388. [Google Scholar] [CrossRef]
- Singh, R.; Kaushik, S.; Wang, Y.; Xiang, Y.; Novak, I.; Komatsu, M.; Tanaka, K.; Cuervo, A.M.; Czaja, M.J. Autophagy regulates lipid metabolism. Nature 2009, 458, 1131–1135. [Google Scholar] [CrossRef]
- Yang, L.; Li, P.; Fu, S.; Calay, E.S.; Hotamisligil, G.S. Defective hepatic autophagy in obesity promotes ER stress and causes insulin resistance. Cell Metab. 2010, 11, 467–478. [Google Scholar] [CrossRef]
- Thomes, P.G.; Trambly, C.S.; Fox, H.S.; Tuma, D.J.; Donohue, T.M., Jr. Acute and Chronic Ethanol Administration Differentially Modulate Hepatic Autophagy and Transcription Factor EB. Alcohol. Clin. Exp. Res. 2015, 39, 2354–2363. [Google Scholar] [CrossRef]
- Kharbanda, K.K.; McVicker, D.L.; Zetterman, R.K.; Donohue, T.M., Jr. Ethanol consumption reduces the proteolytic capacity and protease activities of hepatic lysosomes. Biochim. Biophys. Acta 1995, 1245, 421–429. [Google Scholar] [CrossRef]
- Ding, W.X.; Li, M.; Chen, X.; Ni, H.M.; Lin, C.W.; Gao, W.; Lu, B.; Stolz, D.B.; Clemens, D.L.; Yin, X.M. Autophagy reduces acute ethanol-induced hepatotoxicity and steatosis in mice. Gastroenterology 2010, 139, 1740–1752. [Google Scholar] [CrossRef]
- Thomes, P.G.; Benbow, J.H.; Brandon-Warner, E.; Thompson, K.J.; Jacobs, C.; Donohue, T.M., Jr.; Schrum, L.W. Dietary fructose augments ethanol-induced liver pathology. J. Nutr. Biochem. 2017, 43, 141–150. [Google Scholar] [CrossRef]
- Sanchez-Roman, I.; Barja, G. Regulation of longevity and oxidative stress by nutritional interventions: Role of methionine restriction. Exp. Gerontol. 2013, 48, 1030–1042. [Google Scholar] [CrossRef]
- Mancinelli, R.; Carpino, G.; Petrungaro, S.; Mammola, C.L.; Tomaipitinca, L.; Filippini, A.; Facchiano, A.; Ziparo, E.; Giampietri, C. Multifaceted Roles of GSK-3 in Cancer and Autophagy-Related Diseases. Oxid. Med. Cell. Longev. 2017, 2017, 4629495. [Google Scholar] [CrossRef]
- Cingolani, F.; Czaja, M.J. Regulation and Functions of Autophagic Lipolysis. Trends Endocrinol. Metab. 2016, 27, 696–705. [Google Scholar] [CrossRef]
- Zhang, E.; Yin, S.; Song, X.; Fan, L.; Hu, H. Glycycoumarin inhibits hepatocyte lipoapoptosis through activation of autophagy and inhibition of ER stress/GSK-3-mediated mitochondrial pathway. Sci. Rep. 2016, 6, 38138. [Google Scholar] [CrossRef]
- Bogdan, A.R.; Miyazawa, M.; Hashimoto, K.; Tsuji, Y. Regulators of Iron Homeostasis: New Players in Metabolism, Cell Death, and Disease. Trends Biochem. Sci. 2016, 41, 274–286. [Google Scholar] [CrossRef]
- Bai, Y.; Meng, L.; Han, L.; Jia, Y.; Zhao, Y.; Gao, H.; Kang, R.; Wang, X.; Tang, D.; Dai, E. Lipid storage and lipophagy regulates ferroptosis. Biochem. Biophys. Res. Commun. 2018. [Google Scholar] [CrossRef]
- Zhuang, J.; Lu, J.; Wang, X.; Wang, X.; Hu, W.; Hong, F.; Zhao, X.X.; Zheng, Y.L. Purple sweet potato color protects against high-fat diet-induced cognitive deficits through AMPK-mediated autophagy in mouse hippocampus. J. Nutr. Biochem. 2019, 65, 35–45. [Google Scholar] [CrossRef]
- Wang, X.; Zhang, Z.F.; Zheng, G.H.; Wang, A.M.; Sun, C.H.; Qin, S.P.; Zhuang, J.; Lu, J.; Ma, D.F.; Zheng, Y.L. The Inhibitory Effects of Purple Sweet Potato Color on Hepatic Inflammation Is Associated with Restoration of NAD(+) Levels and Attenuation of NLRP3 Inflammasome Activation in High-Fat-Diet-Treated Mice. Molecules 2017, 22, 1315. [Google Scholar] [CrossRef]
- Kim, H.M.; Kim, Y.; Lee, E.S.; Huh, J.H.; Chung, C.H. Caffeic acid ameliorates hepatic steatosis and reduces ER stress in high fat diet-induced obese mice by regulating autophagy. Nutrition 2018, 55–56, 63–70. [Google Scholar] [CrossRef]
- Ding, S.; Jiang, J.; Zhang, G.; Bu, Y.; Zhang, G.; Zhao, X. Resveratrol and caloric restriction prevent hepatic steatosis by regulating SIRT1-autophagy pathway and alleviating endoplasmic reticulum stress in high-fat diet-fed rats. PLoS ONE 2017, 12, e0183541. [Google Scholar] [CrossRef]
- Elmansi, A.M.; El-Karef, A.A.; Shishtawy, M.; Eissa, L.A. Hepatoprotective Effect of Curcumin on Hepatocellular Carcinoma Through Autophagic and Apoptic Pathways. Ann. Hepatol. 2017, 16, 607–618. [Google Scholar] [CrossRef]
- Yan, S.; Huda, N.; Khambu, B.; Yin, X.M. Relevance of autophagy to fatty liver diseases and potential therapeutic applications. Amino Acids 2017, 49, 1965–1979. [Google Scholar] [CrossRef]
- Willett, W.; Rockstrom, J.; Loken, B.; Springmann, M.; Lang, T.; Vermeulen, S.; Garnett, T.; Tilman, D.; DeClerck, F.; Wood, A.; et al. Food in the Anthropocene: The EAT-Lancet Commission on healthy diets from sustainable food systems. Lancet 2019, 393, 447–492. [Google Scholar] [CrossRef]
- Han, Y.; Onori, P.; Meng, F.; DeMorrow, S.; Venter, J.; Francis, H.; Franchitto, A.; Ray, D.; Kennedy, L.; Greene, J.; et al. Prolonged exposure of cholestatic rats to complete dark inhibits biliary hyperplasia and liver fibrosis. Am. J. Physiol. Gastrointest. Liver Physiol. 2014, 307, G894–G904. [Google Scholar] [CrossRef]
- Salemi, S.; Yousefi, S.; Constantinescu, M.A.; Fey, M.F.; Simon, H.U. Autophagy is required for self-renewal and differentiation of adult human stem cells. Cell Res. 2012, 22, 432–435. [Google Scholar] [CrossRef]
- Cheng, Y.; Wang, B.; Zhou, H.; Dang, S.; Jin, M.; Shi, Y.; Hao, L.; Yang, Z.; Zhang, Y. Autophagy is Required for the Maintenance of Liver Progenitor Cell Functionality. Cell. Physiol. Biochem. 2015, 36, 1163–1174. [Google Scholar] [CrossRef]
- Chen, N.; Karantza, V. Autophagy as a therapeutic target in cancer. Cancer Biol. Ther. 2011, 11, 157–168. [Google Scholar] [CrossRef]
- Zong, Y.; Panikkar, A.; Xu, J.; Antoniou, A.; Raynaud, P.; Lemaigre, F.; Stanger, B.Z. Notch signaling controls liver development by regulating biliary differentiation. Development 2009, 136, 1727–1739. [Google Scholar] [CrossRef]
- Hildebrand, D.; Uhle, F.; Sahin, D.; Krauser, U.; Weigand, M.A.; Heeg, K. The Interplay of Notch Signaling and STAT3 in TLR-Activated Human Primary Monocytes. Front. Cell. Infect. Microbiol. 2018, 8, 241. [Google Scholar] [CrossRef]
- Zeng, J.; Jing, Y.; Shi, R.; Pan, X.; Lai, F.; Liu, W.; Li, R.; Gao, L.; Hou, X.; Wu, M.; et al. Autophagy regulates biliary differentiation of hepatic progenitor cells through Notch1 signaling pathway. Cell Cycle 2016, 15, 1602–1610. [Google Scholar] [CrossRef]
- Gonzalez-Rodriguez, A.; Mayoral, R.; Agra, N.; Valdecantos, M.P.; Pardo, V.; Miquilena-Colina, M.E.; Vargas-Castrillon, J.; Lo Iacono, O.; Corazzari, M.; Fimia, G.M.; et al. Impaired autophagic flux is associated with increased endoplasmic reticulum stress during the development of NAFLD. Cell Death Dis. 2014, 5, e1179. [Google Scholar] [CrossRef]
- Conti, S.; Petrungaro, S.; Marini, E.S.; Masciarelli, S.; Tomaipitinca, L.; Filippini, A.; Giampietri, C.; Ziparo, E. A novel role of c-FLIP protein in regulation of ER stress response. Cell Signal. 2016, 28, 1262–1269. [Google Scholar] [CrossRef]
- Marini, E.S.; Giampietri, C.; Petrungaro, S.; Conti, S.; Filippini, A.; Scorrano, L.; Ziparo, E. The endogenous caspase-8 inhibitor c-FLIPL regulates ER morphology and crosstalk with mitochondria. Cell Death Differ. 2015, 22, 1131–1143. [Google Scholar] [CrossRef][Green Version]
- Sasaki, M.; Nakanuma, Y. Bile Acids and Deregulated Cholangiocyte Autophagy in Primary Biliary Cholangitis. Dig. Dis. 2017, 35, 210–216. [Google Scholar] [CrossRef]
- Masyuk, A.I.; Masyuk, T.V.; Lorenzo Pisarello, M.J.; Ding, J.F.; Loarca, L.; Huang, B.Q.; LaRusso, N.F. Cholangiocyte autophagy contributes to hepatic cystogenesis in polycystic liver disease and represents a potential therapeutic target. Hepatology 2018, 67, 1088–1108. [Google Scholar] [CrossRef]
- Montagna, C.; Rizza, S.; Maiani, E.; Piredda, L.; Filomeni, G.; Cecconi, F. To eat, or NOt to eat: S-nitrosylation signaling in autophagy. FEBS J. 2016, 283, 3857–3869. [Google Scholar] [CrossRef]
- Guo, F.; Li, X.; Peng, J.; Tang, Y.; Yang, Q.; Liu, L.; Wang, Z.; Jiang, Z.; Xiao, M.; Ni, C.; et al. Autophagy regulates vascular endothelial cell eNOS and ET-1 expression induced by laminar shear stress in an ex vivo perfused system. Ann. Biomed. Eng. 2014, 42, 1978–1988. [Google Scholar] [CrossRef]
- Yang, Q.; Li, X.; Li, R.; Peng, J.; Wang, Z.; Jiang, Z.; Tang, X.; Peng, Z.; Wang, Y.; Wei, D. Low Shear Stress Inhibited Endothelial Cell Autophagy Through TET2 Downregulation. Ann. Biomed. Eng. 2016, 44, 2218–2227. [Google Scholar] [CrossRef]
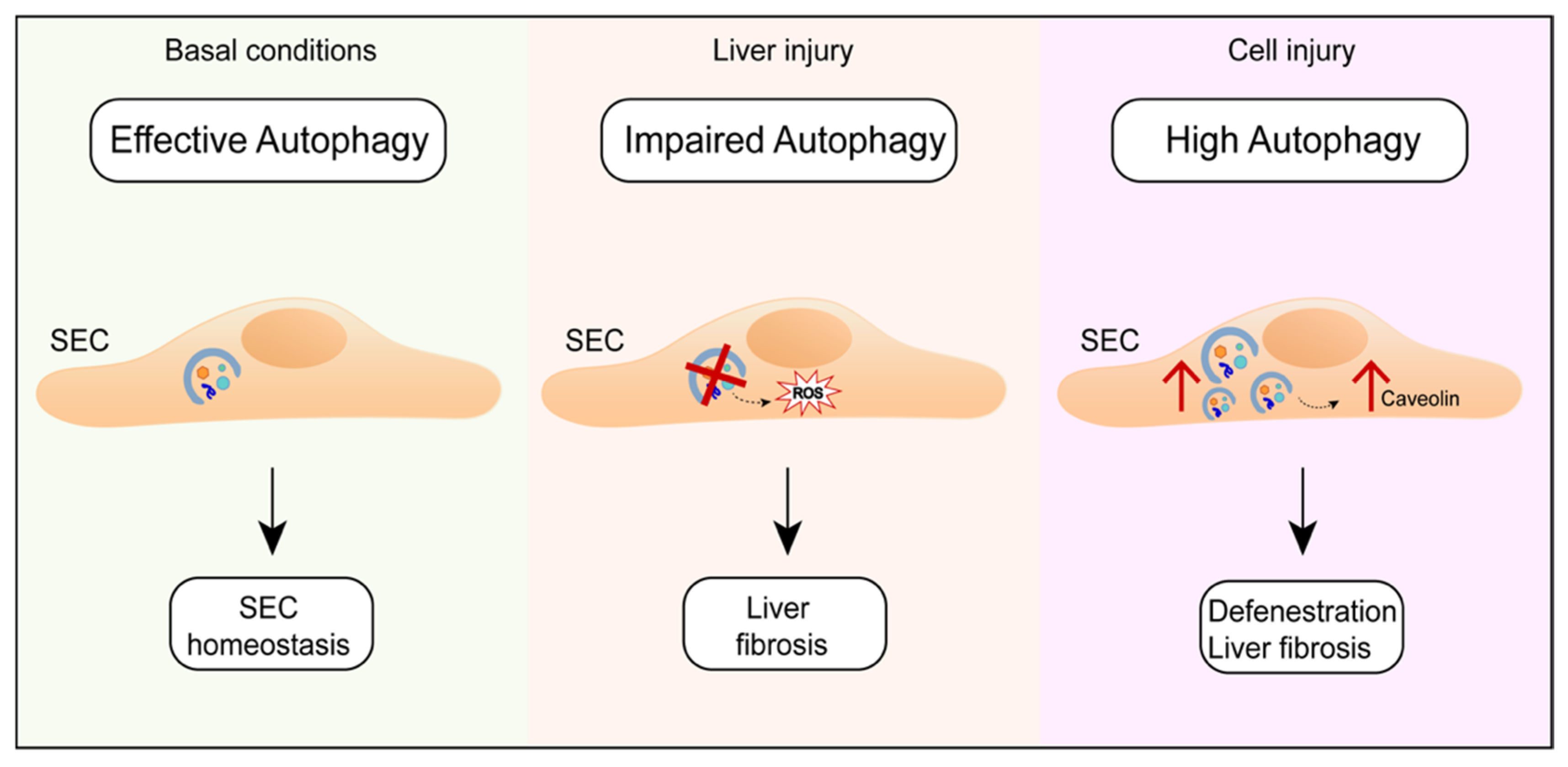
- Ruart, M.; Chavarria, L.; Camprecios, G.; Suarez-Herrera, N.; Montironi, C.; Guixe-Muntet, S.; Bosch, J.; Friedman, S.L.; Garcia-Pagan, J.C.; Hernandez-Gea, V. Impaired endothelial autophagy promotes liver fibrosis by aggravating the oxidative stress response during acute liver injury. J. Hepatol. 2018. [Google Scholar] [CrossRef]
- Luo, X.; Wang, D.; Zhu, X.; Wang, G.; You, Y.; Ning, Z.; Li, Y.; Jin, S.; Huang, Y.; Hu, Y.; et al. Autophagic degradation of caveolin-1 promotes liver sinusoidal endothelial cells defenestration. Cell Death Dis. 2018, 9, 576. [Google Scholar] [CrossRef]
- Boteon, Y.L.; Laing, R.; Mergental, H.; Reynolds, G.M.; Mirza, D.F.; Afford, S.C.; Bhogal, R.H. Mechanisms of autophagy activation in endothelial cell and their targeting during normothermic machine liver perfusion. World J. Gastroenterol. 2017, 23, 8443–8451. [Google Scholar] [CrossRef]
- Ezaki, J.; Matsumoto, N.; Takeda-Ezaki, M.; Komatsu, M.; Takahashi, K.; Hiraoka, Y.; Taka, H.; Fujimura, T.; Takehana, K.; Yoshida, M.; et al. Liver autophagy contributes to the maintenance of blood glucose and amino acid levels. Autophagy 2011, 7, 727–736. [Google Scholar] [CrossRef]
- Stefanadi, E.C.; Dimitrakakis, G.; Antoniou, C.K.; Challoumas, D.; Punjabi, N.; Dimitrakaki, I.A.; Punjabi, S.; Stefanadis, C.I. Metabolic syndrome and the skin: A more than superficial association. Reviewing the association between skin diseases and metabolic syndrome and a clinical decision algorithm for high risk patients. Diabetol. Metab. Syndr. 2018, 10, 9. [Google Scholar] [CrossRef]
- Key, T.J.; Spencer, E.A. Carbohydrates and cancer: An overview of the epidemiological evidence. Eur. J. Clin. Nutr. 2007, 61 (Suppl. 1), S112–S121. [Google Scholar] [CrossRef][Green Version]
- Dobbins, M.; Decorby, K.; Choi, B.C. The Association between Obesity and Cancer Risk: A Meta-Analysis of Observational Studies from 1985 to 2011. ISRN Prev. Med. 2013, 2013, 680536. [Google Scholar] [CrossRef]
- McQuade, J.L.; Daniel, C.R.; Hess, K.R.; Mak, C.; Wang, D.Y.; Rai, R.R.; Park, J.J.; Haydu, L.E.; Spencer, C.; Wongchenko, M.; et al. Association of body-mass index and outcomes in patients with metastatic melanoma treated with targeted therapy, immunotherapy, or chemotherapy: A retrospective, multicohort analysis. Lancet Oncol. 2018, 19, 310–322. [Google Scholar] [CrossRef]
- Sevim, D.G.; Kiratli, H. Serum adiponectin, insulin resistance, and uveal melanoma: Clinicopathological correlations. Melanoma Res. 2016, 26, 164–172. [Google Scholar] [CrossRef]
- Clement, E.; Lazar, I.; Muller, C.; Nieto, L. Obesity and melanoma: Could fat be fueling malignancy? Pigment Cell Melanoma Res. 2017, 30, 294–306. [Google Scholar] [CrossRef]
- Hayes, A.J.; Larkin, J. BMI and outcomes in melanoma: More evidence for the obesity paradox. Lancet Oncol. 2018, 19, 269–270. [Google Scholar] [CrossRef]
- Wang, Y.; Ding, W.X.; Li, T. Cholesterol and bile acid-mediated regulation of autophagy in fatty liver diseases and atherosclerosis. Biochim. Biophys. Acta Mol. Cell Biol. Lipids 2018, 1863, 726–733. [Google Scholar] [CrossRef]
- Kuzu, O.F.; Noory, M.A.; Robertson, G.P. The Role of Cholesterol in Cancer. Cancer Res. 2016, 76, 2063–2070. [Google Scholar] [CrossRef]
- Kuzu, O.F.; Gowda, R.; Sharma, A.; Robertson, G.P. Leelamine mediates cancer cell death through inhibition of intracellular cholesterol transport. Mol. Cancer Ther. 2014, 13, 1690–1703. [Google Scholar] [CrossRef]
- Poirot, M.; Silvente-Poirot, S. The tumor-suppressor cholesterol metabolite, dendrogenin A, is a new class of LXR modulator activating lethal autophagy in cancers. Biochem. Pharmacol. 2018, 153, 75–81. [Google Scholar] [CrossRef]
- Masouminia, M.; Gelfand, R.; Kovanecz, I.; Vernet, D.; Tsao, J.; Salas, R.; Castro, K.; Loni, L.; Rajfer, J.; Gonzalez-Cadavid, N.F. Dyslipidemia Is a Major Factor in Stem Cell Damage Induced by Uncontrolled Long-Term Type 2 Diabetes and Obesity in the Rat, as Suggested by the Effects on Stem Cell Culture. J. Sex. Med. 2018, 15, 1678–1697. [Google Scholar] [CrossRef]
- Lim, S.; Taskinen, M.R.; Boren, J. Crosstalk between nonalcoholic fatty liver disease and cardiometabolic syndrome. Obes. Rev. 2018. [Google Scholar] [CrossRef]
- Korenblat, K.M.; Fabbrini, E.; Mohammed, B.S.; Klein, S. Liver, muscle, and adipose tissue insulin action is directly related to intrahepatic triglyceride content in obese subjects. Gastroenterology 2008, 134, 1369–1375. [Google Scholar] [CrossRef]
- Schneider, J.L.; Suh, Y.; Cuervo, A.M. Deficient chaperone-mediated autophagy in liver leads to metabolic dysregulation. Cell Metab. 2014, 20, 417–432. [Google Scholar] [CrossRef]
- Singh, R.; Cuervo, A.M. Lipophagy: Connecting autophagy and lipid metabolism. Int. J. Cell Biol. 2012, 2012, 282041. [Google Scholar] [CrossRef]
- Cuervo, A.M. Autophagy and aging: Keeping that old broom working. Trends Genet. 2008, 24, 604–612. [Google Scholar] [CrossRef]
- Kaushik, S.; Singh, R.; Cuervo, A.M. Autophagic pathways and metabolic stress. Diabetes Obes. Metab. 2010, 12 (Suppl. 2), 4–14. [Google Scholar] [CrossRef]
- Baldi, A.; Lombardi, D.; Russo, P.; Palescandolo, E.; De Luca, A.; Santini, D.; Baldi, F.; Rossiello, L.; Dell’Anna, M.L.; Mastrofrancesco, A.; et al. Ferritin contributes to melanoma progression by modulating cell growth and sensitivity to oxidative stress. Clin. Cancer Res. 2005, 11, 3175–3183. [Google Scholar] [CrossRef]
- Adams, P. Management of elevated serum ferritin levels. Gastroenterol. Hepatol. 2008, 4, 333–334. [Google Scholar]
- Hou, W.; Xie, Y.; Song, X.; Sun, X.; Lotze, M.T.; Zeh, H.J., 3rd; Kang, R.; Tang, D. Autophagy promotes ferroptosis by degradation of ferritin. Autophagy 2016, 12, 1425–1428. [Google Scholar] [CrossRef]
- Mancias, J.D.; Wang, X.; Gygi, S.P.; Harper, J.W.; Kimmelman, A.C. Quantitative proteomics identifies NCOA4 as the cargo receptor mediating ferritinophagy. Nature 2014, 509, 105–109. [Google Scholar] [CrossRef]
- Tang, M.; Chen, Z.; Wu, D.; Chen, L. Ferritinophagy/ferroptosis: Iron-related newcomers in human diseases. J. Cell. Physiol. 2018, 233, 9179–9190. [Google Scholar] [CrossRef]
- Goodall, E.F.; Haque, M.S.; Morrison, K.E. Increased serum ferritin levels in amyotrophic lateral sclerosis (ALS) patients. J. Neurol. 2008, 255, 1652–1656. [Google Scholar] [CrossRef]
- Nadjar, Y.; Gordon, P.; Corcia, P.; Bensimon, G.; Pieroni, L.; Meininger, V.; Salachas, F. Elevated serum ferritin is associated with reduced survival in amyotrophic lateral sclerosis. PLoS ONE 2012, 7, e45034. [Google Scholar] [CrossRef]
- Luo, Y.; Yang, X.; Shi, Q. The cytochrome P450 inhibitor SKF-525A disrupts autophagy in primary rat hepatocytes. Chem. Biol. Interact. 2016, 255, 55–62. [Google Scholar] [CrossRef]
- Zhu, X.; Ji, M.; Han, Y.; Guo, Y.; Zhu, W.; Gao, F.; Yang, X.; Zhang, C. PGRMC1-dependent autophagy by hyperoside induces apoptosis and sensitizes ovarian cancer cells to cisplatin treatment. Int. J. Oncol. 2017, 50, 835–846. [Google Scholar] [CrossRef]
- Petibone, D.M.; Majeed, W.; Casciano, D.A. Autophagy function and its relationship to pathology, clinical applications, drug metabolism and toxicity. J. Appl. Toxicol. 2017, 37, 23–37. [Google Scholar] [CrossRef]






© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tomaipitinca, L.; Mandatori, S.; Mancinelli, R.; Giulitti, F.; Petrungaro, S.; Moresi, V.; Facchiano, A.; Ziparo, E.; Gaudio, E.; Giampietri, C. The Role of Autophagy in Liver Epithelial Cells and Its Impact on Systemic Homeostasis. Nutrients 2019, 11, 827. https://doi.org/10.3390/nu11040827
Tomaipitinca L, Mandatori S, Mancinelli R, Giulitti F, Petrungaro S, Moresi V, Facchiano A, Ziparo E, Gaudio E, Giampietri C. The Role of Autophagy in Liver Epithelial Cells and Its Impact on Systemic Homeostasis. Nutrients. 2019; 11(4):827. https://doi.org/10.3390/nu11040827
Chicago/Turabian StyleTomaipitinca, Luana, Sara Mandatori, Romina Mancinelli, Federico Giulitti, Simonetta Petrungaro, Viviana Moresi, Antonio Facchiano, Elio Ziparo, Eugenio Gaudio, and Claudia Giampietri. 2019. "The Role of Autophagy in Liver Epithelial Cells and Its Impact on Systemic Homeostasis" Nutrients 11, no. 4: 827. https://doi.org/10.3390/nu11040827
APA StyleTomaipitinca, L., Mandatori, S., Mancinelli, R., Giulitti, F., Petrungaro, S., Moresi, V., Facchiano, A., Ziparo, E., Gaudio, E., & Giampietri, C. (2019). The Role of Autophagy in Liver Epithelial Cells and Its Impact on Systemic Homeostasis. Nutrients, 11(4), 827. https://doi.org/10.3390/nu11040827