Punicalagin Prevents Inflammation in LPS- Induced RAW264.7 Macrophages by Inhibiting FoxO3a/Autophagy Signaling Pathway


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
2.1. Reagents and Antibodies
2.2. Cell Culture
2.3. Cell Viability Assay
2.4. Nitric Oxide (NO) Production Assay
2.5. ELISA of TNF-α and IL-6
2.6. Monodansylcadaverine (MDC) Staining
2.7. Western Blotting
2.8. Statistical Analysis
3. Results
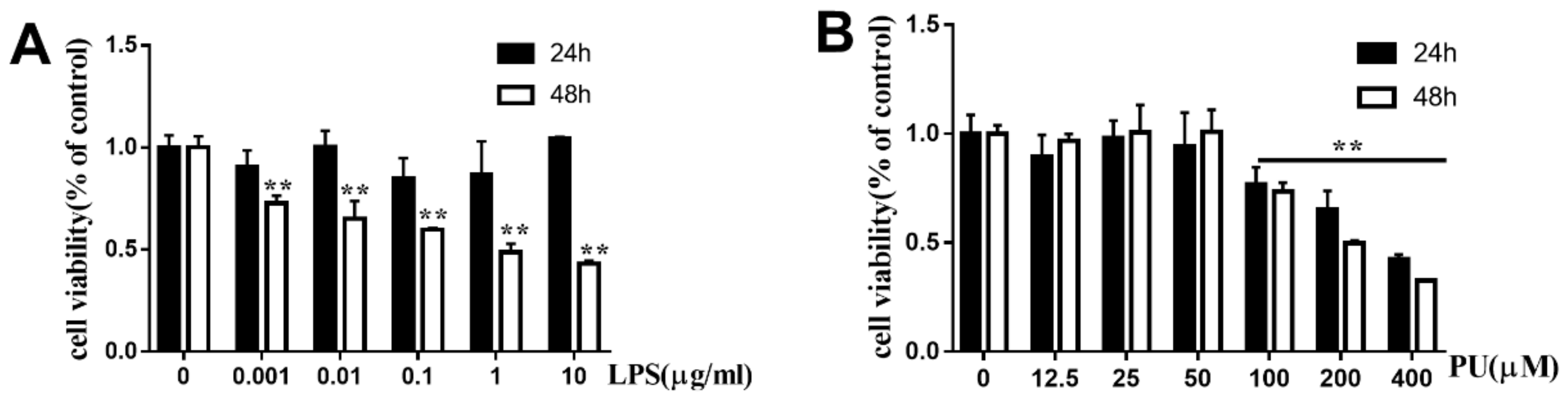
3.1. Effects of PU on the Cell Viability of RAW264.7 Macrophages
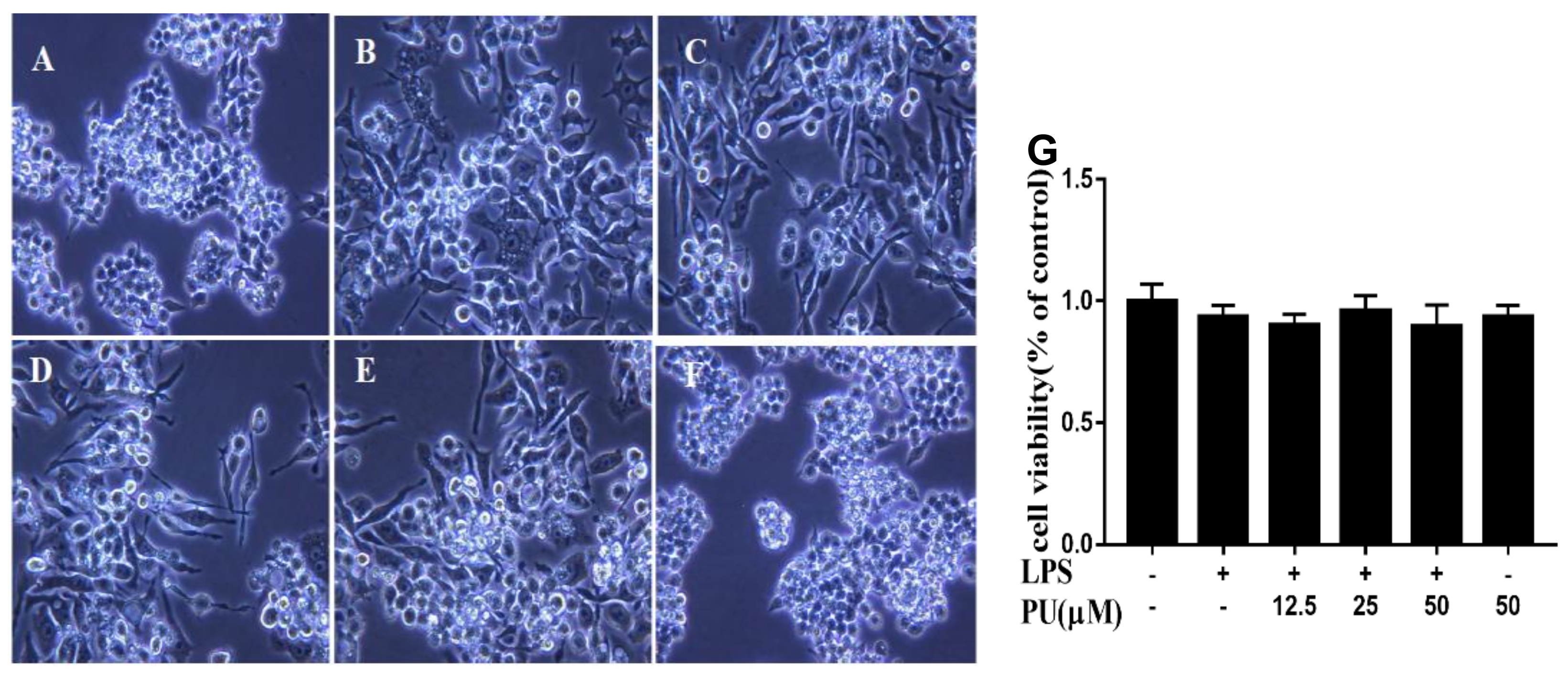
3.2. Effect of PU on Morphology of LPS-Induced RAW264.7 Macrophages
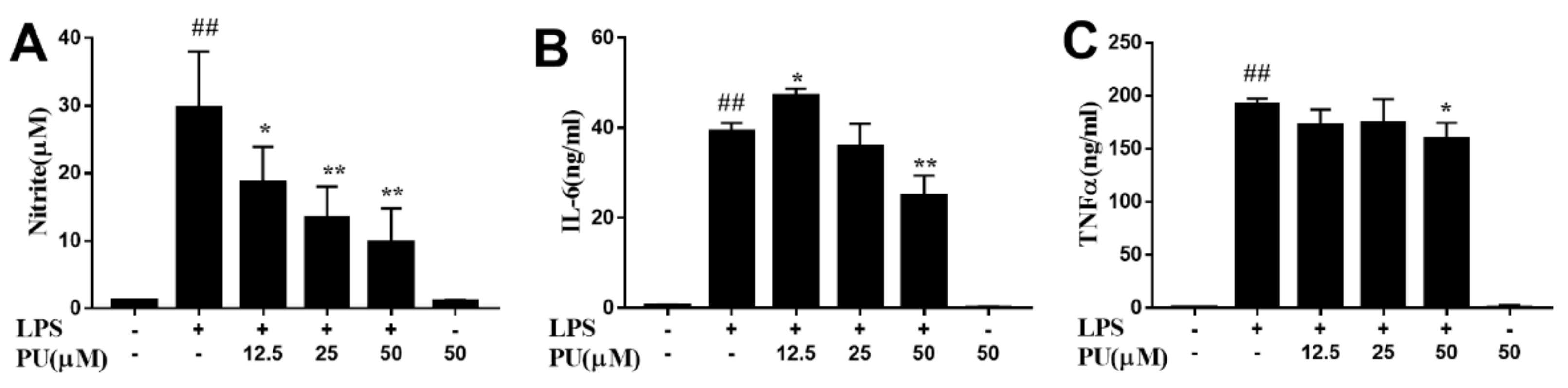
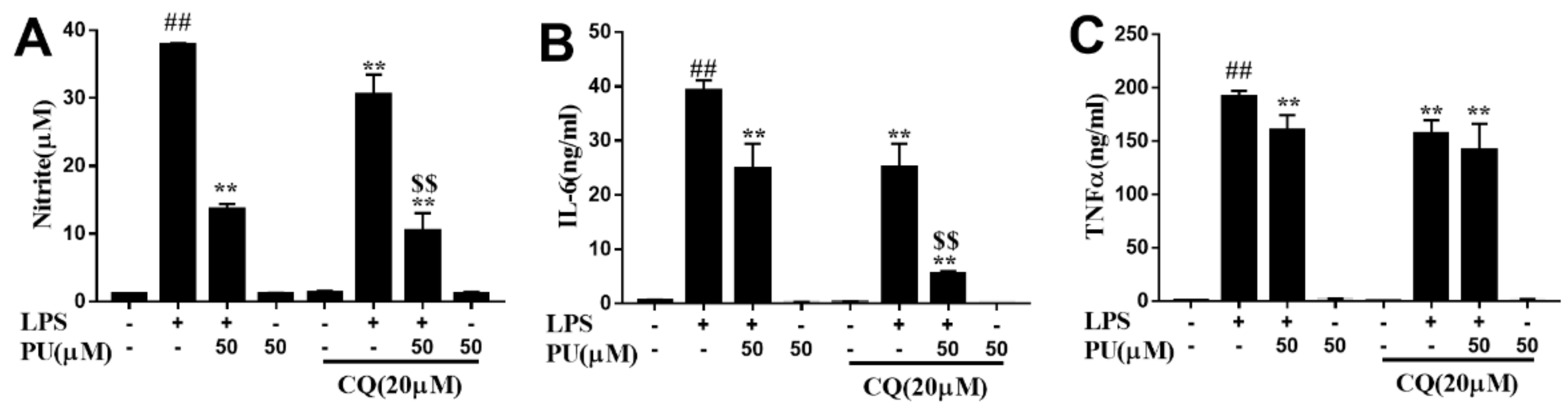
3.3. Effect of PU on NO Production and Pro-Inflammatory Cytokines Production in LPS-Induced RAW264.7 Macrophages
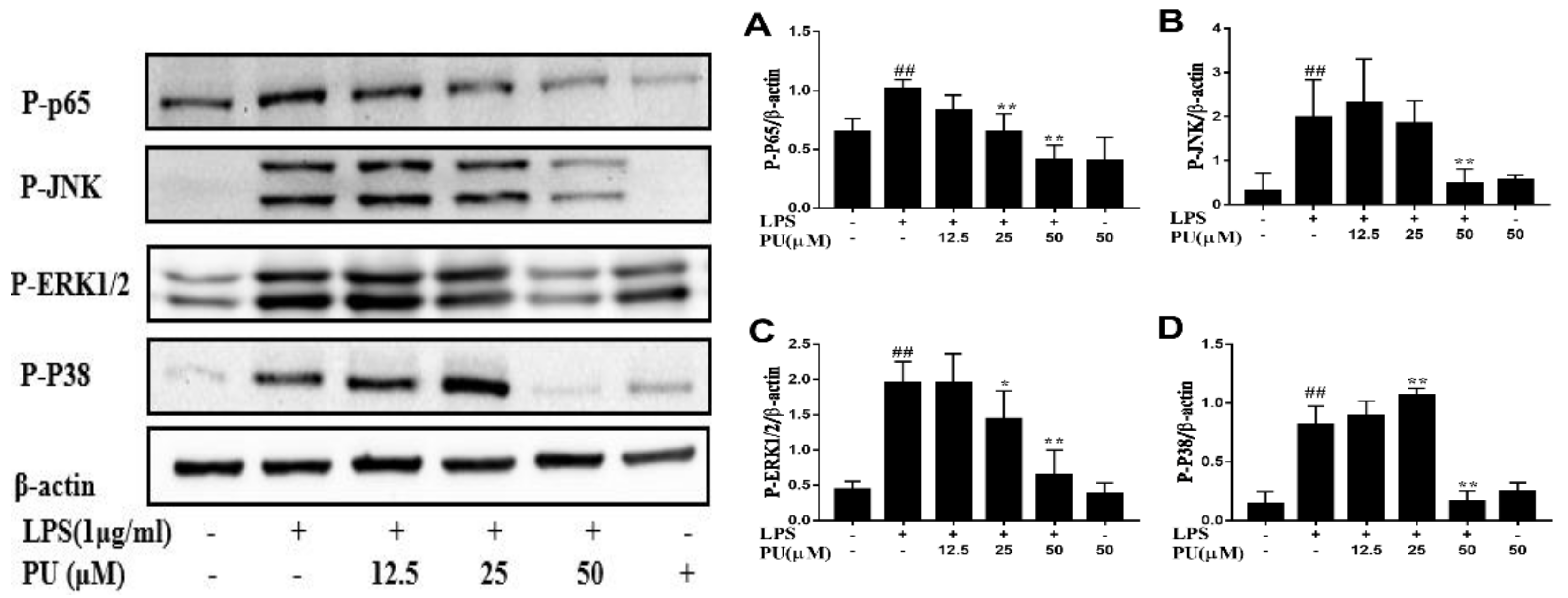
3.4. Inhibition of LPS-Induced NF-κB and MAPK Pathways Activation by PU
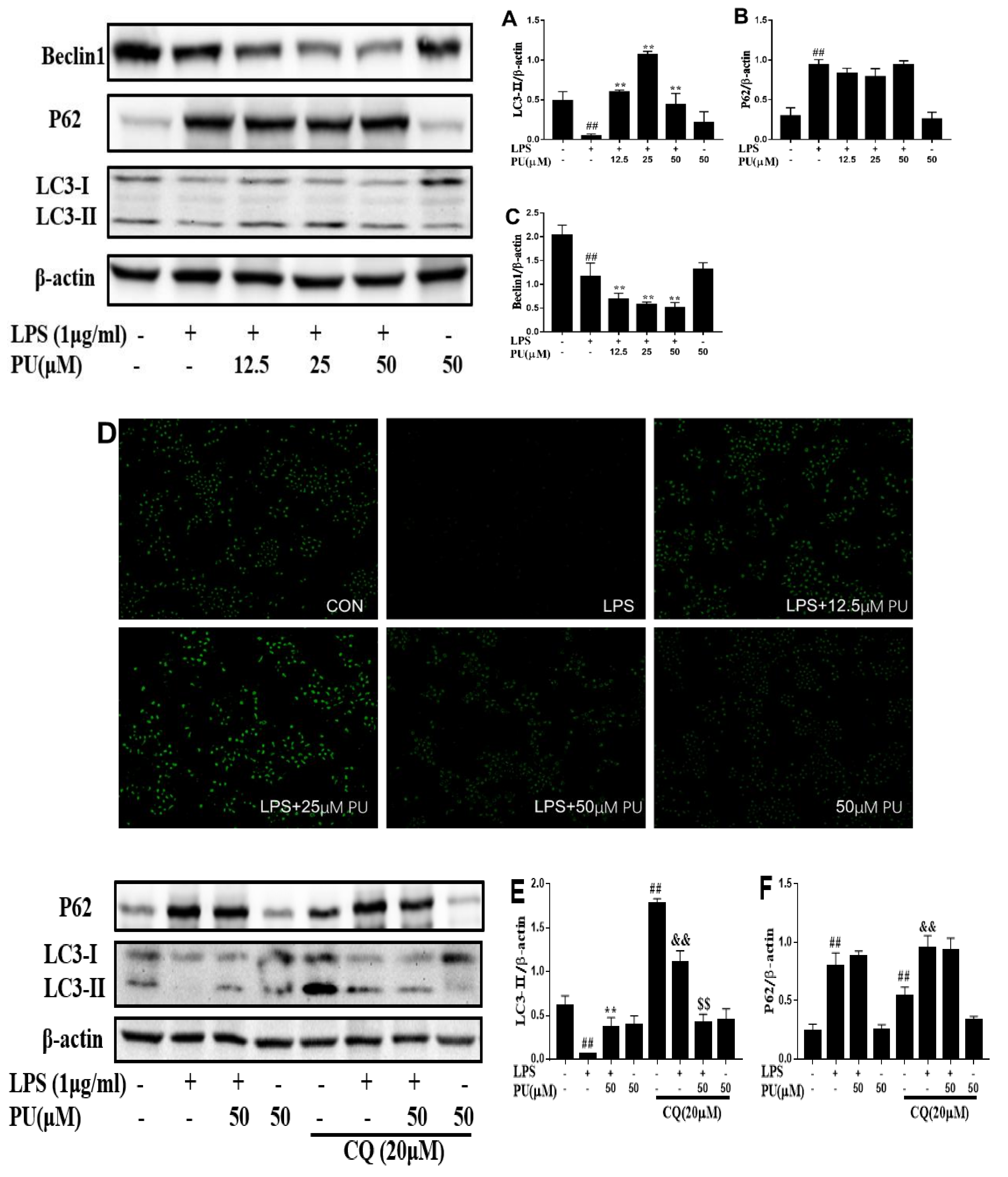
3.5. Effect of PU on Autophagy Inhibition in RAW 264.7 Macrophages
3.6. PU Suppresses Autophagy Via FoxO3a Signaling Pathway
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Acknowledgments
Conflicts of Interest
References
- Gu, L.; Kelm, M.A.; Hammerstone, J.F.; Beecher, G.; Holden, J.; Haytowitz, D.; Gebhardt, S.; Prior, R.L. Concentrations of proanthocyanidins in common foods and estimations of normal consumption. J. Nutr. 2004, 134, 613–617. [Google Scholar] [CrossRef] [PubMed]
- Bernini, R.; Barontini, M.; Cis, V.; Carastro, I.; Tofani, D.; Chiodo, R.A.; Lupattelli, P.; Incerpi, S. Synthesis and Evaluation of the Antioxidant Activity of Lipophilic Phenethyl Trifluoroacetate Esters by in Vitro ABTS, DPPH and in Cell-Culture DCF Assays. Molecules 2018, 23, 208. [Google Scholar] [CrossRef] [PubMed]
- Bernini, R.; Gilardini Montani, M.S.; Merendino, N.; Romani, A.; Velotti, F. Hydroxytyrosol-Derived Compounds: A Basis for the Creation of New Pharmacological Agents for Cancer Prevention and Therapy. J. Med. Chem. 2015, 58, 9089–9107. [Google Scholar] [CrossRef] [PubMed]
- Serino, A.; Salazar, G. Protective Role of Polyphenols against Vascular Inflammation, Aging and Cardiovascular Disease. Nutrients 2018, 11, 53. [Google Scholar] [CrossRef]
- Altieri, F.; Cairone, F.; Giamogante, F.; Carradori, S.; Locatelli, M.; Chichiarelli, S.; Cesa, S. Influence of Ellagitannins Extracted by Pomegranate Fruit on Disulfide Isomerase PDIA3 Activity. Nutrients 2019, 11, 186. [Google Scholar] [CrossRef]
- Cerda, B.; Ceron, J.J.; Tomas-Barberan, F.A.; Espin, J.C. Repeated oral administration of high doses of the pomegranate ellagitannin punicalagin to rats for 37 days is not toxic. J. Agric. Food Chem. 2003, 51, 3493–3501. [Google Scholar] [CrossRef]
- Mastrogiovanni, F.; Mukhopadhya, A.; Lacetera, N.; Ryan, M.T.; Romani, A.; Bernini, R.; Sweeney, T. Anti-Inflammatory Effects of Pomegranate Peel Extracts on in Vitro Human Intestinal Caco-2 Cells and Ex Vivo Porcine Colonic Tissue Explants. Nutrients 2019, 11, 548. [Google Scholar] [CrossRef]
- Xu, X.; Yin, P.; Wan, C.; Chong, X.; Liu, M.; Cheng, P.; Chen, J.; Liu, F.; Xu, J. Punicalagin inhibits inflammation in LPS-induced RAW264.7 macrophages via the suppression of TLR4-mediated MAPKs and NF-kappaB activation. Inflammation 2014, 37, 956–965. [Google Scholar] [CrossRef]
- Marques, L.C.; Pinheiro, A.J.; Araujo, J.G.; de Oliveira, R.A.; Silva, S.N.; Abreu, I.C.; de Sousa, E.M.; Fernandes, E.S.; Luchessi, A.D.; Silbiger, V.N.; et al. Anti-Inflammatory Effects of a Pomegranate Leaf Extract in LPS-Induced Peritonitis. Planta Med. 2016, 82, 1463–1467. [Google Scholar] [CrossRef]
- Van den Bossche, J.; O’Neill, L.A.; Menon, D. Macrophage Immunometabolism: Where Are We (Going)? Trends Immunol. 2017, 395–406. [Google Scholar] [CrossRef]
- Takeuchi, O.; Akira, S. Pattern recognition receptors and inflammation. Cell 2010, 140, 805–820. [Google Scholar] [CrossRef] [PubMed]
- Negishi, H.; Fujita, Y.; Yanai, H.; Sakaguchi, S.; Ouyang, X.; Shinohara, M.; Takayanagi, H.; Ohba, Y.; Taniguchi, T.; Honda, K. Evidence for licensing of IFN-gamma-induced IFN regulatory factor 1 transcription factor by MyD88 in Toll-like receptor-dependent gene induction program. Proc. Natl. Acad. Sci. USA 2006, 103, 15136–15141. [Google Scholar] [CrossRef] [PubMed]
- Sharif, O.; Bolshakov, V.N.; Raines, S.; Newham, P.; Perkins, N.D. Transcriptional profiling of the LPS induced NF-kappaB response in macrophages. BMC Immunol. 2007, 8, 1. [Google Scholar] [CrossRef] [PubMed]
- Gilmore, T.D. Introduction to NF-kappaB: players, pathways, perspectives. Oncogene 2006, 25, 6680–6684. [Google Scholar] [CrossRef] [PubMed]
- Kim, K.H.; Lee, M.S. Autophagy—A key player in cellular and body metabolism. Nat. Rev. Endocrinol. 2014, 10, 322–337. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.; Kapoor, A.; Iozzo, R.V. Methods for Monitoring Matrix-Induced Autophagy. Methods Mol. Biol. 2019, 1952, 157–191. [Google Scholar]
- Mizushima, N.; Yoshimori, T. How to interpret LC3 immunoblotting. Autophagy 2007, 3, 542–545. [Google Scholar] [CrossRef]
- Nazio, F.; Cecconi, F. Autophagy up and down by outsmarting the incredible ULK. Autophagy 2017, 13, 967–968. [Google Scholar] [CrossRef]
- Cadwell, K. Crosstalk between autophagy and inflammatory signalling pathways: balancing defence and homeostasis. Nat. Rev. Immunol. 2016, 16, 661–675. [Google Scholar] [CrossRef]
- Saitoh, T.; Fujita, N.; Jang, M.H.; Uematsu, S.; Yang, B.G.; Satoh, T.; Omori, H.; Noda, T.; Yamamoto, N.; Komatsu, M.; et al. Loss of the autophagy protein Atg16L1 enhances endotoxin-induced IL-1beta production. Nature 2008, 456, 264–268. [Google Scholar] [CrossRef]
- Yoshizaki, T.; Kusunoki, C.; Kondo, M.; Yasuda, M.; Kume, S.; Morino, K.; Sekine, O.; Ugi, S.; Uzu, T.; Nishio, Y.; et al. Autophagy regulates inflammation in adipocytes. Biochem. Biophys. Res. Commun. 2012, 417, 352–357. [Google Scholar] [CrossRef] [PubMed]
- Ding, D.; Xu, S.; Zhang, H.; Zhao, W.; Zhang, X.; Jiang, Y.; Wang, P.; Dai, Z.; Zhang, J. 3-Methyladenine and dexmedetomidine reverse lipopolysaccharide-induced acute lung injury through the inhibition of inflammation and autophagy. Exp. Ther. Med. 2018, 15, 3516–3522. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Chen, B.; Longtine, M.S.; Nelson, D.M. Punicalagin promotes autophagy to protect primary human syncytiotrophoblasts from apoptosis. Reproduction 2016, 97–104. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.G.; Huang, M.H.; Li, J.H.; Lai, F.I.; Lee, H.M.; Hsu, Y.N. Punicalagin induces apoptotic and autophagic cell death in human U87MG glioma cells. Acta Pharmacol. Sin. 2013, 34, 1411–1419. [Google Scholar] [CrossRef]
- Vazquez, C.L.; Colombo, M.I. Assays to assess autophagy induction and fusion of autophagic vacuoles with a degradative compartment, using monodansylcadaverine (MDC) and DQ-BSA. Methods Enzymol. 2009, 452, 85–95. [Google Scholar]
- Pasquier, B. Autophagy inhibitors. Cell. Mol. Life Sci. 2016, 73, 985–1001. [Google Scholar] [CrossRef]
- Zhao, J.; Brault, J.J.; Schild, A.; Cao, P.; Sandri, M.; Schiaffino, S.; Lecker, S.H.; Goldberg, A.L. FoxO3 coordinately activates protein degradation by the autophagic/lysosomal and proteasomal pathways in atrophying muscle cells. Cell Metab. 2007, 6, 472–483. [Google Scholar] [CrossRef]
- Ni, H.M.; Du, K.; You, M.; Ding, W.X. Critical role of FoxO3a in alcohol-induced autophagy and hepatotoxicity. Am. J. Pathol. 2013, 183, 1815–1825. [Google Scholar] [CrossRef]
- Tressera-Rimbau, A.; Arranz, S.; Eder, M.; Vallverdu-Queralt, A. Dietary Polyphenols in the Prevention of Stroke. Oxid. Med. Cell. Longev. 2017, 2017, 7467962. [Google Scholar] [CrossRef]
- Mathon, C.; Chater, J.M.; Green, A.; Merhaut, D.J.; Mauk, P.A.; Preece, J.E.; Larive, C.K. Quantification of punicalagins in commercial preparations and pomegranate cultivars, by liquid chromatography-mass spectrometry. J. Sci. Food Agric. 2019, 99, 4036–4042. [Google Scholar] [CrossRef]
- Dobrovolskaia, M.A.; Vogel, S.N. Toll receptors, CD14, and macrophage activation and deactivation by LPS. Microbes Infect. 2002, 4, 903–914. [Google Scholar] [CrossRef]
- Maldonado, R.F.; Sa-Correia, I.; Valvano, M.A. Lipopolysaccharide modification in Gram-negative bacteria during chronic infection. FEMS Microbiol. Rev. 2016, 40, 480–493. [Google Scholar] [CrossRef] [PubMed]
- Netea-Maier, R.T.; Plantinga, T.S.; van de Veerdonk, F.L.; Smit, J.W.; Netea, M.G. Modulation of inflammation by autophagy: Consequences for human disease. Autophagy 2016, 12, 245–260. [Google Scholar] [CrossRef]
- Deretic, V.; Levine, B. Autophagy balances inflammation in innate immunity. Autophagy 2018, 14, 243–251. [Google Scholar] [CrossRef]
- Ravanan, P.; Srikumar, I.F.; Talwar, P. Autophagy: The spotlight for cellular stress responses. Life Sci. 2017, 188, 53–67. [Google Scholar] [CrossRef] [PubMed]
- Kroemer, G.; Jaattela, M. Lysosomes and autophagy in cell death control. Nat. Rev. Cancer 2005, 5, 886–897. [Google Scholar] [CrossRef] [PubMed]
- Matsumoto, G.; Wada, K.; Okuno, M.; Kurosawa, M.; Nukina, N. Serine 403 phosphorylation of p62/SQSTM1 regulates selective autophagic clearance of ubiquitinated proteins. Mol. Cell 2011, 44, 279–289. [Google Scholar] [CrossRef] [PubMed]
- Wang, M.; Wu, J.; Zhou, E.; Chang, X.; Gan, J.; Cheng, T. Forkhead box o3a suppresses lipopolysaccharide-stimulated proliferation and inflammation in fibroblast-like synoviocytes through regulating tripartite motif-containing protein 3. J. Cell. Physiol. 2019, 234, 20139–20148. [Google Scholar] [CrossRef]
- Gomez-Puerto, M.C.; Verhagen, L.P.; Braat, A.K.; Lam, E.W.; Coffer, P.J.; Lorenowicz, M.J. Activation of autophagy by FOXO3 regulates redox homeostasis during osteogenic differentiation. Autophagy 2016, 12, 1804–1816. [Google Scholar] [CrossRef]
- Zhou, M.; Xu, W.; Wang, J.; Yan, J.; Shi, Y.; Zhang, C.; Ge, W.; Wu, J.; Du, P.; Chen, Y. Boosting mTOR-dependent autophagy via upstream TLR4-MyD88-MAPK signalling and downstream NF-kappaB pathway quenches intestinal inflammation and oxidative stress injury. EBioMedicine 2018, 35, 345–360. [Google Scholar] [CrossRef]
- Wang, F.M.; Hu, Z.; Liu, X.; Feng, J.Q.; Augsburger, R.A.; Gutmann, J.L.; Glickman, G.N. Resveratrol represses tumor necrosis factor alpha/c-Jun N-terminal kinase signaling via autophagy in human dental pulp stem cells. Arch. Oral Biol. 2019, 97, 116–121. [Google Scholar] [CrossRef] [PubMed]
- Criollo, A.; Chereau, F.; Malik, S.A.; Niso-Santano, M.; Marino, G.; Galluzzi, L.; Maiuri, M.C.; Baud, V.; Kroemer, G. Autophagy is required for the activation of NFkappaB. Cell Cycle 2012, 11, 194–199. [Google Scholar] [CrossRef] [PubMed]








© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cao, Y.; Chen, J.; Ren, G.; Zhang, Y.; Tan, X.; Yang, L. Punicalagin Prevents Inflammation in LPS- Induced RAW264.7 Macrophages by Inhibiting FoxO3a/Autophagy Signaling Pathway. Nutrients 2019, 11, 2794. https://doi.org/10.3390/nu11112794
Cao Y, Chen J, Ren G, Zhang Y, Tan X, Yang L. Punicalagin Prevents Inflammation in LPS- Induced RAW264.7 Macrophages by Inhibiting FoxO3a/Autophagy Signaling Pathway. Nutrients. 2019; 11(11):2794. https://doi.org/10.3390/nu11112794
Chicago/Turabian StyleCao, Yuan, Jihua Chen, Guofeng Ren, Yahui Zhang, Xiuying Tan, and Lina Yang. 2019. "Punicalagin Prevents Inflammation in LPS- Induced RAW264.7 Macrophages by Inhibiting FoxO3a/Autophagy Signaling Pathway" Nutrients 11, no. 11: 2794. https://doi.org/10.3390/nu11112794
APA StyleCao, Y., Chen, J., Ren, G., Zhang, Y., Tan, X., & Yang, L. (2019). Punicalagin Prevents Inflammation in LPS- Induced RAW264.7 Macrophages by Inhibiting FoxO3a/Autophagy Signaling Pathway. Nutrients, 11(11), 2794. https://doi.org/10.3390/nu11112794