Intrinsic Dissolution Rate Profiling of Poorly Water-Soluble Compounds in Biorelevant Dissolution Media


Abstract
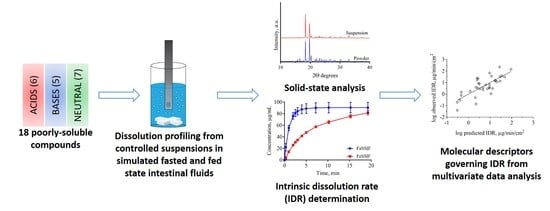
1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Solubility Determination
2.3. Suspensions
2.3.1. Preparation
2.3.2. Characterization
2.4. Small-Scale Dissolution Measurements
2.4.1. Standard Curve
2.4.2. Dissolution from Controlled Suspensions
2.4.3. Dissolution from Discs
2.4.4. Calculation of the Intrinsic Dissolution Rate (IDR)
2.5. Statistics and Physicochemical Analysis
3. Results and Discussion
3.1. Rapid Preparation of Controlled Suspensions by Ultrasonication and their Characterization
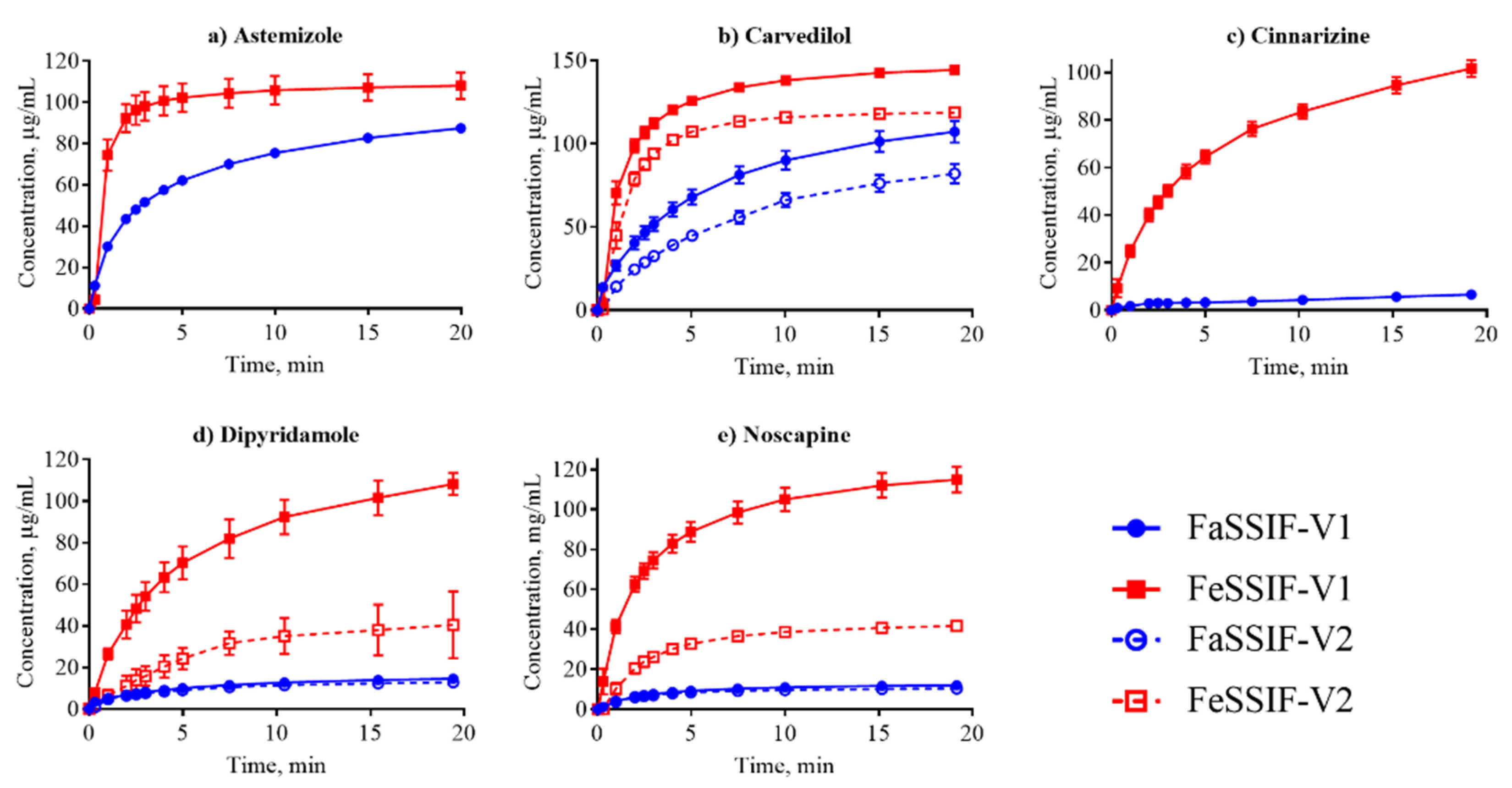
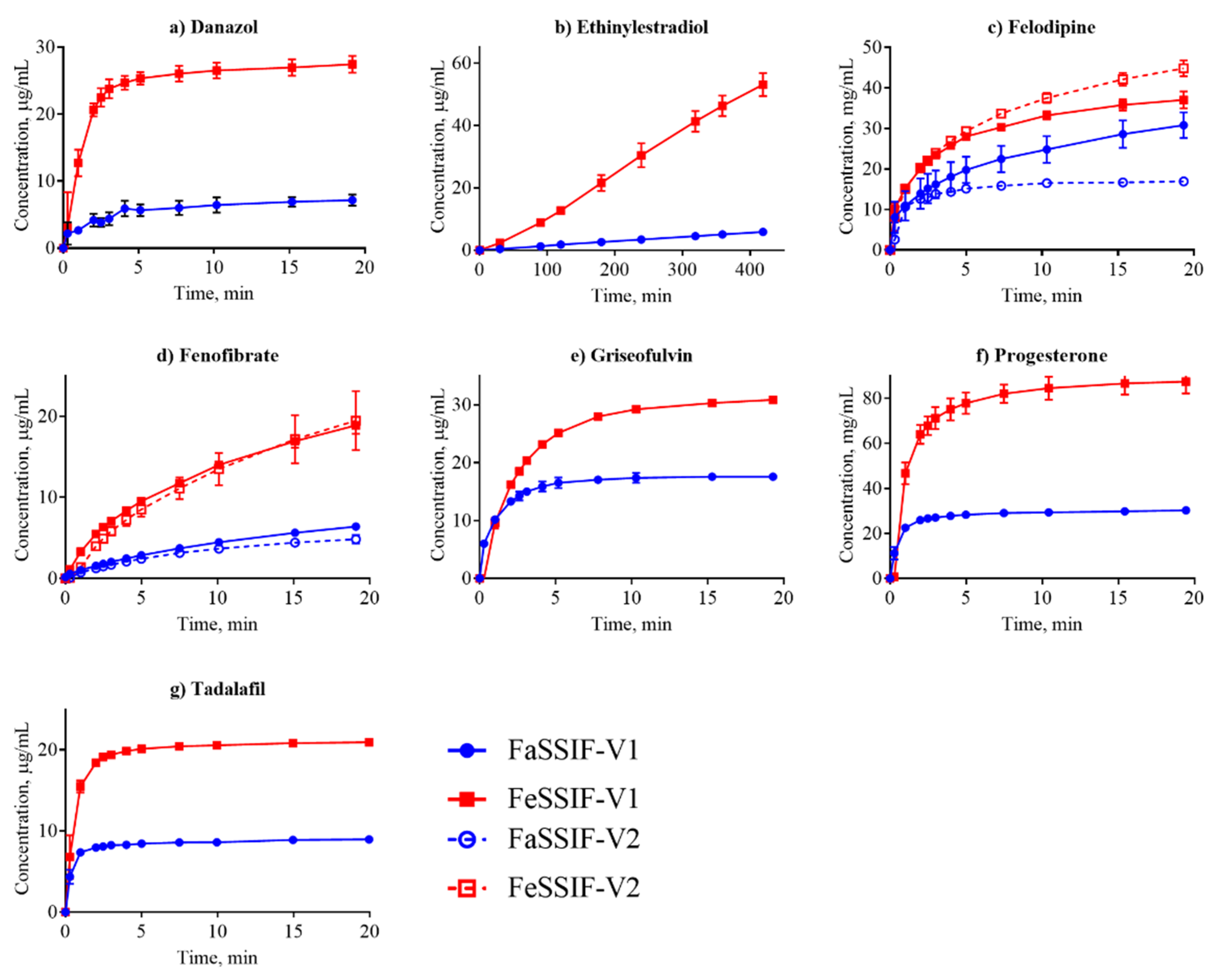
3.2. Dissolution Profiling
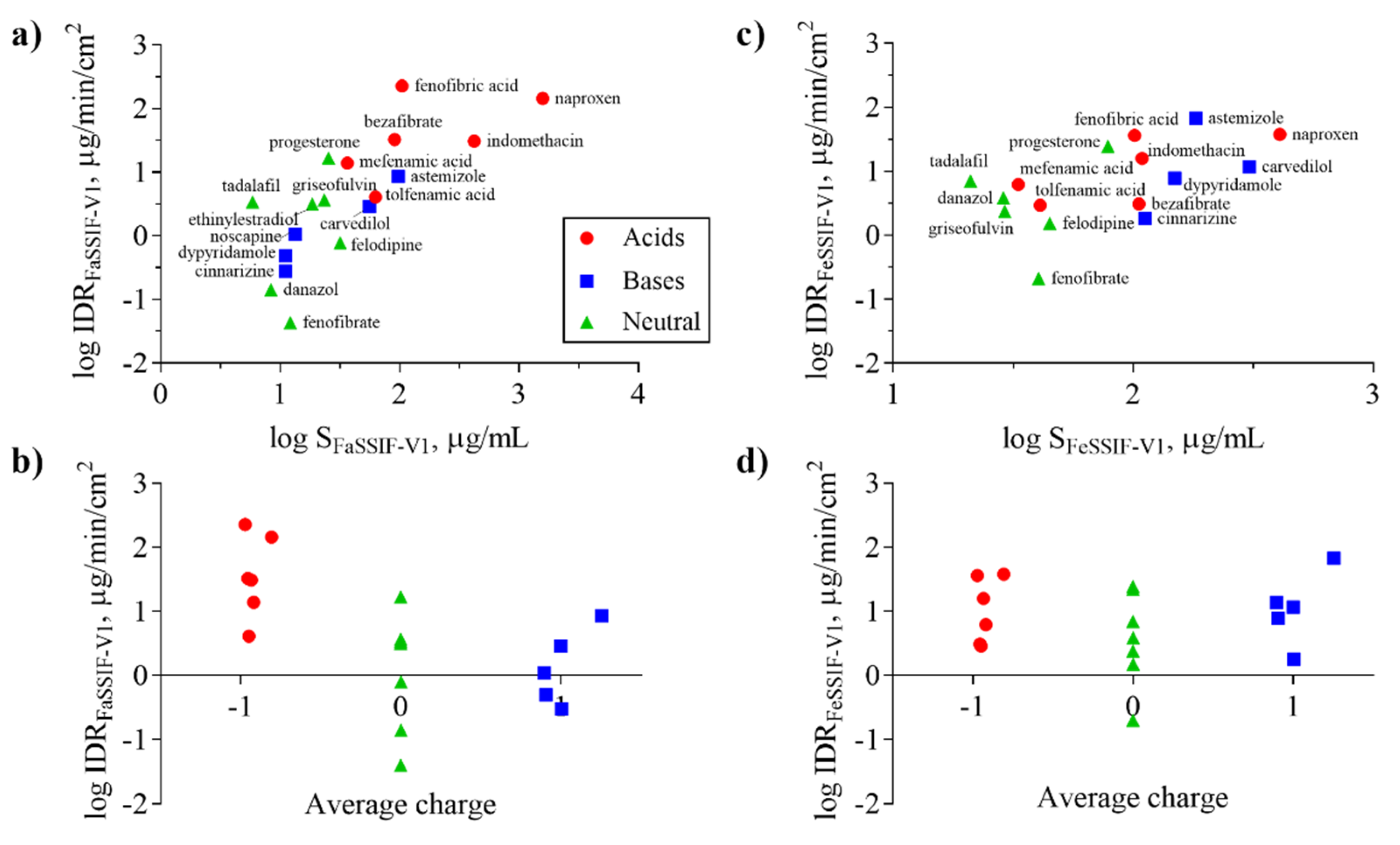
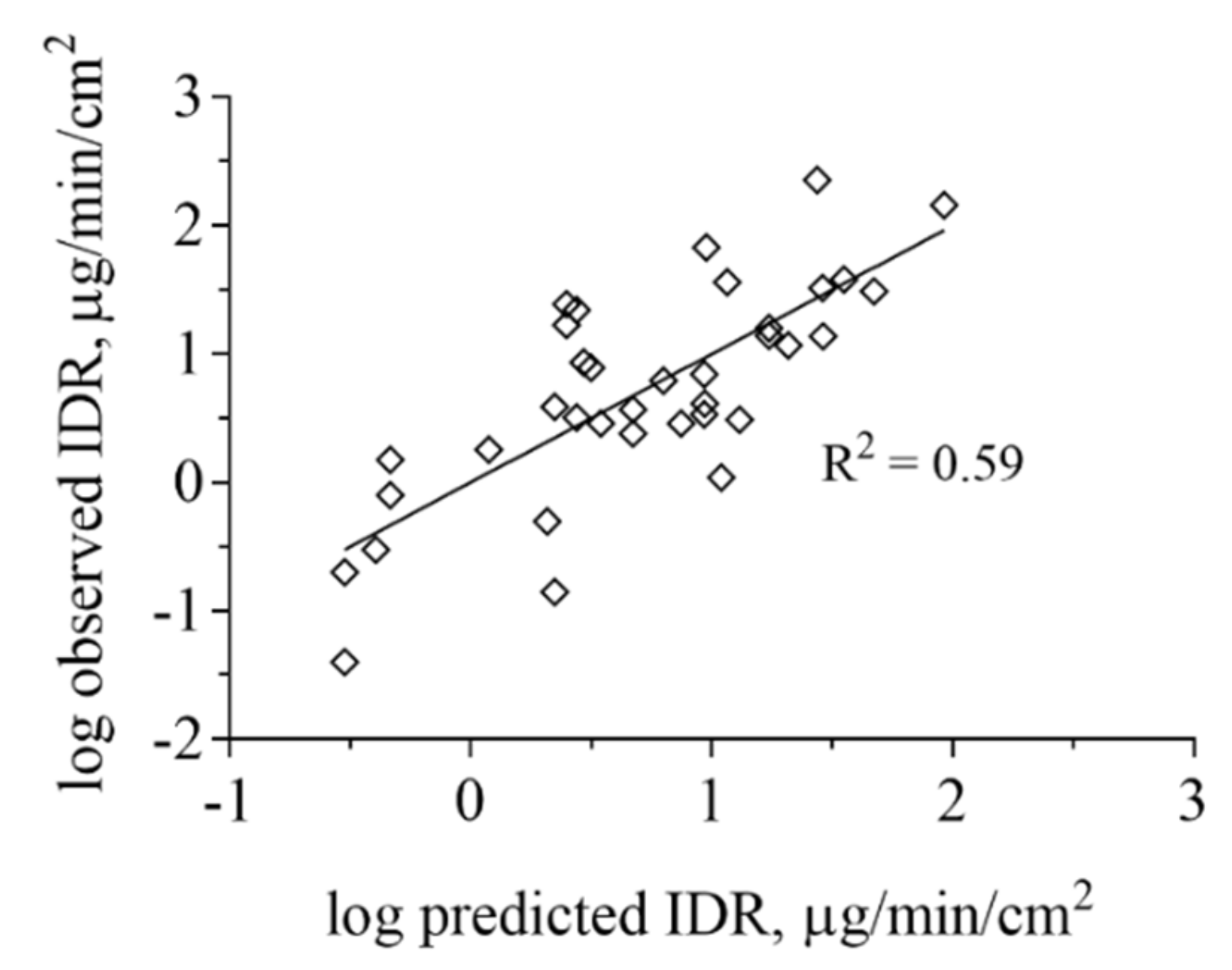
3.3. Impact of Physicochemical Properties on IDR
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Amidon, G.L.; Lennernas, H.; Shah, V.P.; Crison, J.R. A theoretical basis for a biopharmaceutic drug classification: The correlation of in vitro drug product dissolution and in vivo bioavailability. Pharm. Res. 1995, 12, 413–420. [Google Scholar] [CrossRef] [PubMed]
- Ku, M.S. Use of the biopharmaceutical classification system in early drug development. AAPS J. 2008, 10, 208–212. [Google Scholar] [CrossRef] [PubMed]
- Bergström, C.A.S.; Box, K.; Holm, R.; Matthews, W.; McAllister, M.; Müllertz, A.; Rades, T.; Schäfer, K.J.; Teleki, A. Biorelevant intrinsic dissolution profiling in early drug development: Fundamental, methodological, and industrial aspects. Eur. J. Pharm. Biopharm. 2019, 139, 101–114. [Google Scholar] [CrossRef] [PubMed]
- Shekunov, B.; Montgomery, E.R. Theoretical analysis of drug dissolution: I. Solubility and intrinsic dissolution rate. J. Pharm. Sci. 2016, 105, 2685–2697. [Google Scholar] [CrossRef] [PubMed]
- Wlodarski, K.; Sawicki, W.; Haber, K.; Knapik, J.; Wojnarowska, Z.; Paluch, M.; Lepek, P.; Hawelek, L.; Tajber, L. Physicochemical properties of tadalafil solid dispersions—Impact of polymer on the apparent solubility and dissolution rate of tadalafil. Eur. J. Pharm. Biopharm. 2015, 94, 106–115. [Google Scholar] [CrossRef]
- Sami, F.; Philip, B.; Pathak, K. Effect of auxiliary substances on complexation efficiency and intrinsic dissolution rate of gemfibrozil-beta-CD complexes. AAPS Pharm. Sci. Tech. 2010, 11, 27–35. [Google Scholar] [CrossRef]
- Andersson, S.B.E.; Alvebratt, C.; Bergström, C.A.S. Controlled suspensions enable rapid determinations of intrinsic dissolution rate and apparent solubility of poorly water-soluble compounds. Pharm. Res. 2017, 34, 1805–1816. [Google Scholar] [CrossRef]
- Alsenz, J.; Haenel, E.; Anedda, A.; Du Castel, P.; Cirelli, G. Miniaturized INtrinsic DISsolution Screening (MINDISS) assay for preformulation. Eur. J. Pharm. Sci. 2016, 87, 3–13. [Google Scholar] [CrossRef]
- Ostergaard, J. UV imaging in pharmaceutical analysis. J. Pharm. Biomed. Anal. 2018, 147, 140–148. [Google Scholar] [CrossRef]
- Avdeef, A.; Tsinman, K.; Tsinman, O.; Sun, N.; Voloboy, D. Miniaturization of powder dissolution measurement and estimation of particle size. Chem. Biodivers. 2009, 6, 1796–1811. [Google Scholar] [CrossRef]
- Avdeef, A.; Tsinman, O. Miniaturized rotating disk intrinsic dissolution rate measurement: Effects of buffer capacity in comparisons to traditional Wood’s apparatus. Pharm. Res. 2008, 25, 2613–2627. [Google Scholar] [CrossRef] [PubMed]
- Berger, C.M.; Tsinman, O.; Voloboy, D.; Lipp, D.; Stones, S.; Avdeef, A. Technical note: Miniaturized intrinsic dissolution rate (Mini-IDR (TM)) measurement of griseofulvin and carbamazepine. Dissolut. Technol. 2007, 14, 39–41. [Google Scholar] [CrossRef]
- Tsinman, K.; Avdeef, A.; Tsinman, O.; Voloboy, D. Powder dissolution method for estimating rotating disk intrinsic dissolution rates of low solubility drugs. Pharm. Res. 2009, 26, 2093–2100. [Google Scholar] [CrossRef] [PubMed]
- Wood, J.H.; Syarto, J.E.; Letterman, H. Improved holder for intrinsic dissolution rate studies. J. Pharm. Sci. 1965, 54, 1068. [Google Scholar] [CrossRef] [PubMed]
- Fagerberg, J.H.; Tsinman, O.; Sun, N.; Tsinman, K.; Avdeef, A.; Bergström, C.A.S. Dissolution rate and apparent solubility of poorly soluble drugs in biorelevant dissolution media. Mol. Pharm. 2010, 7, 1419–1430. [Google Scholar] [CrossRef] [PubMed]
- Mosharraf, M.; Nyström, C. The effect of particle size and shape on the surface specific dissolution rate of microsized practically insoluble drugs. Int. J. Pharm. 1995, 122, 35–47. [Google Scholar] [CrossRef]
- Andersson, S.B.E.; Alvebratt, C.; Bevernage, J.; Bonneau, D.; da Costa Mathews, C.; Dattani, R.; Edueng, K.; He, Y.; Holm, R.; Madsen, C.; et al. Interlaboratory validation of small-scale solubility and dissolution measurements of poorly water-soluble drugs. J. Pharm. Sci. 2016, 105, 2864–2872. [Google Scholar] [CrossRef]
- Bergström, C.A.S.; Holm, R.; Jørgensen, S.A.; Andersson, S.B.E.; Artursson, P.; Beato, S.; Borde, A.; Box, K.; Brewster, M.; Dressman, J.; et al. Early pharmaceutical profiling to predict oral drug absorption: Current status and unmet needs. Eur. J. Pharm. Sci. 2014, 57, 173–199. [Google Scholar] [CrossRef]
- Persson, L.C.; Porter, C.J.H.; Charman, W.N.; Bergstrom, C.A.S. Computational prediction of drug solubility in lipid based formulation excipients. Pharm. Res. 2013, 30, 3225–3237. [Google Scholar] [CrossRef]
- Alskar, L.C.; Porter, C.J.H.; Bergstrom, C.A.S. Tools for early prediction of drug loading in lipid-based formulations. Mol. Pharm. 2016, 13, 251–261. [Google Scholar] [CrossRef]
- Bergstrom, C.A.; Norinder, U.; Luthman, K.; Artursson, P. Experimental and computational screening models for prediction of aqueous drug solubility. Pharm. Res. 2002, 19, 182–188. [Google Scholar] [CrossRef] [PubMed]
- Wassvik, C.M.; Holmén, A.G.; Bergström, C.A.S.; Zamora, I.; Artursson, P. Contribution of solid-state properties to the aqueous solubility of drugs. Eur. J. Pharm. Sci. 2006, 29, 294–305. [Google Scholar] [CrossRef] [PubMed]
- Lemmerer, A.; Báthori, N.B.; Esterhuysen, C.; Bourne, S.A.; Caira, M.R. Concomitant polymorphs of the antihyperlipoproteinemic bezafibrate. Cryst. Growth Des. 2009, 9, 2646–2655. [Google Scholar] [CrossRef]
- Hiendrawan, S.; Widjojokusumo, E.; Veriansyah, B.; Tjandrawinata, R.R. Pharmaceutical salts of carvedilol: Polymorphism and physicochemical properties. AAPS PharmSciTech 2017, 18, 1417–1425. [Google Scholar] [CrossRef]
- Beattie, K.; Phadke, G.; Novakovic, J. Chapter Four—Carvedilol. In Profiles of Drug Substances, Excipients and Related Methodology; Brittain, H.G., Ed.; Academic Press: Cambridge, MA, USA, 2013; Volume 38, pp. 113–157. [Google Scholar]
- Sassene, P.J.; Knopp, M.M.; Hesselkilde, J.Z.; Koradia, V.; Larsen, A.; Rades, T.; Müllertz, A. Precipitation of a poorly soluble model drug during in vitro lipolysis: Characterization and dissolution of the precipitate. J. Pharm. Sci. 2010, 99, 4982–4991. [Google Scholar] [CrossRef]
- Liversidge, G.G.; Cundy, K.C. Particle size reduction for improvement of oral bioavailability of hydrophobic drugs: I. Absolute oral bioavailability of nanocrystalline danazol in beagle dogs. Int. J. Pharm. 1995, 125, 91–97. [Google Scholar] [CrossRef]
- Berbenni, V.; Marini, A.; Bruni, G.; Maggioni, A.; Cogliati, P. Thermoanalytical and spectroscopic characterization of solid state dipyridamole. J. Therm. Anal. Calorim. 2002, 68, 413–422. [Google Scholar] [CrossRef]
- Wang, S.; Chen, Y.; Gong, T.; Dong, W.; Wang, G.; Li, H.; Wu, S. Solid-liquid equilibrium behavior and thermodynamic analysis of dipyridamole in pure and binary solvents from 293.15 K to 328.15 K. J. Mol. Liq. 2019, 275, 8–17. [Google Scholar] [CrossRef]
- Pheasant, R. Polymorphism of 17-ethinylestradiol. J. Am. Chem. Soc. 1950, 72, 4303–4304. [Google Scholar] [CrossRef]
- Veronez, I.P.; Daniel, J.S.P.; Júnior, C.E.C.; Garcia, J.S.; Trevisan, M.G. Development, characterization, and stability studies of ethinyl estradiol solid dispersion. J. Therm. Anal. Calorim. 2015, 120, 573–581. [Google Scholar] [CrossRef]
- Kestur, U.S.; Ivanesivic, I.; Alonzo, D.E.; Taylor, L.S. Influence of particle size on the crystallization kinetics of amorphous felodipine powders. Powder Technol. 2013, 236, 197–204. [Google Scholar] [CrossRef]
- Heinz, A.; Gordon, K.C.; McGoverin, C.M.; Rades, T.; Strachan, C.J. Understanding the solid-state forms of fenofibrate—A spectroscopic and computational study. Eur. J. Pharm. Biopharm. 2009, 71, 100–108. [Google Scholar] [CrossRef] [PubMed]
- Linn, M.; Collnot, E.-M.; Djuric, D.; Hempel, K.; Fabian, E.; Kolter, K.; Lehr, C.-M. Soluplus® as an effective absorption enhancer of poorly soluble drugs in vitro and in vivo. Eur. J. Pharm. Biopharm. 2012, 45, 336–343. [Google Scholar] [CrossRef] [PubMed]
- Trasi, N.S.; Boerrigter, S.X.M.; Byrn, S.R. Investigation of the milling-induced thermal behavior of crystalline and amorphous griseofulvin. Pharm. Res. 2010, 27, 1377–1389. [Google Scholar] [CrossRef] [PubMed]
- Otsuka, M.; Kaneniwa, N. A kinetic study of the cystallization process of noncrystalline indomethacin under isothermal conditions. Chem. Pharm. Bull. 1988, 36, 4026–4032. [Google Scholar] [CrossRef] [PubMed]
- Romero, S.; Escalera, B.; Bustamante, P. Solubility behavior of polymorphs I and II of mefenamic acid in solvent mixtures. Int. J. Pharm. 1999, 178, 193–202. [Google Scholar] [CrossRef]
- Aguiar, A.J.; Zelmer, J.E. Dissolution behavior of polymorphs of chloramphenicol palmitate and mefenamic acid. J. Pharm. Sci. 1969, 58, 983–987. [Google Scholar] [CrossRef]
- Song, J.-S.; Sohn, Y.-T. Crystal forms of naproxen. Arch. Pharm. Res. 2011, 34, 87. [Google Scholar] [CrossRef]
- Madan, J.; Pandey, R.S.; Jain, V.; Katare, O.P.; Chandra, R.; Katyal, A. Poly (ethylene)-glycol conjugated solid lipid nanoparticles of noscapine improve biological half-life, brain delivery and efficacy in glioblastoma cells. Nanomed. Nanotechnol. 2013, 9, 492–503. [Google Scholar] [CrossRef]
- Fagerberg, J.H.; Bergstrom, C.A. Intestinal solubility and absorption of poorly water soluble compounds: Predictions, challenges and solutions. Ther. Deliv. 2015, 6, 935–959. [Google Scholar] [CrossRef]
- Muramatsu, M.; Iwahashi, M.; Takeuchi, U. Thermodynamic relationship between α- and β-forms of crystalline progesterone. J. Pharm. Sci. 1979, 68, 175–177. [Google Scholar] [CrossRef] [PubMed]
- Wang, F.; Wachter, J.A.; Antosz, F.J.; Berglund, K.A. An investigation of solvent-mediated polymorphic transformation of progesterone using in situ Raman spectroscopy. Org. Process. Res. Dev. 2000, 4, 391–395. [Google Scholar] [CrossRef]
- Mehanna, M.M.; Motawaa, A.M.; Samaha, M.W. In sight into tadalafil—block copolymer binary solid dispersion: Mechanistic investigation of dissolution enhancement. Int. J. Pharm. 2010, 402, 78–88. [Google Scholar] [CrossRef]
- Wei, Y.; Ling, Y.; Su, M.; Qin, L.; Zhang, J.; Gao, Y.; Qian, S. Characterization and stability of amorphous tadalafil and four crystalline polymorphs. Chem. Pharm. Bull. 2018, 66, 1114–1121. [Google Scholar] [CrossRef] [PubMed]
- Mattei, A.; Li, T. Polymorph formation and nucleation mechanism of tolfenamic acid in solution: An investigation of pre-nucleation solute association. Pharm. Res. 2012, 29, 460–470. [Google Scholar] [CrossRef] [PubMed]
- Biorelevant.com Ltd. Available online: https://biorelevant.com/fassif-fessif-fassgf/buy/ (accessed on 23 December 2019).
- Zaki, N.M.; Artursson, P.; Bergstrom, C.A. A modified physiological BCS for prediction of intestinal absorption in drug discovery. Mol. Pharm. 2010, 7, 1478–1487. [Google Scholar] [CrossRef]
- Clarysse, S.; Brouwers, J.; Tack, J.; Annaert, P.; Augustijns, P. Intestinal drug solubility estimation based on simulated intestinal fluids: Comparison with solubility in human intestinal fluids. Eur. J. Pharm. Sci. 2011, 43, 260–269. [Google Scholar] [CrossRef]
- Fagerberg, J.H.; Al-Tikriti, Y.; Ragnarsson, G.; Bergström, C.A.S. Ethanol effects on apparent solubility of poorly soluble drugs in simulated intestinal fluid. Mol. Pharm. 2012, 9, 1942–1952. [Google Scholar] [CrossRef]
- Li, N.; DeGennar, M.D.; Liebenberg, W.; Tiedt, L.R.; Zahn, A.S.; Pishko, M.V.; de Villiers, M.M. Increased dissolution and physical stability of micronized nifedipine particles encapsulated with a biocompatible polymer and surfactants in a wet ball milling process. Pharmazie 2006, 61, 595–603. [Google Scholar]
- Prado, L.D.; Rocha, H.V.A.; Resende, J.A.L.C.; Ferreira, G.B.; de Figuereido Teixeira, A.M.R. An insight into carvedilol solid forms: Effect of supramolecular interactions on the dissolution profiles. CrystEngComm 2014, 16, 3168–3179. [Google Scholar] [CrossRef]
- Jantratid, E.; Janssen, N.; Reppas, C.; Dressman, J.B. Dissolution media simulating conditions in the proximal human gastrointestinal tract: An update. Pharm. Res. 2008, 25, 1663–1676. [Google Scholar] [CrossRef] [PubMed]
- Fuchs, A.; Leigh, M.; Kloefer, B.; Dressman, J.B. Advances in the design of fasted state simulating intestinal fluids: FaSSIF-V3. Eur. J. Pharm. Biopharm. 2015, 94, 229–240. [Google Scholar] [CrossRef] [PubMed]
- Noyes, A.A.; Whitney, W.R. The rate of solution of solid substances in their own solutions. J. Am. Chem. Soc. 1897, 19, 930–934. [Google Scholar] [CrossRef]
- Hasselbalch, K.A. Die Berechnung der Wasserstoffzahl des Blutes aus der freien und gebundenen Kohlensäure desselben, und die Sauerstoffbindung des Blutes als Funktion der Wasserstoffzahl; Julius Springer: Berlin, Germany, 1916. [Google Scholar]
- Mudie, D.M.; Samiei, N.; Marshall, D.J.; Amidon, G.E.; Bergström, C.A.S. Selection of in vivo predictive dissolution media using drug substance and physiological properties. AAPS J. 2020, 22, 34. [Google Scholar] [CrossRef] [PubMed]
- Jinno, J.; Oh, D.M.; Crison, J.R.; Amidon, G.L. Dissolution of ionizable water-insoluble drugs: The combined effect of pH and surfactant. J. Pharm. Sci. 2000, 89, 268–274. [Google Scholar] [CrossRef]
- Balakrishnan, A.; Rege, B.D.; Amidon, G.L.; Polli, J.E. Surfactant-mediated dissolution: Contributions of solubility enhancement and relatively low micelle diffusivity. J. Pharm. Sci. 2004, 93, 2064–2075. [Google Scholar] [CrossRef]
- Okazaki, A.; Mano, T.; Sugano, K. Theoretical dissolution model of poly-disperse drug particles in biorelevant media. J. Pharm. Sci. 2008, 97, 1843–1852. [Google Scholar] [CrossRef]
- Bergstrom, C.A.S.; Charman, W.N.; Porter, C.J.H. Computational prediction of formulation strategies for beyond-rule-of-5 compounds. Adv. Drug Deliv. Rev. 2016, 101, 6–21. [Google Scholar] [CrossRef]
- Jain, N.; Yalkowsky, S.H. Estimation of the aqueous solubility I: Application to organic nonelectrolytes. J. Pharm. Sci. 2001, 90, 234–252. [Google Scholar] [CrossRef]
- Christiansen, M.L.; Holm, R.; Abrahamsson, B.; Jacobsen, J.; Kristensen, J.; Andersen, J.R.; Müllertz, A. Effect of food intake and co-administration of placebo self-nanoemulsifying drug delivery systems on the absorption of cinnarizine in healthy human volunteers. Eur. J. Pharm. Sci. 2016, 84, 77–82. [Google Scholar] [CrossRef]
- Alskär, L.C.; Parrow, A.; Keemink, J.; Johansson, P.; Abrahamsson, B.; Bergström, C.A.S. Effect of lipids on absorption of carvedilol in dogs: Is coadministration of lipids as efficient as a lipid-based formulation? J. Control. Release 2019, 304, 90–100. [Google Scholar] [CrossRef] [PubMed]
- Galipeau, K.; Socki, M.; Socia, A.; Harmon, P.A. Incomplete loading of sodium lauryl sulfate and fasted state simulated intestinal fluid micelles within the diffusion layers of dispersed drug particles during dissolution. J. Pharm. Sci. 2018, 107, 156–169. [Google Scholar] [CrossRef] [PubMed]
- Sailaja, D.; Suhasini, K.L.; Kumar, S.; Gandhi, K.S. Theory of rate of solubilization into surfactant solutions. Langmuir 2003, 19, 4014–4026. [Google Scholar] [CrossRef]
- Hossain, S.; Kabedev, A.; Parrow, A.; Bergstrom, C.A.S.; Larsson, P. Molecular simulation as a computational pharmaceutics tool to predict drug solubility, solubilization processes and partitioning. Eur. J. Pharm. Biopharm. 2019, 137, 46–55. [Google Scholar] [CrossRef]
- Clulow, A.J.; Parrow, A.; Hawley, A.; Khan, J.; Pham, A.C.; Larsson, P.; Bergstrom, C.A.S.; Boyd, B.J. Characterization of solubilizing nanoaggregates present in different versions of simulated intestinal fluid. J. Phys. Chem. B 2017, 121, 10869–10881. [Google Scholar] [CrossRef]
- Vithani, K.; Hawley, A.; Jannin, V.; Pouton, C.; Boyd, B. Solubilisation behaviour of poorly water-soluble drugs during digestion of solid SMEDDS. Eur. J. Pharm. Biopharm. 2018, 130, 236–246. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | Structure | Mw* g/mol | A/B/N | pKa | logP | Tm °C | ΔHf J/g | Crystalline Form As-Received† | Food ‡ |
|---|---|---|---|---|---|---|---|---|---|
| Astemizole |  | 458.6 | B | 8.1 | 5.9 | 174 [22] | 111 [22] | (ZENREP)* | - |
| Bezafibrate |  | 361.9 | A | 3.5 | 4.0 | 182 | 145 | α form [23] | + |
| Carvedilol |  | 406.5 | B | 8.1 | 4.0 | 117 [24] | 120 [24] | form II [25] | + |
| Cinnarizine |  | 368.6 | B | 7.4 | 5.3 | 117 | 118 | ([26]) | + |
| Danazol |  | 337.5 | N | - | 3.6 | 226 | 92 | ([27]) | +/- |
| Dipyridamole |  | 504.7 | B | 6.8; 3.6 | 2.2 | 166 [28] | 56 [28] | form II [29] | + |
| Ethinylestradiol |  | 296.4 | N | - | 3.6 | 183 [30] | 89 [31] | hemihydrate [31] | +/- |
| Felodipine |  | 384.3 | N | - | 4.8 | 142 | 76 | form I [32] | +/- |
| Fenofibrate |  | 360.9 | N | - | 5.3 | 79 | 98 | form I [33] | + |
| Fenofibric acid |  | 318.8 | A | 3.5 | 4.0 | 181 [34] | - | (QANHUJ)* | +/- |
| Griseofulvin |  | 352.8 | N | - | 2.5 | 215 | 122 | ([35]) | + |
| Indomethacin |  | 357.8 | A | 4.1 | 3.8 | 160 [22] | 106 [22] | γ form [36] | +/- |
| Mefenamic acid |  | 241.3 | A | 4.0 | 4.9 | 230 [37] | 158 [37] | form I [38] | + |
| Naproxen |  | 230.3 | A | 4.5 | 3.3 | 156 [22] | 149 [22] | form I [39] | +/- |
| Noscapine |  | 413.4 | B | 6.1 | 2.8 | 176 [40] | - | ([40]) | - |
| Progesterone |  | 314.5 | N | - | 3.9 | 129 [41] | 89 [42] | form I [43] | + |
| Tadalafil |  | 389.4 | N | - | 1.6 | 295 [44] | 113 [44] | form I [45] | +/- |
| Tolfenamic acid |  | 261.7 | A | 4.2 | 5.3 | 212 | 143 | form I [46] | n/a |
| Component | FaSSIF-V1 | FeSSIF-V1 | FaSSIF-V2 | FeSSIF-V2 | |
|---|---|---|---|---|---|
| Taurocholate | mM | 3 | 15 | 3 | 10 |
| Phospholipids | 0.75 | 3.75 | 0.2 | 2 | |
| Sodium | 148 | 319 | 106 | 218 | |
| Chloride | 106 | 203 | 69 | 125 | |
| Phosphate | 29 | ||||
| Acetic acid | 144 | ||||
| Maleic acid | 19 | 55 | |||
| Oleate | 0.8 | ||||
| Glycerol monleate | 5 | ||||
| pH | 6.5 | 5 | 6.5 | 5.8 | |
| Hydrodynamic diameter | nm | 71 | 8 | 32 | 35 |
| Compound | SFaSSIF-V1 μg/mL | SFeSSIF-V1 μg/mL | D/S | IDRFaSSIF-V1 μg/min/cm2 | IDRFeSSIF-V1 μg/min/c2 | IDRFaSSIF-V2 μg/min/cm2 | IDRFeSSIF-V2 μg/min/cm2 |
|---|---|---|---|---|---|---|---|
| Astemizole | 97.9 ± 2.1 [15] | 182 ± 1 [15] | S | 8.6 ± 0.4 | 67.5 ± 14 | 2.0 ± 0.2 | - |
| Bezafibrate | 91.0 ± 8.9 † | 105.7 ‡ | S | 32.7 ± 3.9 | 3.1 ± 0.04 | - | - |
| Carvedilol | 55.9 ± 1.0 [15] | 305 ± 2 [15] | S | 2.9 ± 0.5 | 11.7 ± 0.9 | 1.5 ± 0.2 | 13.6 ± 0.6 |
| Cinnarizine | 11.1 ± 1.2 [7] | 112 ± 2 [15] | S | 0.3 ± 0.01 | 1.8 ± 0.07 | - | - |
| Danazol | 8.4 ± 0.7 [15] | 28.8 ± 0.4 [15] | S | 0.14 ± 0.02 | 3.9 ± 0.4 | - | - |
| Dipyridamole | 11.1 ± 3.0 [48] | 148.9 ± 19.7 [48] | S | 0.5 ± 0.05 | 7.8 ± 2.6 | 0.5 ± 0.03 | 1.7 ± 0.3 |
| Ethinylestradiol | 18.7 ± 1.3 * | - | D | 3.2 ±0.1 | 22.1 ±3.1 | 6.6 ± 0.8 | - |
| Felodipine | 31.8 ± 1.7 [7] | 44.9 ± 9.2 [48] | S | 0.8 ± 0.4 | 1.5 ± 0.06 | 0.6 ± 0.2 | 1.5 ± 0.2 |
| Fenofibrate | 12.2 ± 0.3 [7] | 40.4 ± 2.9 [49] | S | 0.04 ± 0.002 | 0.2 ± 0.008 | 0.05 ± 0.001 | 2.7 ± 0.3 |
| Fenofibric acid | 104.8 ± 1.0 † | 101.4 ± 3.8 † | D | 227 ± 33 | 36.3 ± 5.8 | - | - |
| Griseofulvin | 23.4 ± 1.8 [41] | 29.2 ± 3.4 [41] | S | 3.7 ± 0.6 | 2.4 ± 0.02 | - | - |
| Indomethacin | 421.7 ± 17.6 [7] | 109.0 ± 7.0 [15] | D | 31 ± 1.3 | 16 ± 1.0 | - | - |
| Mefenamic acid | 36.6 ± 0.6 † | 33.2 ± 0.7 † | S | 13.9 ± 2.0 | 6.2 ± 0.9 | 11.9 ± 1.0 | 7.9 ± 2.1 |
| Naproxen | 1584.8 ± 101.4 * | 408.0 ± 35.1 * | D | 144 ± 12 | 38 ± 1.5 | 179 ± 11 | - |
| Noscapine | 13.4 ‡ | - | S | 1.1 ± 0.2 | 13.8 ± 1.3 | 1.2 ± 0.1 | 5.2 ± 0.8 |
| Progesterone | 25.5 ± 1.1 [50] | 78.6 ± 16.2 [41] | S | 16.8 ± 0.5 | 24.5 ± 5.7 | - | - |
| Tadalafil | 5.9 ± 0.7 [7] | 21.0 ± 0.5 † | S | 3.4 ± 0.7 | 7.0 ± 0.9 | 1.3 ± 0.8 | - |
| Tolfenamic acid | 63.0 ± 2.7 [15] | 41.0 ± 0.5 [15] | S | 4.1 ± 0.8 | 2.9 ± 0.2 | 12.9 ± 0.9 | 4.8 ± 0.6 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Teleki, A.; Nylander, O.; Bergström, C.A.S. Intrinsic Dissolution Rate Profiling of Poorly Water-Soluble Compounds in Biorelevant Dissolution Media. Pharmaceutics 2020, 12, 493. https://doi.org/10.3390/pharmaceutics12060493
Teleki A, Nylander O, Bergström CAS. Intrinsic Dissolution Rate Profiling of Poorly Water-Soluble Compounds in Biorelevant Dissolution Media. Pharmaceutics. 2020; 12(6):493. https://doi.org/10.3390/pharmaceutics12060493
Chicago/Turabian StyleTeleki, Alexandra, Olivia Nylander, and Christel A.S. Bergström. 2020. "Intrinsic Dissolution Rate Profiling of Poorly Water-Soluble Compounds in Biorelevant Dissolution Media" Pharmaceutics 12, no. 6: 493. https://doi.org/10.3390/pharmaceutics12060493
APA StyleTeleki, A., Nylander, O., & Bergström, C. A. S. (2020). Intrinsic Dissolution Rate Profiling of Poorly Water-Soluble Compounds in Biorelevant Dissolution Media. Pharmaceutics, 12(6), 493. https://doi.org/10.3390/pharmaceutics12060493