Insulin Sensitivity and Glucose Homeostasis Can Be Influenced by Metabolic Acid Load



Abstract
1. Introduction
2. Metabolic Acidosis Disrupts Insulin Sensitivity
3. Muscle Metabolism and Lean Mass Are Influenced by Acid–Base Balance
4. Association between Acid load and Glucose Homeostasis
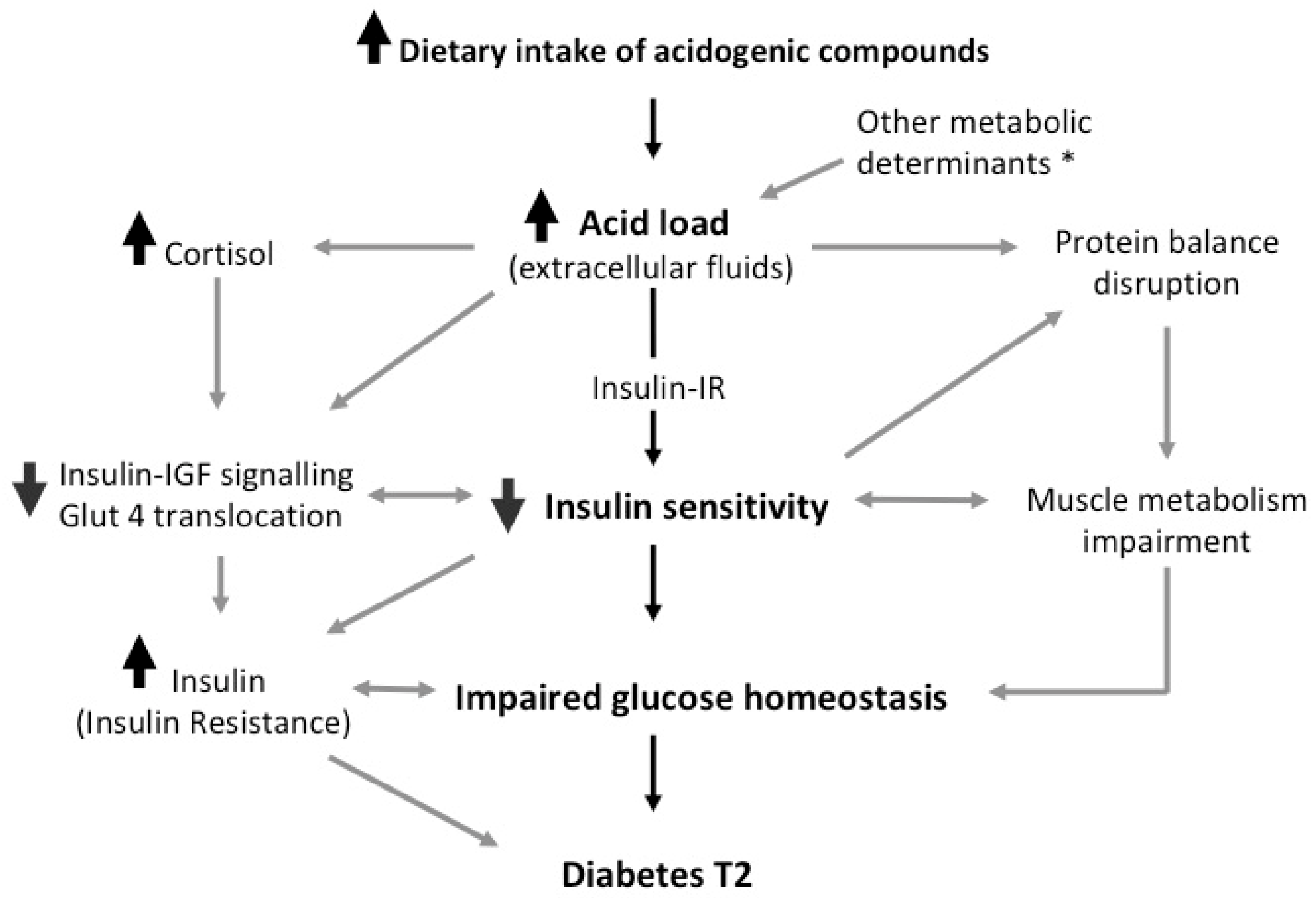
5. Discussion
5.1. Parameters Estimation
5.2. Nutritional and Metabolic Confounders
5.3. Investigation Methodology
6. Conclusions
Author Contributions
Conflicts of Interest
References
- Sung, K.C.; Jeong, W.S.; Wild, S.H.; Byrne, C.D. Combined influence of insulin resistance, overweight/obesity, and fatty liver as risk factors for type 2 diabetes. Diabetes Care 2012, 35, 717–722. [Google Scholar] [CrossRef] [PubMed]
- Cooper, A.J.; Sharp, S.J.; Lentjes, M.A.; Luben, R.N.; Khaw, K.T.; Wareham, N.J.; Forouhi, N.G. A prospective study of the association between quantity and variety of fruit and vegetable intake and incident type 2 diabetes. Diabetes Care 2012, 35, 1293–1300. [Google Scholar] [CrossRef] [PubMed]
- Ley, S.H.; Hamdy, O.; Mohan, V.; Hu, F.B. Prevention and management of type 2 diabetes: Dietary components and nutritional strategies. Lancet 2014, 383, 1999–2007. [Google Scholar] [CrossRef]
- Sharma, B.R.; Kim, H.J.; Rhyu, D.Y. Caulerpa lentillifera inhibits protein-tyrosine phosphatase 1B and protects pancreatic beta cell via its insulin mimetic effect. Food Sci. Biotechnol. 2017, 26, 495–499. [Google Scholar] [CrossRef]
- Della Guardia, L.; Roggi, C.; Cena, H. Diet-induced acidosis and alkali supplementation. Int. J. Food Sci. Nutr. 2016, 67, 754–761. [Google Scholar] [CrossRef] [PubMed]
- Adeva, M.M.; Souto, G. Diet-induced metabolic acidosis. Clin. Nutr. 2011, 30, 416–421. [Google Scholar] [CrossRef] [PubMed]
- Frassetto, L.A.; Todd, K.M.; Morris, R.C., Jr.; Sebastian, A. Estimation of net endogenous noncarbonic acid production in humans from diet potassium and protein contents. Am. J. Clin. Nutr. 1998, 68, 576–583. [Google Scholar] [CrossRef] [PubMed]
- Remer, T. Influence of nutrition on acid-base balance—Metabolic aspects. Eur. J. Nutr. 2001, 40, 214–220. [Google Scholar] [CrossRef] [PubMed]
- Remer, T.; Dimitriou, T.; Manz, F. Dietary potential renal acid load and renal net acid excretion in healthy, free-living children and adolescents. Am. J. Clin. Nutr. 2003, 77, 1255–1260. [Google Scholar] [CrossRef] [PubMed]
- Sebastian, A.; Frassetto, L.A.; Sellmeyer, D.E.; Merriam, R.L.; Morris, R.C., Jr. Estimation of the net acid load of the diet of ancestral preagricultural Homo sapiens and their hominid ancestors. Am. J. Clin. Nutr. 2002, 76, 1308–1316. [Google Scholar] [CrossRef] [PubMed]
- Pizzorno, J.; Frassetto, L.A.; Katzinger, J. Diet induced acidosis: Is it real and clinically relevant? Br. J. Nutr. 2010, 103, 1185–1194. [Google Scholar] [CrossRef] [PubMed]
- Liu, Z.M.; Leung, J.; Wong, S.Y.; Wong, C.K.; Chan, R.; Woo, J. Greater fruit intake was associated with better bone mineral status among Chinese elderly men and women: Results of Hong Kong Mr. Os and Ms. Os studies. J. Am. Med. Dir. Assoc. 2015, 16, 309–315. [Google Scholar] [CrossRef] [PubMed]
- Xu, H.; Jia, T.; Huang, X.; Risérus, U.; Cederholm, T.; Arnlöv, J.; Sjögren, P.; Lindholm, B.; Carrero, J.J. Dietary acid load, insulin sensitivity and risk of type 2 diabetes in community-dwelling older men. Diabetologia 2014, 57, 1561–1568. [Google Scholar] [CrossRef] [PubMed]
- Kiefte-de Jong, J.C.; Li, Y.; Chen, M.; Curhan, G.C.; Mattei, J.; Malik, V.S.; Forman, J.P.; Franco, O.H.; Hu, F.B. Diet-dependent acid load and type 2 diabetes: Pooled results from three prospective cohort studies. Diabetologia 2017, 60, 270–279. [Google Scholar] [CrossRef] [PubMed]
- Fagherazzi, G.; Vilier, A.; Bonnet, F.; Lajous, M.; Balkau, B.; Boutron-Rualt, M.C.; Clavel-Chapelon, F. Dietary acid load and risk of type 2 diabetes: The E3N-EPIC cohort study. Diabetologia 2014, 57, 313–320. [Google Scholar] [CrossRef] [PubMed]
- Garibotto, G.; Sofia, A.; Russo, R.; Paoletti, E.; Bonanni, A.; Parodi, E.L.; Viazzi, F.; Verzola, D. Insulin sensitivity of muscle protein metabolism is altered in patients with chronic kidney disease and metabolic acidosis. Kidney Int. 2015, 88, 1419–1426. [Google Scholar] [CrossRef] [PubMed]
- Bellasi, A.; Di Micco, L.; Santoro, D.; Marzocco, S.; De Simone, E.; Cozzolino, M.; Di Lullo, L.; Guastaferro, P.; Di Iorio, B. Correction of metabolic acidosis improves insulin resistance in chronic kidney disease. BMC Nephrol. 2016, 17, 158. [Google Scholar] [CrossRef] [PubMed]
- DeFronzo, R.A.; Beckles, A.D. Glucose intolerance following chronic metabolic acidosis in man. Am. J. Physiol. 1979, 236, E328–E334. [Google Scholar] [CrossRef] [PubMed]
- Igarashi, M.; Yamatani, K.; Fukase, N.; Daimon, M.; Ohnuma, H.; Ogawa, A.; Tominaga, M.; Sasaki, H. Effect of acidosis on insulin binding and glucose uptake in isolated rat adipocytes. Tohoku J. Exp. Med. 1993, 169, 205–213. [Google Scholar] [CrossRef] [PubMed]
- Whittaker, J.; Cuthbert, C.; Hammond, V.A.; Alberti, K.G. The effects of metabolic acidosis in vivo on insulin binding to isolated rat adipocytes. Metabolism 1982, 31, 553–557. [Google Scholar] [CrossRef]
- Hayata, H.; Miyazaki, H.; Niisato, N.; Yokoyama, N.; Marunaka, Y. Lowered extracellular pH is involved in the pathogenesis of skeletal muscle insulin resistance. Biochem. Biophys. Res. Commun. 2014, 445, 170–174. [Google Scholar] [CrossRef] [PubMed]
- Esche, J.; Shi, L.; Sánchez-Guijo, A.; Hartmann, M.F.; Wudy, S.A.; Remer, T. Higher diet-dependent renal acid load associates with higher glucocorticoid secretion and potentially bioactive free glucocorticoids in healthy children. Kidney Int. 2016, 90, 325–333. [Google Scholar] [CrossRef] [PubMed]
- Andrews, R.C.; Walker, B.R. Glucocorticoids and insulin resistance: Old hormones, new targets. Clin. Sci. Lond. 1999, 96, 513–523. [Google Scholar] [CrossRef] [PubMed]
- Yuen, K.C.J.; Chong, L.E.; Riddle, M.C. Influence of glucocorticoids and growth hormone on insulin sensitivity in humans. Diabet. Med. 2013, 30, 651–663. [Google Scholar] [CrossRef] [PubMed]
- Crawford, S.O.; Hoogeveen, R.C.; Brancati, F.L.; Astor, B.C.; Ballantyne, C.M.; Schmidt, M.I.; Young, J.H. Association of blood lactate with type 2 diabetes: The Atherosclerosis Risk in Communities Carotid MRI Study. Int. J. Epidemiol. 2010, 39, 1647–1655. [Google Scholar] [CrossRef] [PubMed]
- Cupisti, A.; Meola, M.; D’Alessandro, C.; Bernabini, G.; Pasquali, E.; Carpi, A.; Barsotti, G. Insulin resistance and low urinary citrate excretion in calcium stone formers. Biomed. Pharmacother. 2007, 61, 86–90. [Google Scholar] [CrossRef] [PubMed]
- Frassetto, L.A.; Lanham-New, S.A.; Macdonald, H.M.; Remer, T.; Sebastian, A.; Tucker, K.L.; Tylavsky, F.A. Standardizing terminology for estimating the diet-dependent net acid load to the metabolic system. J. Nutr. 2007, 137, 1491–1492. [Google Scholar] [CrossRef] [PubMed]
- Choi, C.S.; Kim, Y.B.; Lee, F.N.; Zabolotny, J.M.; Kahn, B.B.; Youn, J.H. Lactate induces insulin resistance in skeletal muscle by suppressing glycolysis and impairing insulin signaling. Am. J. Physiol. Endocrinol. Metab. 2002, 283, E233–E240. [Google Scholar] [CrossRef] [PubMed]
- Whittaker, J.; Cuthbert, C.; Hammond, V.; Alberti, K.G. Impaired insulin binding to isolated adipocytes in experimental diabetic ketoacidosis. Diabetologia 1981, 21, 563–568. [Google Scholar] [PubMed]
- Cuthbert, C.; Alberti, K.G. Acidemia and insulin resistance in the diabetic ketoacidotic rat. Metabolism 1978, 27, 1903–1916. [Google Scholar] [CrossRef]
- Franch, H.A.; Raissi, S.; Wang, X.; Zheng, B.; Bailey, J.L.; Price, S.R. Acidosis impairs insulin receptor substrate-1-associated phosphoinositide 3-kinase signaling in muscle cells: Consequences on proteolysis. Am. J. Physiol. Ren. Physiol. 2004, 287, F700–F706. [Google Scholar] [CrossRef] [PubMed]
- Pedersen, S.B.; Fuglsig, S.; Sjøgren, P.; Richelsen, B. Identification of steroid receptors in human adipose tissue. Eur. J. Clin. Investig. 1996, 26, 1051–1056. [Google Scholar] [CrossRef]
- Berne, R.M.; Levy, M.N.; Koeppen, B.M.; Stanton, B.A. Berne & Levy Physiology, 6th ed.; Elsevier Inc.: New York, NY, USA, 2009. [Google Scholar]
- Rebuffé-Scrive, M.; Brönnegard, M.; Nilsson, A.; Eldh, J.; Gustafsson, J.A.; Björntorp, P. Steroid hormone receptors in human adipose tissues. J. Clin. Endocrinol. Metab. 1990, 71, 1215–1219. [Google Scholar] [CrossRef] [PubMed]
- Maurizi, G.; Della Guardia, L.; Maurizi, A.; Poloni, A. Adipocytes properties and crosstalk with immune system in obesity-related inflammation. J. Cell. Physiol. 2018, 233, 88–97. [Google Scholar] [CrossRef] [PubMed]
- Hamm, L.L.; Ambühl, P.M.; Alpern, R.J. Role of glucocorticoids in acidosis. Am. J. Kidney Dis. 1999, 34, 960–965. [Google Scholar] [CrossRef]
- Bailey, J.L.; Mitch, W.E. Twice-told tales of metabolic acidosis, glucocorticoids, and protein wasting: What do results from rats tell us about patients with kidney disease? Semin. Dial. 2000, 13, 227–231. [Google Scholar] [CrossRef] [PubMed]
- Buehlmeier, J.; Remer, T.; Frings-Meuthen, P.; Maser-Gluth, C.; Heer, M. Glucocorticoid activity and metabolism with NaCl-induced low-grade metabolic acidosis and oral alkalization: Results of two randomized controlled trials. Endocrine 2016, 52, 139–147. [Google Scholar] [CrossRef] [PubMed]
- Challa, A.; Chan, W.; Krieg, R.J., Jr.; Thabet, M.A.; Liu, F.; Hintz, R.L.; Chan, J.C. Effect of metabolic acidosis on the expression of insulin-like growth factor and growth hormone receptor. Kidney Int. 1993, 44, 1224–1227. [Google Scholar] [CrossRef] [PubMed]
- Maurer, M.; Riesen, W.; Muser, J.; Hulter, H.N.; Krapf, R. Neutralization of Western diet inhibits bone resorption independently of K intake and reduces cortisol secretion in humans. Am. J. Physiol. Renal Physiol. 2003, 284, F32–F40. [Google Scholar] [CrossRef] [PubMed]
- Disthabanchong, S.; Niticharoenpong, K.; Radinahamed, P.; Stitchantrakul, W.; Ongphiphadhanakul, B.; Hongeng, S. Metabolic acidosis lowers circulating adiponectin through inhibition of adiponectin gene transcription. Nephrol. Dial. Transplant. 2010, 26, 592–598. [Google Scholar] [CrossRef] [PubMed]
- Patel, S.A.; Hoehn, K.L.; Lawrence, R.T.; Sawbridge, L.; Talbot, N.A.; Tomsig, J.L.; Turner, N.; Cooney, G.J.; Whitehead, J.P.; Kraegen, E.W.; et al. Overexpression of the adiponectin receptor AdipoR1 in rat skeletal muscle amplifies local insulin sensitivity. Endocrinology 2012, 153, 5231–5246. [Google Scholar] [CrossRef] [PubMed]
- Han, S.J.; Boyko, E.J.; Fujimoto, W.Y.; Kahn, S.E.; Leonetti, D.L.J. Low Plasma Adiponectin Concentrations Predict Increases in Visceral Adiposity and Insulin Resistance. Clin. Endocrinol. Metab. 2017, 102, 4626–4633. [Google Scholar] [CrossRef] [PubMed]
- Pocai, A.; Morgan, K.; Buettner, C.; Gutierrez-Juarez, R.; Obici, S.; Rossetti, L. Central leptin acutely reverses diet-induced hepatic insulin resistance. Diabetes 2005, 54, 3182–3189. [Google Scholar] [CrossRef] [PubMed]
- Perry, R.J.; Zhang, X.-M.; Zhang, D.; Kumashiro, N.; Camporez, J.-P.G.; Cline, G.W.; Shulman, G.I. Mechanism for the Anti-Diabetic Effect of Leptin. Nat. Med. 2014, 20, 759–763. [Google Scholar] [CrossRef] [PubMed]
- Perry, R.J.; Petersen, K.F.; Shulman, G.I. Pleotropic effects of leptin to reverse insulin resistance and diabetic ketoacidosis. Diabetologia 2016, 59, 933–937. [Google Scholar] [CrossRef] [PubMed]
- Zheng, F.; Qiu, X.; Yin, S.; Li, Y. Changes in serum leptin levels in chronic renal failure patients with metabolic acidosis. J. Ren. Nutr. 2001, 11, 207–211. [Google Scholar] [CrossRef]
- Teta, D.; Bevington, A.; Brown, J.; Pawluczyk, I.; Harris, K.; Walls, J. Acidosis downregulates leptin production from cultured adipocytes through a glucose transport-dependent post-transcriptional mechanism. J. Am. Soc. Nephrol. 2003, 14, 2248–2254. [Google Scholar] [CrossRef] [PubMed]
- Yang, J. Enhanced skeletal muscle for effective glucose homeostasis. Prog. Mol. Biol. Transl. Sci. 2014, 121, 133–163. [Google Scholar] [PubMed]
- Shishikura, K.; Tanimoto, K.; Sakai, S.; Tanimoto, Y.; Terasaki, J.; Hanafusa, T. Association between skeletal muscle mass and insulin secretion in patients with type 2 diabetes mellitus. Endocr. J. 2014, 61, 281–287. [Google Scholar] [CrossRef] [PubMed]
- Aune, D.; Norat, T.; Leitzmann, M.; Tonstad, S.; Vatten, L.J. Physical activity and the risk of type 2 diabetes: A systematic review and dose-response meta-analysis. Eur. J. Epidemiol. 2015, 30, 529–542. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Hu, Z.; Hu, J.; Du, J.; Mitch, W.E. Insulin resistance accelerates muscle protein degradation: Activation of the ubiquitin-proteasome pathway by defects in muscle cell signaling. Endocrinology 2006, 147, 4160–4168. [Google Scholar] [CrossRef] [PubMed]
- Lofberg, E.; Gutierrez, A.; Anderstam, B.; Wernerman, J.; Bergström, J.; Price, S.R.; Mitch, W.E.; Alvestrand, A. Effect of bicarbonate on muscle protein in patients receiving hemodialysis. Am. J. Kidney Dis. 2006, 48, 419–429. [Google Scholar] [CrossRef] [PubMed]
- Bailey, J.L.; Mitch, W.E. Mechanisms of protein degradation: What do the rat studies tell us. J. Nephrol. 2000, 13, 89–95. [Google Scholar] [PubMed]
- Mitch, W.E.; Medina, R.; Grieber, S.; May, R.C.; England, B.K.; Price, S.R.; Bailey, J.L.; Goldberg, A.L. Metabolic acidosis stimulates muscle protein degradation by activating the adenosine triphosphate-dependent pathway involving ubiquitin and proteasomes. J. Clin. Investig. 1994, 93, 2127–2133. [Google Scholar] [CrossRef] [PubMed]
- Lyon, R.C.; Lange, S.; Sheikh, F. Breaking down protein degradation mechanisms in cardiac muscle. Trends Mol. Med. 2013, 19, 239–249. [Google Scholar] [CrossRef] [PubMed]
- Price, S.R.; Bailey, J.L.; Wang, X.; Jurkovitz, C.; England, B.K.; Ding, X.; Phillips, L.S.; Mitch, W.E. Muscle wasting in insulinopenic rats results from activation of the ATP-dependent, ubiquitin-proteasome proteolytic pathway by a mechanism including gene transcription. J. Clin. Investig. 1996, 98, 1703–1708. [Google Scholar] [CrossRef] [PubMed]
- Clemmons, D.R. Role of IGF-I in skeletal muscle mass maintenance. Trends Endocrinol. Metab. 2009, 20, 349–356. [Google Scholar] [CrossRef] [PubMed]
- Zanou, N.; Gailly, P. Skeletal muscle hypertrophy and regeneration: Interplay between the myogenic regulatory factors (MRFs) and insulin-like growth factors (IGFs) pathways. Cell. Mol. Life Sci. 2013, 70, 4117–4130. [Google Scholar] [CrossRef] [PubMed]
- Brungger, M.; Hulter, H.N.; Krapf, R. Effect of chronic metabolic acidosis on the growth hormone/IGF-1 endocrine axis: New cause of growth hormone insensitivity in humans. Kidney Int. 1997, 51, 216–221. [Google Scholar] [CrossRef] [PubMed]
- Ordonez, F.A.; Santos, F.; Martınez, V.; Garcıa, E.; Fernandez, P.; Rodrıguez, J.; Fernandez, M.; Alvarez, J.; Ferrando, S. Resistance to growth hormone and insulin-like growth factor-I in acidotic rats. Pediatr. Nephrol. 2000, 14, 720–725. [Google Scholar] [CrossRef] [PubMed]
- Ceglia, L.; Harris, S.; Abrams, S.A.; Rasmussen, H.M.; Dallal, G.E.; Dawson-Huges, B. Potassium Bicarbonate Attenuates the Urinary Nitrogen Excretion That Accompanies an Increase in Dietary Protein and May Promote Calcium Absorption. J. Clin. Endocrinol. Metab. 2009, 94, 645–653. [Google Scholar] [CrossRef] [PubMed]
- Welch, A.A.; MacGregor, A.J.; Skinner, J.; Spector, T.D.; Moayyeri, A.; Cassidy, A. A higher alkaline dietary load is associated with greater indexes of skeletal muscle mass in women. Osteoporos. Int. 2013, 24, 1899–1908. [Google Scholar] [CrossRef] [PubMed]
- Chan, R.; Leung, J.; Woo, J. Association between Estimated Net Endogenous Acid Production and Subsequent Decline in Muscle Mass Over Four Years in Ambulatory Older Chinese People in Hong Kong: A Prospective Cohort Study. J. Gerontol. A Biol. Sci. Med. Sci. 2015, 70, 905–911. [Google Scholar] [CrossRef] [PubMed]
- Frassetto, L.; Morris, R.C., Jr.; Sebastian, A. Potassium bicarbonate reduces urinary nitrogen excretion in postmenopausal women. J. Clin. Endocrinol. Metab. 1997, 82, 254–259. [Google Scholar] [CrossRef] [PubMed]
- Dawson-Hughes, B.; Castaneda-Sceppa, C.; Harris, S.S.; Palermo, N.J.; Cloutier, G.; Ceglia, L.; Dallal, G.E. Impact of supplementation with bicarbonate on lower-extremity muscle performance in older men and women. Osteoporos. Int. 2010, 21, 1171–1179. [Google Scholar] [CrossRef] [PubMed]
- Mandel, E.I.; Curhan, G.C.; Hu, F.B.; Taylor, E.N. Plasma bicarboante and risk of type 2 diabetes mellitus. Can. Med. Assoc. J. 2012, 184, E719–E725. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, S.; Inokuchi, T.; Kobayashi, T.; Ka, T.; Tsutsumi, Z.; Moriwaki, Y.; Yamamoto, T. Relationship between insulin resistance and low urinary pH in patients with gout, and effects of PPAR-alpha agonists on urine pH. Horm. Metab. Res. 2007, 39, 511–514. [Google Scholar] [CrossRef] [PubMed]
- Farwell, W.R.; Taylor, E.N. Serum bicarbonate, anion gap and insulin resistance in the National Health and Nutrition Examination Survey. Diabet. Med. 2008, 25, 798–804. [Google Scholar] [CrossRef] [PubMed]
- Akter, S.; Kurotani, K.; Kashino, I.; Goto, A.; Mizoue, T.; Noda, M.; Sawada, N.; Tsugane, S. High Dietary Acid Load Score Is Associated with Increased Risk of Type 2 Diabetes in Japanese Men: The Japan Public Health Center-based Prospective Study. J. Nutr. 2016, 146, 1076–1083. [Google Scholar] [CrossRef] [PubMed]
- Akter, S.; Eguchi, M.; Kuwahara, K.; Kochi, T.; Ito, R.; Kurotani, K.; Tsuruoka, H.; Nanri, A.; Kabe, I.; Mizoue, T. High dietary acid load is associated with insulin resistance: The Furukawa Nutrition and Health Study. Clin. Nutr. 2016, 35, 453–459. [Google Scholar] [CrossRef] [PubMed]
- Kozan, P.; Blythe, J.C.; Greenfield, J.R.; Samocha-Bonet, D. The Effect of Buffering High Acid Load Meal with Sodium Bicarbonate on Postprandial Glucose Metabolism in Humans-A Randomized Placebo-Controlled Study. Nutrients 2017, 9, 861. [Google Scholar] [CrossRef] [PubMed]
- Harris, S.S.; Dawson-Hughes, B. No effect of bicarbonate treatment on insulin sensitivity and glucose control in non-diabetic older adults. Endocrine 2010, 38, 221–226. [Google Scholar] [CrossRef] [PubMed]
- Palmer, B.F.; Clegg, D.J. Electrolyte and Acid-Base Disturbances in Patients with Diabetes Mellitus. N. Engl. J. Med. 2015, 373, 548–559. [Google Scholar] [CrossRef] [PubMed]
- Weisberg, L.S. Lactic Acidosis in a Patient with Type 2 Diabetes Mellitus. Clin. J. Am. Soc. Nephrol. 2015, 10, 1476–1483. [Google Scholar] [CrossRef] [PubMed]
- Hamm, L.L.; Nakhoul, N.; Hering-Smith, K.S. Acid-Base Homeostasis. Clin. J. Am. Soc. Nephrol. 2015, 10, 232–242. [Google Scholar] [CrossRef] [PubMed]
- Jia, T.; Huang, X.; Qureshi, A.R.; Xu, H.; Ärnlöv, J.; Lindholm, B.; Cederholm, T.; Stenvinkel, P.; Risérus, U.; Carrero, J.J. Validation of insulin sensitivity surrogate indices and prediction of clinical outcomes in individuals with and without impaired renal function. Kidney Int. 2014, 86, 383–391. [Google Scholar] [CrossRef] [PubMed]
- Wallace, T.M.; Levy, J.C.; Matthews, D.R. Use and abuse of HOMA modeling. Diabetes Care 2004, 27, 1487–1495. [Google Scholar] [CrossRef] [PubMed]
- Kang, E.S.; Yun, Y.S.; Park, S.W.; Kim, H.J.; Ahn, C.W.; Song, Y.D.; Cha, B.S.; Lim, S.K.; Kim, K.R.; Lee, H.C. Limitation of the validity of the homeostasis model assessment as an index of insulin resistance in Korea. Metabolism 2005, 54, 206–211. [Google Scholar] [CrossRef] [PubMed]
- Ferrara, C.M.; Goldberg, A.P. Limited value of the homeostasis model assessment to predict insulin resistance in older men with impaired glucose tolerance. Diabetes Care 2001, 24, 245–249. [Google Scholar] [CrossRef] [PubMed]
- Miki, A.; Hashimoto, Y.; Tanaka, M.; Kobayashi, Y.; Wada, S.; Kuwahata, M.; Kido, Y.; Yamazaki, M.; Fukui, M. Urinary pH reflects dietary acid load in patients with type 2 diabetes. J. Clin. Biochem. Nutr. 2017, 61, 74–77. [Google Scholar] [CrossRef] [PubMed]
- Stice, E.; Palmrose, C.A.; Burger, K.S. Elevated BMI and Male Sex Are Associated with Greater Underreporting of Caloric Intake as Assessed by Doubly Labeled Water. J. Nutr. 2015, 145, 2412–2418. [Google Scholar] [CrossRef] [PubMed]
- Frassetto, L.A.; Shi, L.; Schloetter, M.; Sebastian, A.; Remer, T. Established dietary estimates of net acid production do not predict measured net acid excretion in patients with Type 2 diabetes on Paleolithic Hunter-Gatherer-type diets. Eur. J. Clin. Nutr. 2013, 67, 899–903. [Google Scholar] [CrossRef] [PubMed]
- Palmer, B.F. Regulation of Potassium Homeostasis. Clin. J. Am. Soc. Nephrol. 2015, 10, 1050–1060. [Google Scholar] [CrossRef] [PubMed]
- Orr-Walker, B.J.; Horne, A.M.; Evans, M.C.; Grey, A.B.; Murray, M.A.F.; McNeil, A.R.; Reid, I.R. Hormone replacement therapy causes a respiratory alkalosis in normal postmenopausal women. J. Clin. Endocrinol. Metab. 1999, 84, 1997–2001. [Google Scholar] [CrossRef] [PubMed]
- Khairallah, P.; Scialla, J.J. Role of Acid-Base Homeostasis in Diabetic Kidney Disease. Curr. Diabetes Rep. 2017, 17, 28. [Google Scholar] [CrossRef] [PubMed]
- Maalouf, N.M.; Cameron, M.A.; Moe, O.W.; Sakhaee, K. Metabolic basis for low urine pH in type 2 diabetes. Clin. J. Am. Soc. Nephrol. 2010, 5, 1277–1281. [Google Scholar] [CrossRef] [PubMed]
- Keane, D.; Newsholme, P. Saturated and unsaturated (including arachidonic acid) non-esterified fatty acid modulation of insulin secretion from pancreatic beta-cells. Biochem. Soc. Trans. 2008, 36, 955–958. [Google Scholar] [CrossRef] [PubMed]
- Baudrand, R.; Campino, C.; Carvajal, C.A.; Olivieri, O.; Guidi, G.; Faccini, G.; Vöhringer, P.A.; Cerda, J.; Owen, G.; Kalergis, A.M.; et al. High sodium intake is associated with increased glucocorticoid production, insulin resistance and metabolic syndrome. Clin. Endocrinol. 2014, 80, 677–684. [Google Scholar] [CrossRef] [PubMed]
- Sluijs, I.; Beulens, J.W.; van der, A.D.; Spijkerman, A.M.; Grobbee, D.E.; van der Schouw, Y.T. Dietary intake of total, animal, and vegetable protein and risk of type 2 diabetes in the European Prospective Investigation into Cancer and Nutrition (EPIC)-NL study. Diabetes Care 2010, 33, 43–48. [Google Scholar] [CrossRef] [PubMed]
- Männistö, S.; Kontto, J.; Kataja-Tuomola, M.; Albanes, D.; Virtamo, J. High processed meat consumption is a risk factor of type 2 diabetes in the Alpha-Tocopherol, Beta-Carotene Cancer Prevention study. Br. J. Nutr. 2010, 103, 1817–1822. [Google Scholar] [CrossRef] [PubMed]
- Krisai, P.; Leib, S.; Aeschbacher, S.; Kofler, T.; Assadian, M.; Maseli, A.; Todd, J.; Estis, J.; Risch, M.; Risch, L.; et al. Relationships of iron metabolism with insulin resistance and glucose levels in young and healthy adults. Eur. J. Intern. Med. 2016, 32, 31–37. [Google Scholar] [CrossRef] [PubMed]
- Huth, C.; Beuerle, S.; Zierer, A.; Heier, M.; Herder, C.; Kaiser, T.; Koenig, W.; Kronenberg, F.; Oexle, K.; Rathmann, W.; et al. Biomarkers of iron metabolism are independently associated with impaired glucose metabolism and type 2 diabetes: The KORA F4 study. Eur. J. Endocrinol. 2015, 173, 643–653. [Google Scholar] [CrossRef] [PubMed]
- Cheung, C.L.; Cheung, T.T.; Lam, K.S.; Cheung, B.M. High ferritin and low transferrin saturation are associated with pre-diabetes among a national representative sample of U.S. adults. Clin. Nutr. 2013, 32, 1055–1060. [Google Scholar] [CrossRef] [PubMed]
- Mobbs, C.V.; Mastaitis, J.; Isoda, F.; Poplawski, M. Treatment of Diabetes and Diabetic Complications with a Ketogenic Diet. J. Child Neurol. 2013, 28, 1009–1014. [Google Scholar] [CrossRef] [PubMed]
- Feinman, R.D.; Pogozelski, W.; Astrup, A.; Bernstein, R.K.; Fine, E.J.; Westman, E.C.; Accurso, A.; Frassetto, L.; Gower, B.A.; McFarlane, S.I.; et al. Dietary carbohydrate restriction as the first approach in diabetes management: Critical review and evidence base. Nutrition 2015, 31, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Weickert, M.O.; Pfeiffer, A.F.H. Impact of Dietary Fiber Consumption on Insulin Resistance and the Prevention of Type 2 Diabetes. J. Nutr. 2018, 148, 7–12. [Google Scholar] [CrossRef] [PubMed]
- De Carvalho, C.M.; de Paula, T.P.; Viana, L.V.; Machado, V.M.; de Almeida, J.C.; Azevedo, M.J. Plasma glucose and insulin responses after consumption of breakfasts with different sources of soluble fiber in type 2 diabetes patients: A randomized crossover clinical trial. Am. J. Clin. Nutr. 2017, 106, 1238–1245. [Google Scholar] [CrossRef] [PubMed]
- Livesey, G.; Tagami, H. Interventions to lower the glycemic response to 26 carbohydrate foods with a low-viscosity fiber (resistant maltodextrin): Metaanalysis of randomized controlled trials. Am. J. Clin. Nutr. 2009, 89, 114–125. [Google Scholar] [CrossRef] [PubMed]
- Trujillo, J.M.; Nuffer, W. GLP-1 receptor agonists for type 2 diabetes mellitus: Recent developments and emerging agents. Pharmacotherapy 2014, 34, 1174–1186. [Google Scholar] [CrossRef] [PubMed]
- Eelderink, C.; Noort, M.W.; Sozer, N.; Koehorst, M.; Holst, J.J.; Deacon, C.F.; Rehfeld, J.F.; Poutanen, K.; Vonk, R.J.; Oudhuis, L.; et al. Difference in postprandial GLP-1 response despite similar glucose kinetics after consumption of wheat breads with different particle size in healthy men. Eur. J. Nutr. 2017, 56, 1063–1076. [Google Scholar] [CrossRef] [PubMed]
- Kirkman, M.S.; Briscoe, V.J.; Clark, N.; Florez, H.; Haas, L.B.; Halter, J.B.; Huang, E.S.; Korytkowski, M.T.; Munshi, M.N.; Odegard, P.S.; et al. Diabetes in older adults. Diabetes Care 2012, 35, 2650–2664. [Google Scholar] [CrossRef] [PubMed]
- McAuley, K.; Mann, J. Thematic review series: Patient-oriented research. Nutritional determinants of insulin resistance. J. Lipid Res. 2006, 47, 1668–1676. [Google Scholar] [CrossRef] [PubMed]
- Goto, A.; Morita, A.; Goto, M.; Sasaki, S.; Miyachi, M.; Aiba, N.; Kato, M.; Terauchi, Y.; Noda, M.; Watanabe, S.; et al. Validity of diabetes self-reports in the Saku diabetes study. J. Epidemiol. 2013, 23, 295–300. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
| Author | Equation |
|---|---|
| Frassetto et al. | NEAP (mEq/day) = [0.91 × protein (g/day) − 0.57 × potassium (mEq/day)] + 21 NEAP (mEq/day) = [54.5 × protein (g/day)/potassium (mEq/day)] − 10.2 |
| Remer et al. | NEAPest (mEq/day) = PRAL * + OAest |
| Sebastian et al. | NEAPest (mEq/day) = PRAL + OAest § |
| Author | Subjects 1 | Age (year) | Study Type | Variables Measured | Results | Duration/Design |
|---|---|---|---|---|---|---|
| Welch et al., 2013 [63] | 2689 women | 18–79 | Cross-sectional | Fat mass Fat-free mass PRAL | Lower quartile of PRAL correlates with a less preserved fat-free mass | - |
| Chan, 2015 [64] | 3122 men and women | >65 | Cohort Prospective | Axial muscle mass Energy-adjusted NEAP | Participants in the highest quartile of energy-adjusted estimated NEAP lost significantly more muscle mass than those in the lowest | 4 years |
| Frassetto et al., 1997 [65] | 14 postmenopausal women | 51–77 | Intervention clinical trial | NAE Nitrogen excretion | Alkali supplementation reduced NAE and nitrogen excretion | 18 days 60–120 mmol/day of KHCO3 |
| Ceglia et al., 2009 [62] | 19 men and women | 54–82 | Double-blind, randomized, placebo-controlled study | IGF-I Urinary nitrogen Urinary calcium | KHCO3 reduced the rise in urinary nitrogen excretion that accompanied an increase in protein intake | 90 mmol/day of KHCO3 41 days 2 |
| Dawson-Hughes, 2010 [66] | 71 men 91 women | >50 | Double-blind, placebo-controlled trial | NAE Nitrogen excretion Muscle power Training endurance | KHCO3 reduced NAE and nitrogen excretion. In women, bicarbonate increased double leg press power at 70% one repetition maximum by 13% | 67.5 mmol/day of KHCO3 for 3 months |
| Author | Subjects 1 | Age (year) | Study Type | Variables Measured | Results | Duration/Design |
|---|---|---|---|---|---|---|
| Farwell et al., 2008 [69] | 1496 women | >12 | Cross-sectional | HCO3− Insulin resistance via both HOMA-IR and MFFM | Lower anion gap and bicarbonates correlate with increased insulin resistance | - |
| Mandel et al., 2012 [67] | 630 (and 730 controls) (nurses) | 30–55 | Prospective nested case-control | HCO3− Self-Reported T2D diagnosis | Lower bicarbonates correlate with increased diabetes T2 incidence | 10 years |
| Fagherazzi et al., 2014 [15] | 66, 485 women (teachers) | mean 53 | Cohort retrospective | PRAL NEAP Self-reported T2D 2 | Highest PRAL-NEAP quartile shows higher incidence of diabetes T2 compared to lowest | 14 years |
| Kiefte-de Jong et al., 2016 [14] | 67,433 women 3 84,310 women 35,743 men | 30–55 25–42 40–75 | Cohort retrospective | PRAL NEAP A:P ratio 4 T2D | Highest PRAL-NEAP and A:P quartile shows higher incidence of diabetes T2 compared to lowest | 24 years |
| Akter, et al., 2016 [71] | 1536 men 169 women (manifacture workers) | 19–69 | Cross-sectional | PRAL, NEAP HOMA-IR HOMA-β HbA1c Fasting glucose | PRAL and NEAP associated with HOMA-IR 5 NEAP positively associates with HOMA-β No association with fasting glucose and HbA1c | - |
| Akter, et al., 2016 [70] | 27,809 men 36,851 women | 45–75 | Cohort retrospective | PRAL, NEAP Self reported T2D diagnosis | Only PRAL associates with T2D incidence in men < 50 year-old | 10 years |
| Xu et al., 2014 [13] | 911 men | 70–71 | Cohort Prospective | PRAL, NEAP Insulin resistance T2D 6 | No association of PRAL-NEAP with insulin sensitivity, β-cell function or diabetes incidence | 18 years |
| Kozan et al., 2017 [72] | 20 men 10 women | 24–44 | Placebo-controlled, crossover trial | C-peptide Insulin Fasting glucose Glucose (0–180′) GLP-1 | No effect of NaHCO3 on postprandial insulin, plasma glucose, C-peptide and GLP-1 compared to placebo | 0-180 min - placebo - NaHCO3 (1680 mg) |
| Harris et al., 2010 [73] | 153 men and women 6 | >50 mean 64 | Randomized, placebo-controlled trial | HOMA-IR Insulin Fasting glucose | No effect of either NaHCO3 or KHCO3 on insulin, plasma glucose and HOMA-IR compared to placebo | 84 days - placebo or 67.5 mmol/day of - KCl - NaHCO3 - KHCO3 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Della Guardia, L.; Thomas, M.A.; Cena, H. Insulin Sensitivity and Glucose Homeostasis Can Be Influenced by Metabolic Acid Load. Nutrients 2018, 10, 618. https://doi.org/10.3390/nu10050618
Della Guardia L, Thomas MA, Cena H. Insulin Sensitivity and Glucose Homeostasis Can Be Influenced by Metabolic Acid Load. Nutrients. 2018; 10(5):618. https://doi.org/10.3390/nu10050618
Chicago/Turabian StyleDella Guardia, Lucio, Michael Alex Thomas, and Hellas Cena. 2018. "Insulin Sensitivity and Glucose Homeostasis Can Be Influenced by Metabolic Acid Load" Nutrients 10, no. 5: 618. https://doi.org/10.3390/nu10050618
APA StyleDella Guardia, L., Thomas, M. A., & Cena, H. (2018). Insulin Sensitivity and Glucose Homeostasis Can Be Influenced by Metabolic Acid Load. Nutrients, 10(5), 618. https://doi.org/10.3390/nu10050618