Baicalein as a Potential Inhibitor against BACE1 and AChE: Mechanistic Comprehension through In Vitro and Computational Approaches

 ,
, 

Abstract
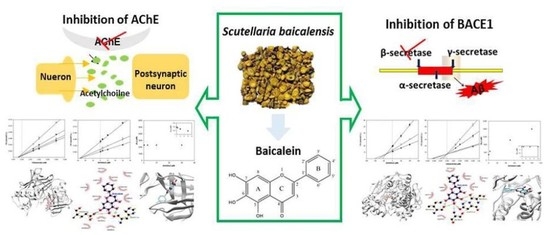
1. Introduction
2. Materials and Methods
2.1. Chemicals and Reagent
2.2. Enzymatic Assay for Biological Evaluation
2.3. Kinetic Studies of Baicalein on BACE1 and AChE
2.4. In Silico Molecular Docking Studies and Drug-Likeness Prediction
2.5. Statistical Analysis
3. Results and Discussion
3.1. In Vitro Inhibitory Activity of Baicalein against BACE1 and AChE
3.2. Enzyme Kinetic Analysis of BACE1 and AChE Inhibition
3.3. Selectivity of Baicalein against BACE1
3.4. In Silico Docking Simulation of Baicalein and Its Drug-Likeness Prediction
4. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Selkoe, D.J.; Hardy, J. The amyloid hypothesis of Alzheimer’s disease at 25 years. EMBO Mol. Med. 2016, 8, 595–608. [Google Scholar] [CrossRef]
- Vassar, R.; Kuhn, P.H.; Haass, C.; Kennedy, M.E.; Rajendran, L.; Wong, P.C.; Lichtenthaler, S.F. Function, therapeutic potential and cell biology of BACE proteases: Current status and future prospects. J. Neurochem. 2014, 130, 4–28. [Google Scholar] [CrossRef] [PubMed]
- Hussain, I.; Hawkins, J.; Harrison, D.; Hille, C.; Wayne, G.; Cutler, L.; Buck, T.; Walter, D.; Demont, E.; Howes, C.; et al. Oral administration of a potent and selective non-peptidic BACE-1 inhibitor decreases beta-cleavage of amyloid precursor protein and amyloid-beta production in vivo. J. Neurochem. 2007, 100, 802–809. [Google Scholar] [CrossRef]
- Roberds, S.L.; Anderson, J.; Basi, G.; Bienkowski, M.J.; Branstetter, D.G.; Chen, K.S.; Freedman, S.B.; Frigon, N.L.; Games, D.; Hu, K.; et al. BACE knockout mice are healthy despite lacking the primary beta-secretase activity in brain: Implications for Alzheimer’s disease therapeutics. Hum. Mol. Genet. 2001, 10, 1317–1324. [Google Scholar] [CrossRef] [PubMed]
- Sisodia, S.S.; St George-Hyslop, P.H. Gamma-Secretase, Notch, Abeta and Alzheimer’s disease: Where do the presenilins fit in? Nat. Rev. Neurosci. 2002, 3, 281–290. [Google Scholar] [CrossRef]
- Chang, W.P.; Huang, X.; Downs, D.; Cirrito, J.R.; Koelsch, G.; Holtzman, D.M.; Ghosh, A.K.; Tang, J. Beta-secretase inhibitor GRL-8234 rescues age-related cognitive decline in APP transgenic mice. FASEB J. 2011, 25, 775–784. [Google Scholar] [CrossRef] [PubMed]
- Inestrosa, N.C.; Dinamarca, M.C.; Alvarez, A. Amyloid-cholinesterase interactions. Implications for Alzheimer’s disease. FEBS J. 2008, 275, 625–632. [Google Scholar] [CrossRef] [PubMed]
- Vyas, N.A.; Singh, S.B.; Kumbhar, A.S.; Ranade, D.S.; Walke, G.R.; Kulkarni, P.P.; Jani, V.; Sonavane, U.B.; Joshi, R.R.; Rapole, S. Acetylcholinesterase and Aβ aggregation inhibition by heterometallic ruthenium(II)-platinum(II) polypyridyl complexes. Inorg. Chem. 2018, 57, 7524–7535. [Google Scholar] [CrossRef]
- Mushtaq, G.; Greig, N.H.; Khan, J.A.; Kamal, M.A. Status of acetylcholinesterase and butyrylcholinesterase in Alzheimer’s disease and type 2 diabetes mellitus. CNS Neurol. Disord. Drug Targets 2014, 13, 1432–1439. [Google Scholar] [CrossRef] [PubMed]
- Sharom, F.J. The P-glycoprotein multidrug transporter. Essays Biochem. 2011, 50, 161–178. [Google Scholar] [CrossRef]
- Liu, J.; Hou, J.; Jiang, C.; Li, G.; Lu, H.; Meng, F.; Shi, L. Deep sequencing of the Scutellaria baicalensis georgi transcriptome reveals flavonoid biosynthetic profiling and organ-specific gene expression. PLoS ONE 2015, 10, e0136397. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Zhao, J.; Hölscher, C. Therapeutic potential of baicalein in Alzheimer’s disease and Parkinson’s disease. CNS Drugs 2017, 31, 639–652. [Google Scholar] [CrossRef]
- Lebeau, A.; Esclaire, F.; Rostène, W.; Pélaprat, D. Baicalein protects cortical neurons from beta-amyloid (25–35) induced toxicity. Neuroreport 2001, 12, 2199–2202. [Google Scholar] [CrossRef]
- Chen, S.F.; Hsu, C.W.; Huang, W.H.; Wang, J.Y. Post-injury baicalein improves histological and functional outcomes and reduces inflammatory cytokines after experimental traumatic brain injury. Br. J. Pharmacol. 2008, 155, 1279–1296. [Google Scholar] [CrossRef]
- Zhou, L.; Tan, S.; Shan, Y.L.; Wang, Y.G.; Cai, W.; Huang, X.H.; Liao, X.Y.; Li, H.Y.; Zhang, L.; Zhang, B.J.; et al. Baicalein improves behavioral dysfunction induced by Alzheimer’s disease in rats. Neuropsychiatr. Dis. Treat. 2016, 12, 3145–3152. [Google Scholar] [CrossRef]
- Tsai, T.H.; Liu, S.C.; Tsai, P.L.; Ho, L.K.; Shum, A.Y.; Chen, C.F. The effects of the cyclosporin A, a P-glycoprotein inhibitor, on the pharmacokinetics of baicalein in the rat: A microdialysis study. Br. J. Pharmacol. 2002, 137, 1314–1320. [Google Scholar] [CrossRef] [PubMed]
- Fong, S.Y.; Wong, Y.C.; Zuo, Z. Development of a SPE-LC/MS/MS method for simultaneous quantification of baicalein, wogonin, oroxylin A and their glucuronides baicalin, wogonoside and oroxyloside in rats and its application to brain uptake and plasma pharmacokinetic studies. J. Pharm. Biomed. Anal. 2014, 97, 9–23. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.; Youn, K.; Lim, G.; Lee, J.; Jun, M. In Silico docking and In Vitro approaches towards BACE1 and cholinesterases inhibitory effect of citrus flavanones. Molecules 2018, 23, 1509. [Google Scholar] [CrossRef] [PubMed]
- Youn, K.; Jun, M. Biological evaluation and docking analysis of potent BACE1 inhibitors from Boesenbergia rotunda. Nutrients 2019, 11, 662. [Google Scholar] [CrossRef]
- Li, K.; Fan, H.; Yin, P.; Yang, L.; Xue, Q.; Li, X.; Sun, L.; Liu, Y. Structure-activity relationship of eight high content flavonoids analyzed with a preliminary assign-score method and their contribution to antioxidant ability of flavonoids-rich extract from Scutellaria baicalensis shoots. Arab. J. Chem. 2018, 11, 159–170. [Google Scholar] [CrossRef]
- Mouton-Liger, F.; Paquet, C.; Dumurgier, J.; Bouras, C.; Pradier, L.; Gray, F.; Hugon, J. Oxidative stress increases BACE1 protein levels through activation of the PKR-eIF2α pathway. Biochim. Biophys. Acta 2012, 1822, 885–896. [Google Scholar] [CrossRef]
- Choi, J.S.; Islam, M.N.; Ali, M.Y.; Kim, E.J.; Kim, Y.M.; Jung, H.A. Effects of C-glycosylation on anti-diabetic, anti-Alzheimer’s disease and anti-inflammatory potential of apigenin. Food Chem. Toxicol. 2014, 64, 27–33. [Google Scholar] [CrossRef]
- Cho, J.K.; Ryu, Y.B.; Curtis-Long, M.J.; Kim, J.Y.; Kim, D.; Lee, S.; Lee, W.S.; Park, K.H. Inhibition and structural reliability of prenylated flavones from the stem bark of Morus lhou on β-secretase (BACE-1). Bioorg. Med. Chem. Lett. 2011, 21, 2945–2948. [Google Scholar] [CrossRef] [PubMed]
- Shimizu, H.; Tosaki, A.; Kaneko, K.; Hisano, T.; Sakurai, T.; Nukina, N. Crystal structure of an active form of BACE1, an enzyme responsible for amyloid beta protein production. Mol. Cell Biol. 2008, 28, 3663–3671. [Google Scholar] [CrossRef]
- De Ferrari, G.V.; Mallender, W.D.; Inestrosa, N.C.; Rosenberry, T.L. Thioflavin T is a fluorescent probe of the acetylcholinesterase peripheral site that reveals conformational interactions between the peripheral and acylation sites. J. Biol. Chem. 2001, 276, 23282–23287. [Google Scholar] [CrossRef]
- Choy, Y.B.; Prausnitz, M.R. The rule of five for non-oral routes of drug delivery: Ophthalmic, inhalation and transdermal. Pharm. Res. 2011, 28, 943–948. [Google Scholar] [CrossRef]
- Heo, H.J.; Kim, D.O.; Choi, S.J.; Shin, D.H.; Lee, C.Y. Potent inhibitory effect of flavonoids in Scutellaria baicalensis on amyloid beta protein-induced neurotoxicity. J. Agric. Food Chem. 2004, 52, 4128–4132. [Google Scholar] [CrossRef]
- Zhang, S.; Ye, J.; Dong, G. Neuroprotective effect of baicalein on hydrogen peroxide-mediated oxidative stress and mitochondrial dysfunction in PC12 cells. J. Mol. Neurosci. 2010, 40, 311–320. [Google Scholar] [CrossRef]
- Zhang, S.Q.; Obregon, D.; Ehrhart, J.; Deng, J.; Tian, J.; Hou, H.; Giunta, B.; Sawmiller, D.; Tan, J. Baicalein reduces β-amyloid and promotes nonamyloidogenic amyloid precursor protein processing in an Alzheimer’s disease transgenic mouse model. J. Neurosci. Res. 2013, 91, 1239–1246. [Google Scholar] [CrossRef]
- Gu, X.H.; Xu, L.J.; Liu, Z.Q.; Wei, B.; Yang, Y.J.; Xu, G.G.; Yin, X.P.; Wang, W. The flavonoid baicalein rescues synaptic plasticity and memory deficits in a mouse model of Alzheimer’s disease. Behav. Brain Res. 2016, 311, 309–321. [Google Scholar] [CrossRef]
- Sowndhararajan, K.; Deepa, P.; Kim, M.; Park, S.J.; Kim, S. Baicalein as a potent neuroprotective agent: A review. Biomed. Pharmacother. 2017, 95, 1021–1032. [Google Scholar] [CrossRef]
- Pang, H.; Xue, W.; Shi, A.; Li, M.; Li, Y.; Cao, G.; Yan, B.; Dong, F.; Xiao, W.; He, G.; et al. Multiple-ascending-dose pharmacokinetics and safety evaluation of BAICALEIN chewable tablets in healthy Chinese volunteers. Clin. Drug Investig. 2016, 36, 713–724. [Google Scholar] [CrossRef] [PubMed]
- Pardridge, W.M. Transport of small molecules through the blood-brain barrier: Biology and methodology. Adv. Drug Delivery Rev. 1995, 15, 5–36. [Google Scholar] [CrossRef]
- Akao, T.; Kawabata, K.; Yanagisawa, E.; Ishihara, K.; Mizuhara, Y.; Wakui, Y.; Sakashita, Y.; Kobashi, K. Baicalin, the predominant flavone glucuronide of scutellariae radix, is absorbed from the rat gastrointestinal tract as the aglycone and restored to its original form. J. Pharm. Pharmacol. 2000, 52, 1563–1568. [Google Scholar] [CrossRef]
- Tian, S.; He, G.; Song, J.; Wang, S.; Xin, W.; Zhang, D.; Du, G. Pharmacokinetic study of baicalein after oral administration in monkeys. Fitoterapia 2012, 83, 532–540. [Google Scholar] [CrossRef] [PubMed]
- Lai, M.Y.; Hsiu, S.L.; Chen, C.C.; Hou, Y.C.; Chao, P.D. Urinary pharmacokinetics of baicalein, wogonin and their glycosides after oral administration of Scutellariae Radix in humans. Biol. Pharm. Bull. 2003, 26, 79–83. [Google Scholar] [CrossRef] [PubMed]
- Li, C.; Lin, G.; Zuo, Z. Pharmacological effects and pharmacokinetics properties of Radix Scutellariae and its bioactive flavones. Biopharm. Drug Dispos. 2011, 32, 427–445. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compounds | BACE1 | AChE | ||||
|---|---|---|---|---|---|---|
| IC50 1 | Ki Value 2 | Inhibition Type 3 | IC50 | Ki Value | Inhibition Type | |
| Baicalein | 23.71 ± 1.91 | 27.6 | Non-competitive | 45.95 ± 3.44 | 35.9 | Competitive |
| Resveratrol 4 | 15.04 ± 0.87 | |||||
| Galantamine 5 | 1.30 ± 0.01 | |||||
| Baicalein (μM) | TACE | Trypsin | Chymotrypsin | Elastase |
|---|---|---|---|---|
| 50 | 8.31 ± 0.89 | 3.40 ± 0.76 | 8.42 ± 0.76 | 10.29 ± 1.04 |
| 100 | 9.45 ± 0.71 | −6.95 ± 0.87 | 4.48 ± 0.76 | 9.00 ± 0.45 |
| Enzymes | Lowest Energy (Kcal/moL) | No. of H-Bond 1 | Residues | Bond Distance (Å) | Van der Waals Interacting Residues |
|---|---|---|---|---|---|
| BACE1 | −8.60 | 6 | Ser35 | 3.15 | Val69, Tyr71, Trp76, Phe108, Ile118, Ala127, Arg128 |
| Ser36 | 3.27 | ||||
| Asn37 | 2.85 and 2.88 | ||||
| Ile126 | 2.66 and 2.89 | ||||
| AChE | −8.70 | 3 | Glu202 | 2.88 and 2.94 | Tyr72, Asp74, Trp86, Asn87, Gly120, Gly121, Tyr124, Ser125, Ser203, Tyr337, Gly448 |
| His447 | 2.96 | ||||
| P-gp | −8.40 | 3 | Ser975 | 3.08 and 3.33 | Met68, Phe71, Phe332, Phe724, Phe728, Leu971, Phe974, Val978 |
| Ser725 | 2.85 |
| Compounds | No. of Violations | MW (g/moL) | H-Bond Acceptor | H-Bond Donor | Log Po/w 1 | TPSA 2 (Å2) | No. of rotb 3 |
|---|---|---|---|---|---|---|---|
| Baicalein | 0 | 270.24 | 5 | 3 | 2.682 | 90.89 | 1 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Han, J.; Ji, Y.; Youn, K.; Lim, G.; Lee, J.; Kim, D.H.; Jun, M. Baicalein as a Potential Inhibitor against BACE1 and AChE: Mechanistic Comprehension through In Vitro and Computational Approaches. Nutrients 2019, 11, 2694. https://doi.org/10.3390/nu11112694
Han J, Ji Y, Youn K, Lim G, Lee J, Kim DH, Jun M. Baicalein as a Potential Inhibitor against BACE1 and AChE: Mechanistic Comprehension through In Vitro and Computational Approaches. Nutrients. 2019; 11(11):2694. https://doi.org/10.3390/nu11112694
Chicago/Turabian StyleHan, Jin, Yeongseon Ji, Kumju Youn, GyuTae Lim, Jinhyuk Lee, Dong Hyun Kim, and Mira Jun. 2019. "Baicalein as a Potential Inhibitor against BACE1 and AChE: Mechanistic Comprehension through In Vitro and Computational Approaches" Nutrients 11, no. 11: 2694. https://doi.org/10.3390/nu11112694
APA StyleHan, J., Ji, Y., Youn, K., Lim, G., Lee, J., Kim, D. H., & Jun, M. (2019). Baicalein as a Potential Inhibitor against BACE1 and AChE: Mechanistic Comprehension through In Vitro and Computational Approaches. Nutrients, 11(11), 2694. https://doi.org/10.3390/nu11112694