
Brain-Targeted Intranasal Delivery of Zotepine Microemulsion: Pharmacokinetics and Pharmacodynamics

 , , and
, , and
Abstract
1. Introduction
2. Materials and Methods
2.1. Materials
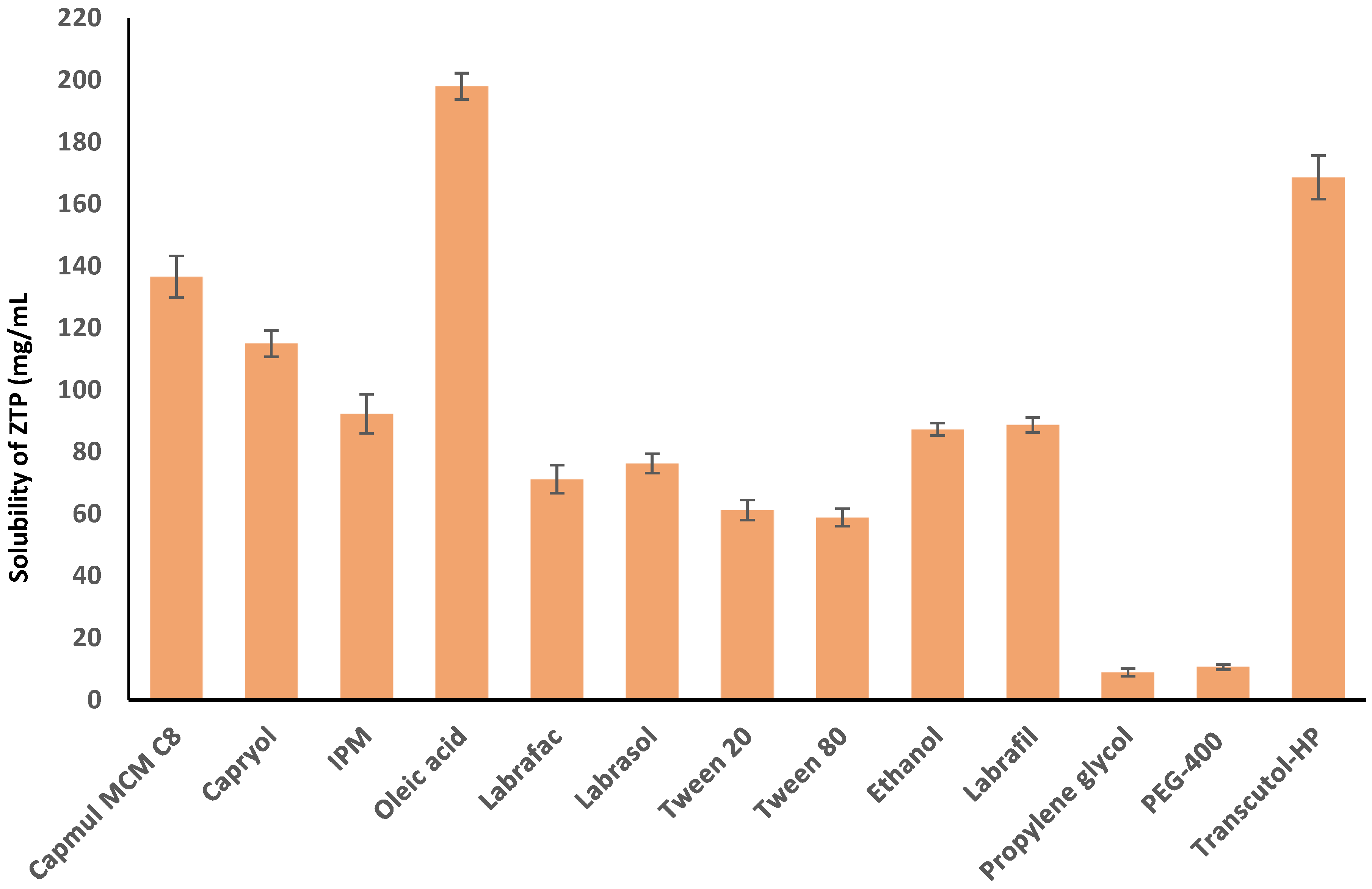
2.2. Solubility of ZTP in Various Vehicles
2.3. Formulation Development
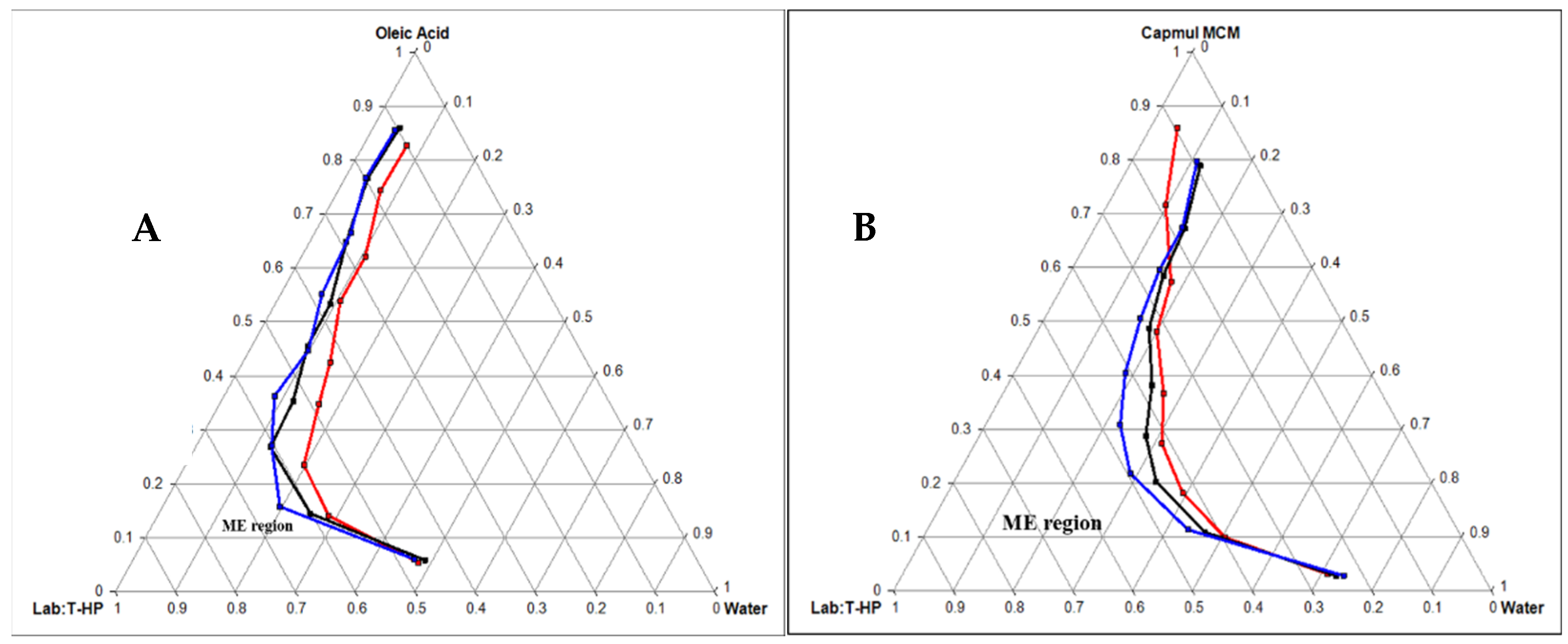
2.3.1. Construction of Pseudo Ternary Phase Diagram
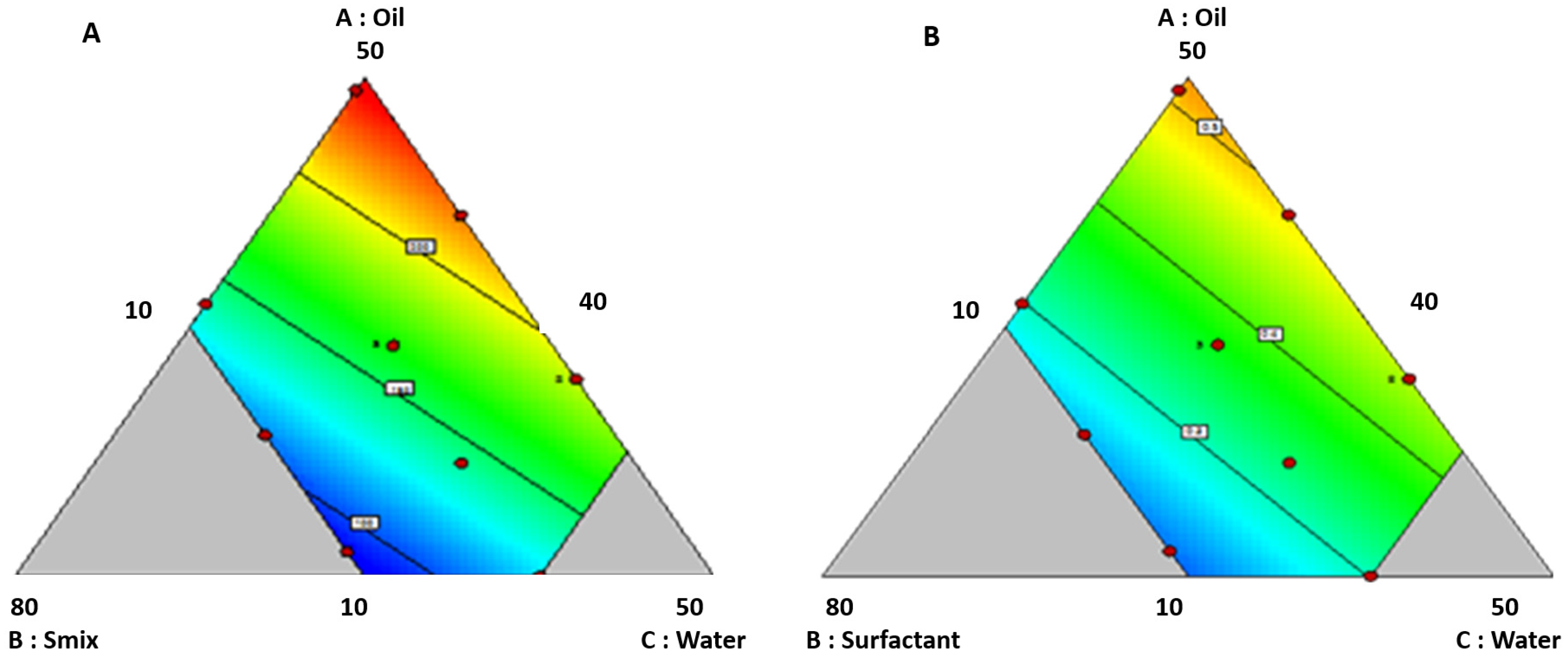
2.3.2. Optimization of ZTP Loaded ME by Quality by Design (QbD) Approach
2.3.3. Preparation of ZTP-ME
2.4. Physicochemical Characterization of ZTP-ME
2.4.1. Determination of Globule Size, PDI, and Zeta Potential (ZP)
2.4.2. Drug Content and pH
2.4.3. Freeze-Thaw Stability and Long-Term Stability Studies
2.5. Ex Vivo Studies
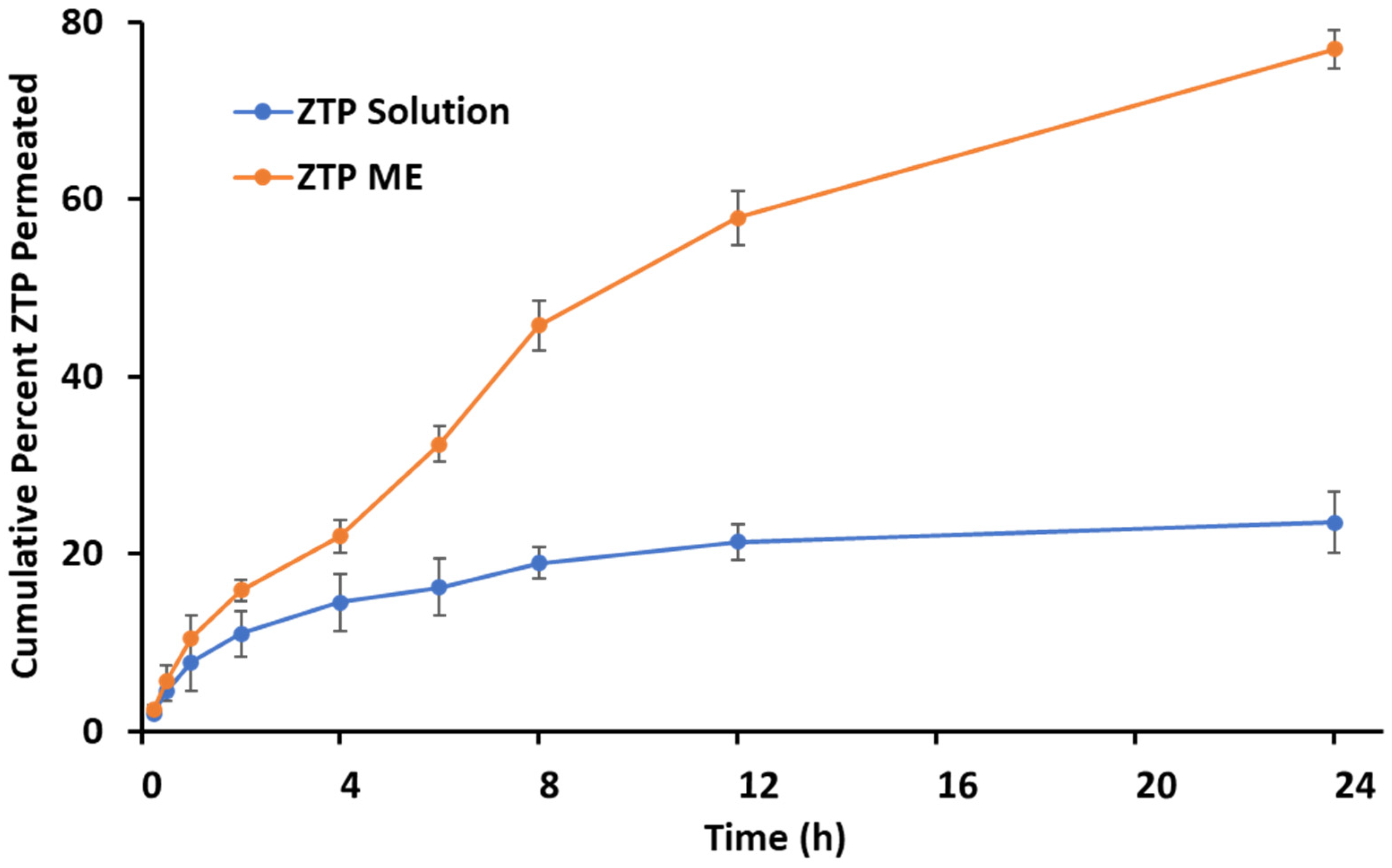
2.5.1. Ex Vivo Permeation
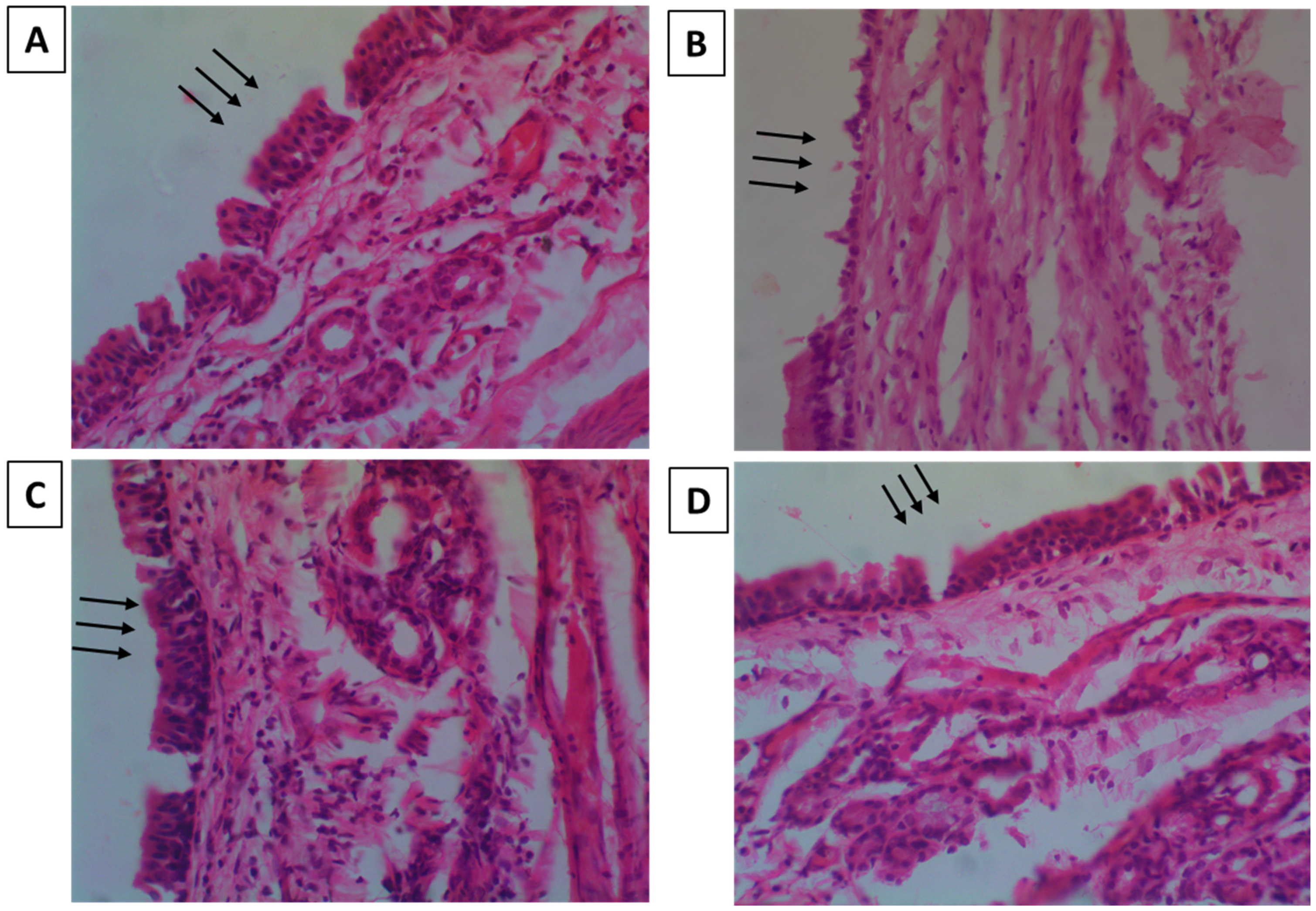
2.5.2. Nasal-Cilia Toxicity by Histopathology
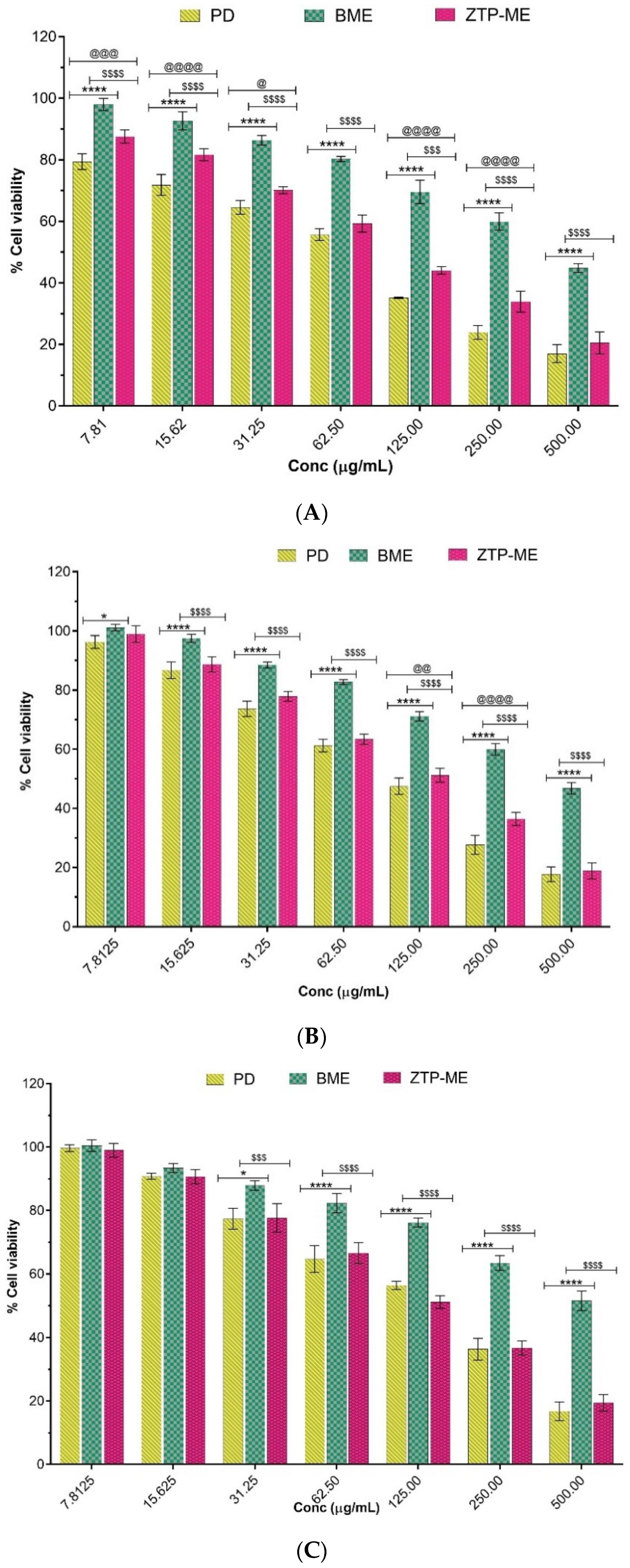
2.6. In Vitro Cytotoxicity
2.7. In Vivo Pharmacokinetic Study
2.7.1. Animal Experimentation
2.7.2. Sample Collection, and Processing
2.7.3. Bioanalytical Method Development and Validation
2.7.4. Pharmacokinetic Parameters
- BIV = area under curve0–t in the brain resulted from intravenous administration;
- PIV = area under curve0–t in the blood resulted from intravenous administration;
- BIN = area under curve0–t in the brain resulted from intranasal administration;
- PIN = area under curve0–t in the blood resulted from intranasal administration.
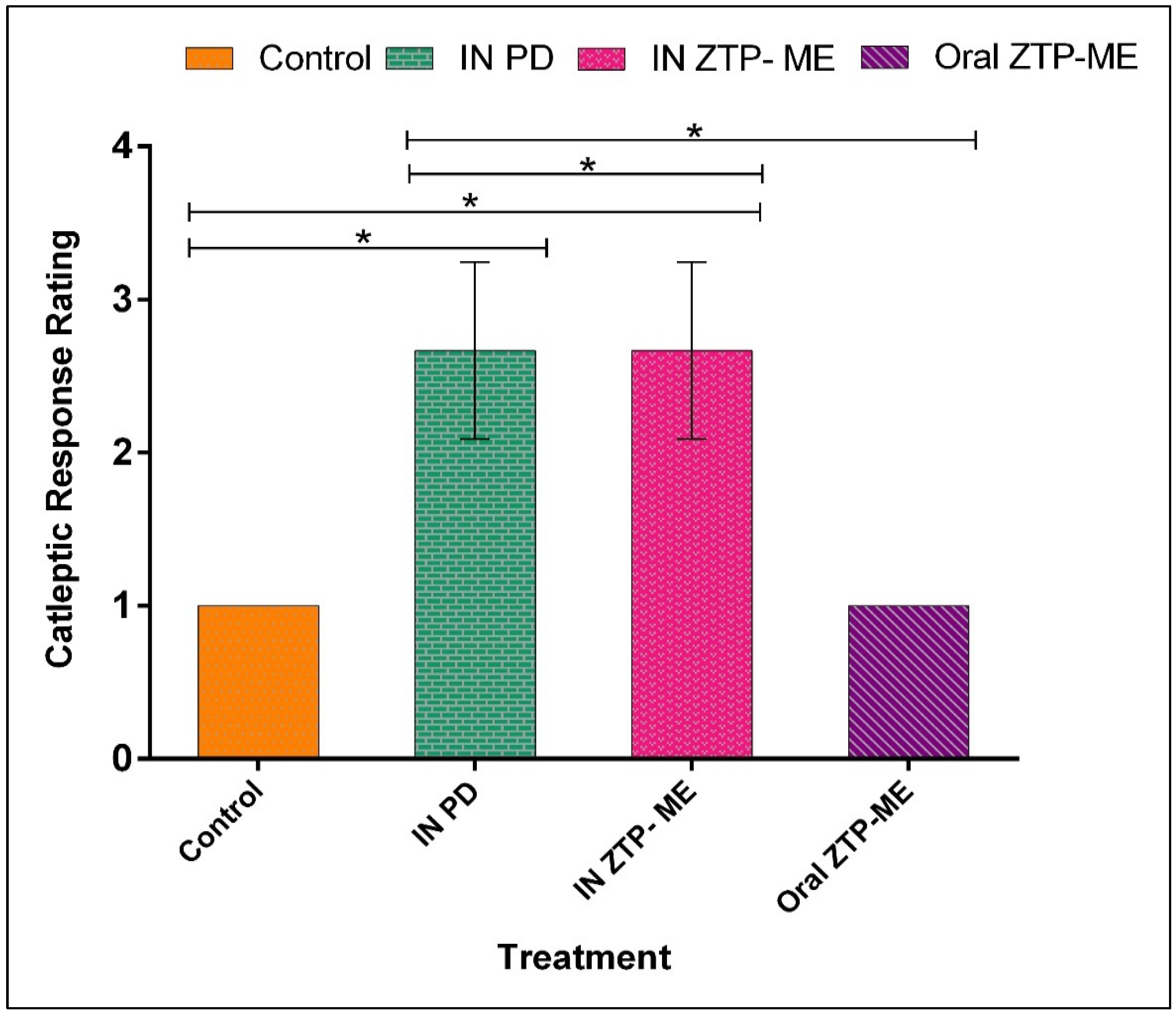
2.8. In Vivo Anti-Schizophrenic by Catalepsy Test
2.9. Statistical Analysis
3. Results and Discussion
3.1. Solubility of ZTP in Various Vehicles
3.2. Formulation Development
Pseudo Ternary Phase Diagram
3.3. Optimization of ZTP-ME by QbD
3.4. Physicochemical Characterization of ZTP-ME
3.4.1. Globule Size, PDI, and Zeta Potential (ZP)
3.4.2. pH and Drug Content
3.4.3. Freeze-Thaw Stability and Long-Term Stability Studies
3.5. Ex Vivo Studies
3.5.1. Ex Vivo Permeation
3.5.2. Nasal-Cilia Toxicity by Histopathology
3.6. In Vitro Cytotoxicity
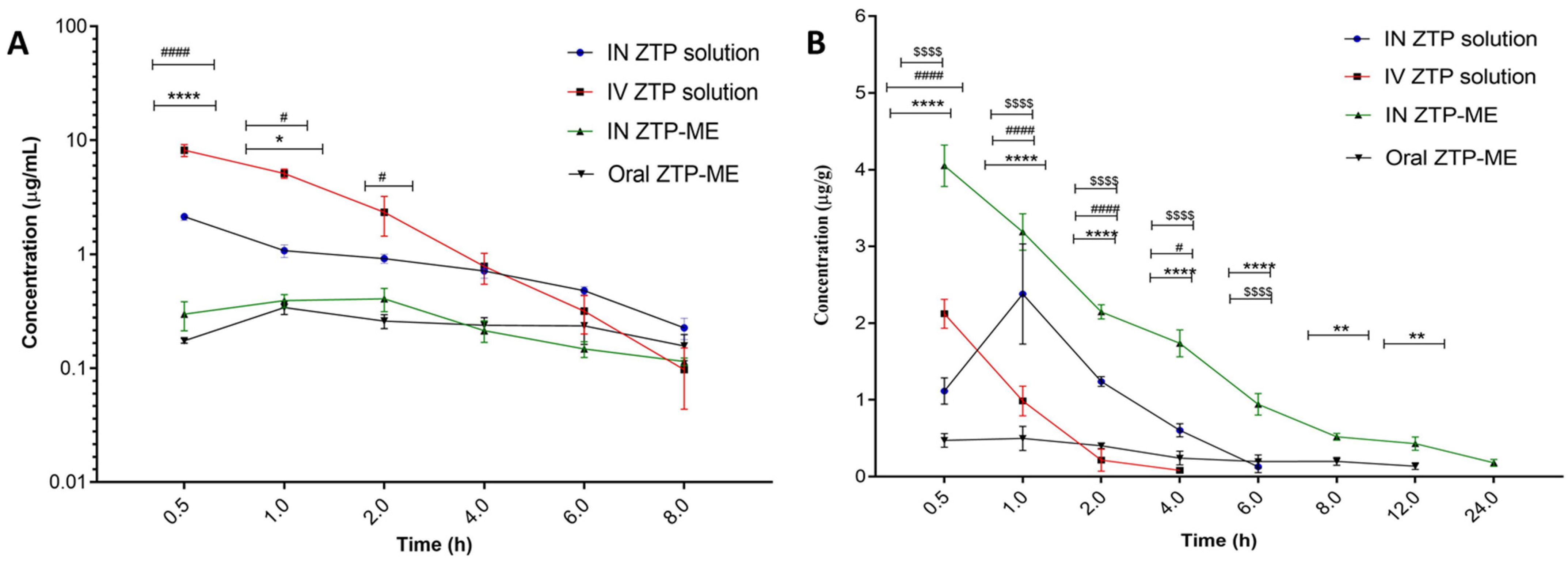
3.7. In Vivo Pharmacokinetic Study
3.7.1. Bioanalytical Method Development and Validation
3.7.2. Pharmacokinetics in Plasma and Brain
3.8. In Vivo Anti-Schizophrenic by Catalepsy Test
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
References
- Stępnicki, P.; Kondej, M.; Kaczor, A.A. Current concepts and treatments of schizophrenia. Molecules 2018, 23, 2087. [Google Scholar] [CrossRef] [PubMed]
- Prakash, A.; Lamb, H.M. Zotepine: A review of its pharmacodynamic and pharmacokinetic properties and therapeutic efficacy in the management of schizophrenia. CNS Drugs 1998, 9, 280. [Google Scholar] [CrossRef]
- Shobo, M.; Kondo, Y.; Yamada, H.; Mihara, T.; Yamamoto, N.; Katsuoka, M.; Harada, K.; Ni, K.; Matsuoka, N. Norzotepine, a Major Metabolite of Zotepine, Exerts Atypical Antipsychotic-Like and Antidepressant-Like Actions through Its Potent Inhibition of Norepinephrine Reuptake. J. Pharmacol. Exp. Ther. 2010, 333, 772–781. [Google Scholar] [CrossRef] [PubMed]
- Schotte, A.; Janssen, P.F.M.; Gommeren, W.; Luyten, W.H.M.L.; Van Gompel, P.; Lesage, A.S.; De Loore, K.; Leysen, J.E. Risperidone compared with new and reference antipsychotic drugs: In vitro and in vivo receptor binding. Psychopharmacology 1996, 124, 57–73. [Google Scholar] [CrossRef] [PubMed]
- Monahan, M. Pimavanserin (NuplazidTM) for the Treatment of Parkinson’s Disease Psychosis. Ment. Health Clin. 2018, 7, 230–234. [Google Scholar]
- Banala, N.; Suram, D.; Dudhipala, N.; Nagaraj, B. Design, development and in vivo pharmacokinetic evaluation of zotepine loaded solid lipid nanoparticles for enhanced oral bioavailability. Acta Pharm. Sci. 2021, 59, 385–404. [Google Scholar] [CrossRef]
- Setthacheewakul, S.; Mahattanadul, S.; Phadoongsombut, N.; Pichayakorn, W.; Wiwattanapatapee, R. Development and evaluation of self-microemulsifying liquid and pellet formulations of curcumin, and absorption studies in rats. Eur. J. Pharm. Biopharm. 2010, 76, 475–485. [Google Scholar] [CrossRef]
- Banks, W.A. Characteristics of compounds that cross the blood-brain barrier. BMC Neurol. 2009, 9, S3. [Google Scholar] [CrossRef]
- Miller, D.S. Regulation of P-glycoprotein and other ABC drug transporters at the blood–brain barrier. Trends Pharmacol. Sci. 2010, 31, 246–254. [Google Scholar] [CrossRef]
- Cooper, S.J.; Butler, A.; Tweed, J.; Welch, C.; Raniwalla, J. Zotepine in the prevention of recurrence: A randomised, double-blind, placebo-controlled study for chronic schizophrenia. Psychopharmacology 2000, 150, 237–243. [Google Scholar] [CrossRef]
- Gareri, P.; De Fazio, P.; De Fazio, S.; Marigliano, N.; Ferreri Ibbadu, G.; De Sarro, G. Adverse Effects of Atypical Antipsychotics in the Elderly. Drugs Aging 2006, 23, 937–956. [Google Scholar] [CrossRef] [PubMed]
- Leucht, S.; Cipriani, A.; Spineli, L.; Mavridis, D.; Örey, D.; Richter, F.; Samara, M.; Barbui, C.; Engel, R.R.; Geddes, J.R.; et al. Comparative efficacy and tolerability of 15 antipsychotic drugs in schizophrenia: A multiple-treatments meta-analysis. Lancet 2013, 382, 951–962. [Google Scholar] [CrossRef]
- Md, S.; Khan, R.A.; Mustafa, G.; Chuttani, K.; Baboota, S.; Sahni, J.K.; Ali, J. Bromocriptine loaded chitosan nanoparticles intended for direct nose to brain delivery: Pharmacodynamic, Pharmacokinetic and Scintigraphy study in mice model. Eur. J. Pharm. Sci. 2013, 48, 393–405. [Google Scholar] [CrossRef] [PubMed]
- Mistry, A.; Stolnik, S.; Illum, L. Nanoparticles for direct nose-to-brain delivery of drugs. Int. J. Pharm. 2009, 379, 146–157. [Google Scholar] [CrossRef] [PubMed]
- Bhavna; Md, S.; Ali, M.; Ali, R.; Bhatnagar, A.; Baboota, S.; Ali, J. Donepezil nanosuspension intended for nose to brain targeting: In vitro and in vivo safety evaluation. Int. J. Biol. Macromol. 2014, 67, 418–425. [Google Scholar] [CrossRef]
- Popescu, C.; Manda, P.; Juluri, A.; Leon, Z.; Repka, M.A.; Murthy, N. Preparation and Characterization of Zotepine Solid Didpersions by Cyclodextrin Complexation. In Proceedings of the AAPS Annual Meeting, San Antonio, TX, USA, 10–14 November 2013. [Google Scholar]
- Dalvadi, H.; Patel, N.; Parmar, K. Systematic development of design of experiments (DoE) optimised self-microemulsifying drug delivery system of Zotepine. J. Microencapsul. 2017, 34, 308–318. [Google Scholar] [CrossRef]
- Pailla, S.R.; Talluri, S.; Rangaraj, N.; Ramavath, R.; Challa, V.S.; Doijad, N.; Sampathi, S. Intranasal Zotepine Nanosuspension: Intended for improved brain distribution in rats. DARU J. Pharm. Sci. 2019, 27, 541–556. [Google Scholar] [CrossRef]
- Nagaraj, B.; Tirumalesh, C.; Dinesh, S.; Narendar, D. Zotepine loaded lipid nanoparticles for oral delivery: Development, characterization, and in vivo pharmacokinetic studies. J. Pharm. Sci. 2020, 6, 37. [Google Scholar] [CrossRef]
- Piao, H.; Balakrishnan, P.; Cho, H.; Kim, H.; Kim, Y.S.; Chung, S.J.; Shim, C.K.; Kim, D. Preparation and evaluation of fexofenadine microemulsions for intranasal delivery. Int. J. Pharm. 2010, 395, 309–316. [Google Scholar] [CrossRef]
- Zhang, Q.; Jiang, X.; Jiang, W.; Lu, W.; Su, L.; Shi, Z. Preparation of nimodipine-loaded microemulsion for intranasal delivery and evaluation on the targeting efficiency to the brain. Int. J. Pharm. 2004, 275, 85–96. [Google Scholar] [CrossRef]
- Goos, P.; Jones, B.; Syafitri, U. I-Optimal Design of Mixture Experiments. J. Am. Stat. Assoc. 2016, 111, 899–911. [Google Scholar] [CrossRef]
- Dhingani, A.; Patel, J.; Garala, K.; Raval, M.; Dharamsi, A. Quality by Design Approach for Development of W/O Type Microemulsion-Based Transdermal Systems for Atenolol. J. Dispers. Sci. Technol. 2014, 35, 619–640. [Google Scholar] [CrossRef]
- Komati, S.; Swain, S.; Rao, M.E.B.; Jena, B.R.; Unnam, S.; Dasi, V. QbD-based design and characterization of mucoadhesive microspheres of quetiapine fumarate with improved oral bioavailability and brain biodistribution potential. Bull. Fac. Pharm. Cairo Univ. 2018, 56, 129–145. [Google Scholar] [CrossRef]
- Rangaraj, N.; Pailla, S.R.; Chowta, P.; Sampathi, S. Fabrication of Ibrutinib Nanosuspension by Quality by Design Approach: Intended for Enhanced Oral Bioavailability and Diminished Fast Fed Variability. AAPS PharmSciTech 2019, 20, 326. [Google Scholar] [CrossRef]
- Shah, B.; Khunt, D.; Misra, M.; Padh, H. Non-invasive intranasal delivery of quetiapine fumarate loaded microemulsion for brain targeting: Formulation, physicochemical and pharmacokinetic consideration. Eur. J. Pharm. Sci. 2016, 91, 196–207. [Google Scholar] [CrossRef]
- Shah, B.M.; Misra, M.; Shishoo, C.J.; Padh, H. Nose to brain microemulsion-based drug delivery system of rivastigmine: Formulation and ex-vivo characterization. Drug Deliv. 2015, 22, 918–930. [Google Scholar] [CrossRef]
- Shinde, R.L.; Bharkad, G.P.; Devarajan, P.V. Intranasal microemulsion for targeted nose to brain delivery in neurocysticercosis: Role of docosahexaenoic acid. Eur. J. Pharm. Biopharm. 2015, 96, 363–379. [Google Scholar] [CrossRef]
- European Medicines Agency. ICH Topic Q1A (R2) Stability Testing of New Drug Substances and Products. 2003. Available online: www.ich.org (accessed on 7 April 2022).
- Samson, G.; García De La Calera, A.; Dupuis-Girod, S.; Faure, F.; Decullier, E.; Paintaud, G.; Vignault, C.; Scoazec, J.Y.; Pivot, C.; Plauchu, H.; et al. Ex vivo study of bevacizumab transport through porcine nasal mucosa. Eur. J. Pharm. Biopharm. 2012, 80, 465–469. [Google Scholar] [CrossRef]
- Shah, B.; Khunt, D.; Bhatt, H.; Misra, M.; Padh, H. Application of quality by design approach for intranasal delivery of rivastigmine loaded solid lipid nanoparticles: Effect on formulation and characterization parameters. Eur. J. Pharm. Sci. 2015, 78, 54–66. [Google Scholar] [CrossRef]
- Patel, R.B.; Patel, M.R.; Bhatt, K.K.; Patel, B.G. Formulation consideration and characterization of microemulsion drug delivery system for transnasal administration of carbamazepine. Bull. Fac. Pharm. Cairo Univ. 2013, 51, 243–253. [Google Scholar] [CrossRef]
- Gonçalves, V.S.S.; Matias, A.A.; Poejo, J.; Serra, A.T.; Duarte, C.M.M. Application of RPMI 2650 as a cell model to evaluate solid formulations for intranasal delivery of drugs. Int. J. Pharm. 2016, 515, 1–10. [Google Scholar] [CrossRef] [PubMed]
- So, M.Y.; Tian, Z.; Phoon, Y.S.; Sha, S.; Antoniou, M.N.; Zhang, J.; Wu, R.S.S.; Tan-Un, K.C. Gene Expression Profile and Toxic Effects in Human Bronchial Epithelial Cells Exposed to Zearalenone. PLoS ONE 2014, 9, e96404. [Google Scholar] [CrossRef] [PubMed]
- Nigam, K.; Kaur, A.; Tyagi, A.; Manda, K.; Gabrani, R.; Dang, S. Baclofen-Loaded Poly (D,L-Lactide-Co-Glycolic Acid) Nanoparticles for Neuropathic Pain Management: In Vitro and In Vivo Evaluation. Rejuvenation Res. 2019, 22, 235–245. [Google Scholar] [CrossRef] [PubMed]
- PatilSatish, K.; WaghKalpesh, S.; Kamlesh, M.; BaviskarDheeraj, T.; JainDinesh, K.; Pharmacy, C.; Academy, I.P.S.; Nagar, R. Development and evaluation of solid lipid nanoparticles containing anti-migraine drug. World J. Pharm. Sci. 2014, 2, 1014–1021. [Google Scholar]
- Vyas, T.K.; Babbar, A.K.; Sharma, R.K.; Singh, S.; Misra, A. Preliminary brain-targeting studies on intranasal mucoadhesive microemulsions of sumatriptan. AAPS PharmSciTech 2006, 7, E49–E57. [Google Scholar] [CrossRef] [PubMed]
- Youssef, N.A.H.A.; Kassem, A.A.; Farid, R.M.; Ismail, F.A.; EL-Massik, M.A.E.; Boraie, N.A. A novel nasal almotriptan loaded solid lipid nanoparticles in mucoadhesive in situ gel formulation for brain targeting: Preparation, characterization and in vivo evaluation. Int. J. Pharm. 2018, 548, 609–624. [Google Scholar] [CrossRef] [PubMed]
- Yasir, M.; Sara, U.V.S. Solid lipid nanoparticles for nose to brain delivery of haloperidol: In vitro drug release and pharmacokinetics evaluation. Acta Pharm. Sin. B 2014, 4, 454–463. [Google Scholar] [CrossRef]
- Mahajan, H.S.; Mahajan, M.S.; Nerkar, P.P.; Agrawal, A. Nanoemulsion-based intranasal drug delivery system of saquinavir mesylate for brain targeting. Drug Deliv. 2014, 21, 148–154. [Google Scholar] [CrossRef]
- Yasir, M.; Sara, U.; Som, I. Haloperidol Loaded Solid Lipid Nanoparticles for Nose to Brain Delivery: Stability and In vivo Studies. J. Nanomed. Nanotechnol. 2015. [Google Scholar] [CrossRef]
- Sanberg, P.R.; Bunsey, M.D.; Giordano, M.; Norman, A.B. The Catalepsy Test: Its Ups and Downs. Behav. Neurosci. 1988, 102, 748–759. [Google Scholar] [CrossRef]
- Wong, Y.C.; Zuo, Z. Brain Disposition and Catalepsy After Intranasal Delivery of Loxapine: Role of Metabolism in PK/PD of Intranasal CNS Drugs. Pharm. Res. 2013, 30, 2368–2384. [Google Scholar] [CrossRef] [PubMed]
- Adams, M.; Brandon, E.; Chartoff, R.H.; Idzerda, R.L.; Dorsa, D.M.; McKnight, G. Loss of haloperidol induced gene expression and catalepsy in protein kinase A-deficient mice. Proc. Natl. Acad. Sci. USA 1997, 94, 12157–12161. [Google Scholar] [CrossRef] [PubMed]
- Piazza, J.; Hoare, T.; Molinaro, L.; Terpstra, K.; Bhandari, J.; Selvaganapathy, P.R.; Gupta, B.; Mishra, R. Haloperidol-loaded intranasally administered lectin functionalized poly (ethylene glycol)–block-poly (D, L)-lactic-co-glycolic acid (PEG–PLGA) nanoparticles for the. Eur. J. Pharm. Biopharm. 2014, 87, 30–39. [Google Scholar] [CrossRef] [PubMed]
- Patel, K.; Sarma, V.; Vavia, P. Design and evaluation of Lumefantrine—Oleic acid self nanoemulsifying ionic complex for enhanced dissolution. DARU J. Pharm. Sci. 2013, 21, 27. [Google Scholar] [CrossRef] [PubMed]
- Bandivadeka, M.M.; Pancholi, S.S.; Kaul-Ghanekar, R.; Choudhari, A.; Koppikar, S. Self-microemulsifying smaller molecular volume oil (Capmul MCM) using non-ionic surfactants: A delivery system for poorly water-soluble drug. Drug Dev. Ind. Pharm. 2012, 38, 883–892. [Google Scholar] [CrossRef] [PubMed]
- Santos, P.; Watkinson, A.C.; Hadgraft, J.; Lane, M.E. Application of Microemulsions in Dermal and Transdermal Drug Delivery. Skin Pharmacol. Physiol. 2008, 21, 246–259. [Google Scholar] [CrossRef]
- He, C.X.; He, Z.G.; Gao, J.Q. Microemulsions as drug delivery systems to improve the solubility and the bioavailability of poorly water-soluble drugs. Expert Opin. Drug Deliv. 2010, 7, 445–460. [Google Scholar] [CrossRef]
- Talegaonkar, S.; Negi, L.M. Nanoemulsion in Drug Targeting. In Targeted Drug Delivery: Concepts and Design; Springer: Berlin/Heidelberg, Germany, 2015; pp. 433–459. [Google Scholar]
- Choi, D.H.; Kim, Y.S.; Kim, D.D.; Jeong, S.H. QbD based development and evaluation of topical microemulsion-based hydrogel against superficial fungal infections. J. Pharm. Investig. 2019, 49, 87–103. [Google Scholar] [CrossRef]
- Bhattacharjee, S. DLS and zeta potential—What they are and what they are not? J. Control. Release 2016, 235, 337–351. [Google Scholar] [CrossRef]
- Fabricant, N.D. Significance of the pH of nasal secretions insitu: Further studies. Arch. Otolaryngol. 1941, 34, 297–301. [Google Scholar] [CrossRef]
- Malik, M.A.; Wani, M.Y.; Hashim, M.A. Microemulsion method: A novel route to synthesize organic and inorganic nanomaterials. 1st Nano Update. Arab. J. Chem. 2012, 5, 397–417. [Google Scholar] [CrossRef]
- Bunchongprasert, K.; Shao, J. Cytotoxicity and permeability enhancement of Capmul®MCM in nanoemulsion formulation. Int. J. Pharm. 2019, 561, 289–295. [Google Scholar] [CrossRef]
- Holmes, E.H.; Devalapally, H.; Li, L.; Perdue, M.L.; Ostrander, G.K. Permeability Enhancers Dramatically Increase Zanamivir Absolute Bioavailability in Rats: Implications for an Orally Bioavailable Influenza Treatment. PLoS ONE 2013, 8, e61853. [Google Scholar] [CrossRef] [PubMed]
- Espinoza, L.C.; Silva-Abreu, M.; Clares, B.; Rodríguez-Lagunas, M.J.; Halbaut, L.; Cañas, M.A.; Calpena, A.C. Formulation strategies to improve nose-to-brain delivery of donepezil. Pharmaceutics 2019, 11, 64. [Google Scholar] [CrossRef] [PubMed]
- Moghadam, S.H.; Saliaj, E.; Wettig, S.D.; Dong, C.; Ivanova, M.V.; Huzil, J.T.; Foldvari, M. Effect of chemical permeation enhancers on stratum corneum barrier lipid organizational structure and interferon alpha permeability. Mol. Pharm. 2013, 10, 2248–2260. [Google Scholar] [CrossRef]
- Noda, K.; Suzuki, A.; Okui, M.; Noguchi, H.; Nishiura, M.; Nishiura, N. Pharmacokinetics and metabolism of 2-chloro-11-(2-dimethylaminoethoxy)-dibenzo[b,f]thiepine (zotepine) in rat, mouse, dog and man. Arzneimittel-Forschung/Drug Res. 1979, 29, 1595–1600. [Google Scholar]
- Mahmoud, E.A.; Bendas, E.R.; Mohamed, M.I. Preparation and Evaluation of Self-nanoemulsifying Tablets of Carvedilol. AAPS PharmSciTech 2009, 10, 183–192. [Google Scholar] [CrossRef]
- Nornoo, A.O.; Zheng, H.A.; Lopes, L.B.; Johnson-Restrepo, B.; Kannan, K.; Reed, R. Oral microemulsions of paclitaxel: In situ and pharmacokinetic studies. Eur. J. Pharm. Biopharm. 2009, 71, 310–317. [Google Scholar] [CrossRef]
- Cornaire, G.; Woodley, J.; Hermann, P.; Cloarec, A.; Arellano, C.; Houin, G. Impact of excipients on the absorption of P-glycoprotein substrates in vitro and in vivo. Int. J. Pharm. 2004, 278, 119–131. [Google Scholar] [CrossRef]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Trials | (%w/w) |
Smix (1:1) (%w/w) |
Aqueous Phase (%w/w) |
Response 1 Size (nm) |
Response 2 PDI | Response 3 ZP (mV) |
|---|---|---|---|---|---|---|
| 1 | 31.86 | 58.13 | 10.22 | 156.8 ± 3.23 | 0.282 ± 0.015 | 37.5 ± 1.52 |
| 2 | 19.09 | 49.90 | 31.12 | 152.9 ±1.23 | 0.368 ± 0.087 | 36.2 ± 2.45 |
| 3 | 21.35 | 60.32 | 18.64 | 90.1 ± 3.43 | 0.171 ± 0.023 | 40.8 ± 3.35 |
| 4 | 25.82 | 40.13 | 34.17 | 180.5 ± 4.23 | 0.409 ± 0.015 | 36.0 ± 4.12 |
| 5 | 38.99 | 40.22 | 21.006 | 238.3 ± 5.65 | 0.547 ± 0.035 | 35.1 ± 1.35 |
| 6 | 10.13 | 50.23 | 40.12 | 124.6 ± 3.52 | 0.212 ± 0.013 | 38.7 ± 2.35 |
| 7 | 25.82 | 40.76 | 34.17 | 194.3 ± 0.91 | 0.416 ± 0.077 | 36.4 ± 3.29 |
| 8 | 12.87 | 60.24 | 28.09 | 92.81 ± 2.81 | 0.347 ± 0.098 | 37.3 ± 1.99 |
| 9 | 28.55 | 49.08 | 22.35 | 139.7 ± 1.98 | 0.216 ± 0.033 | 40.1 ± 1.66 |
| 10 | 49.22 | 41.23 | 10.65 | 229.2 ± 2.98 | 0.584 ± 0.011 | 43.2 ± 4.35 |
| 11 | 28.55 | 49.08 | 22.35 | 163.1 ± 3.54 | 0.393± 0.017 | 41.1 ± 3.54 |
| 12 | 28.55 | 49.08 | 22.35 | 173.4 ± 1.34 | 0.352 ± 0.013 | 36.6 ± 3.33 |
| Temperature | Duration | PS (nm) | PDI | ZP (mV) | Drug Content (%) |
|---|---|---|---|---|---|
| 30 ± 2 °C/65% RH | 0 day | 124.6 ± 7.33 | 0.21 ± 0.013 | 38.7 ± 2.35 | 98.92 ± 0.74 |
| 15 days | 118.5 ± 5.91 | 0.18 ± 0.032 | 31.9 ± 2.83 | 99.42 ± 0.53 | |
| 1 month | 116.6 ± 5.42 | 0.15 ± 0.052 | 29.7 ± 3.62 | 98.67 ± 0.24 | |
| 3 months | 121.2 ±9.29 | 0.19 ± 0.064 | 36.1 ± 4.18 | 97.24 ± 0.47 | |
| 6 months | 120.3 ± 3.48 | 0.17 ± 0.037 | 33.5 ± 3.28 | 97.73 ± 0.63 |
| Samples | Neuro 2A | Beas 2B | RPMI 2650 |
|---|---|---|---|
| PD | 142.46 ± 9.88 | 88.08 ± 5.29 | 93.55 ± 13.91 |
| BME | 193.93 ± 8.10 | 155.9 ± 8.91 | 188.66 ± 7.13 |
| ZTP-ME | 98.75 ± 10.90 | 99.54 ± 12.41 | 89.43 ± 8.27 |
| Parameter | Organ | IV ZTP | IN ZTP | IN ZTP-ME | Oral ZTP- ME |
|---|---|---|---|---|---|
| Cmax (µg/mL) | Plasma | 8.18 ± 0.97 | 2.13 ± 0.14 | 0.41 ± 0.07 | 0.34 ± 0.04 |
| Brain | 2.37 ± 0.65 | 1.90 ± 0.37 | 4.04 ± 0.26 | 0.57 ± 0.04 | |
| Tmax(h) | Plasma | 0.5 ± 0.12 | 0.5 ± 0.23 | 1.66 ± 0.65 | 1± 0.47 |
| Brain | 0.5 ± 0.32 | 1.02 ± 0.21 | 0.5 ± 0.26 | 2.01 ± 0.25 | |
| AUC0–24 (µg*h/mL) | Plasma | 14.27 ±2.49 | 6.76 ± 0.24 | 2.98 ± 0.05 | 2.38 ± 0.71 |
| Brain | 4.40 ± 0.36 | 5.87 ± 0.47 | 18.63 ±1.33 *** | 3.10 ± 0.92 | |
| t1/2 (h) | Plasma | 2.41 ± 0.96 | 2.49 ± 0.21 | 7.94 ±1.82 | 11.08 ± 1.87 |
| Brain | 0.83 ± 0.21 | 4.21 ± 1.33 | 5.31 ± 0.48 | 8.35 ± 0.29 | |
| MRT (h) | Plasma | 2.06 ± 0.44 | 3.956 ± 0.28 | 12.21 ± 2.91 | 15.94 ± 2.62 |
| Brain | 0.97 ± 0.15 | 3.04 ± 0.24 | 7.99 ± 0.94 | 14.49 ± 2.38 | |
| DTE % | - | - | 520.70 | 3754.97 **** | - |
| DTP | - | - | 80.6 | 97.54 | - |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pailla, S.R.; Sampathi, S.; Junnuthula, V.; Maddukuri, S.; Dodoala, S.; Dyawanapelly, S. Brain-Targeted Intranasal Delivery of Zotepine Microemulsion: Pharmacokinetics and Pharmacodynamics. Pharmaceutics 2022, 14, 978. https://doi.org/10.3390/pharmaceutics14050978
Pailla SR, Sampathi S, Junnuthula V, Maddukuri S, Dodoala S, Dyawanapelly S. Brain-Targeted Intranasal Delivery of Zotepine Microemulsion: Pharmacokinetics and Pharmacodynamics. Pharmaceutics. 2022; 14(5):978. https://doi.org/10.3390/pharmaceutics14050978
Chicago/Turabian StylePailla, Sravanthi Reddy, Sunitha Sampathi, Vijayabhaskarreddy Junnuthula, Sravya Maddukuri, Sujatha Dodoala, and Sathish Dyawanapelly. 2022. "Brain-Targeted Intranasal Delivery of Zotepine Microemulsion: Pharmacokinetics and Pharmacodynamics" Pharmaceutics 14, no. 5: 978. https://doi.org/10.3390/pharmaceutics14050978
APA StylePailla, S. R., Sampathi, S., Junnuthula, V., Maddukuri, S., Dodoala, S., & Dyawanapelly, S. (2022). Brain-Targeted Intranasal Delivery of Zotepine Microemulsion: Pharmacokinetics and Pharmacodynamics. Pharmaceutics, 14(5), 978. https://doi.org/10.3390/pharmaceutics14050978