In Vitro–In Vivo Correlations Based on In Vitro Dissolution of Parent Drug Diltiazem and Pharmacokinetics of Its Metabolite


 , and
, and
Abstract
:
1. Introduction
2. Materials and Methods
2.1. Chemicals and Reagents
2.2. In Vitro Dissolution Studies
2.3. Clinical Study
2.4. Sample Treatment
2.5. Preparation of Standard Solutions and Quality Control Samples
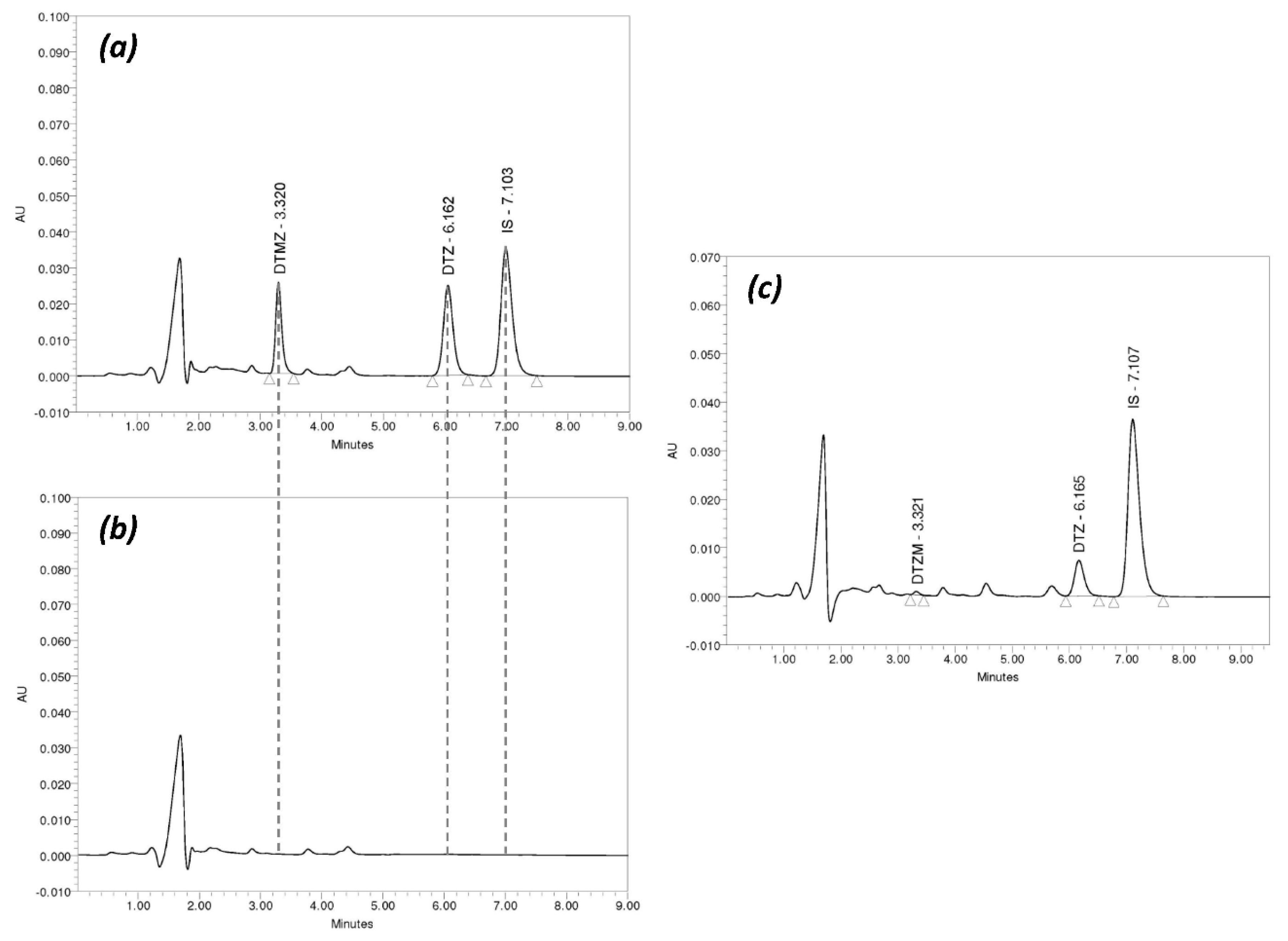
2.6. Chromatographic Analysis
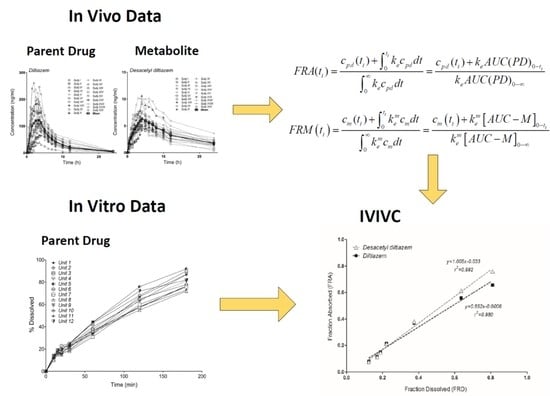
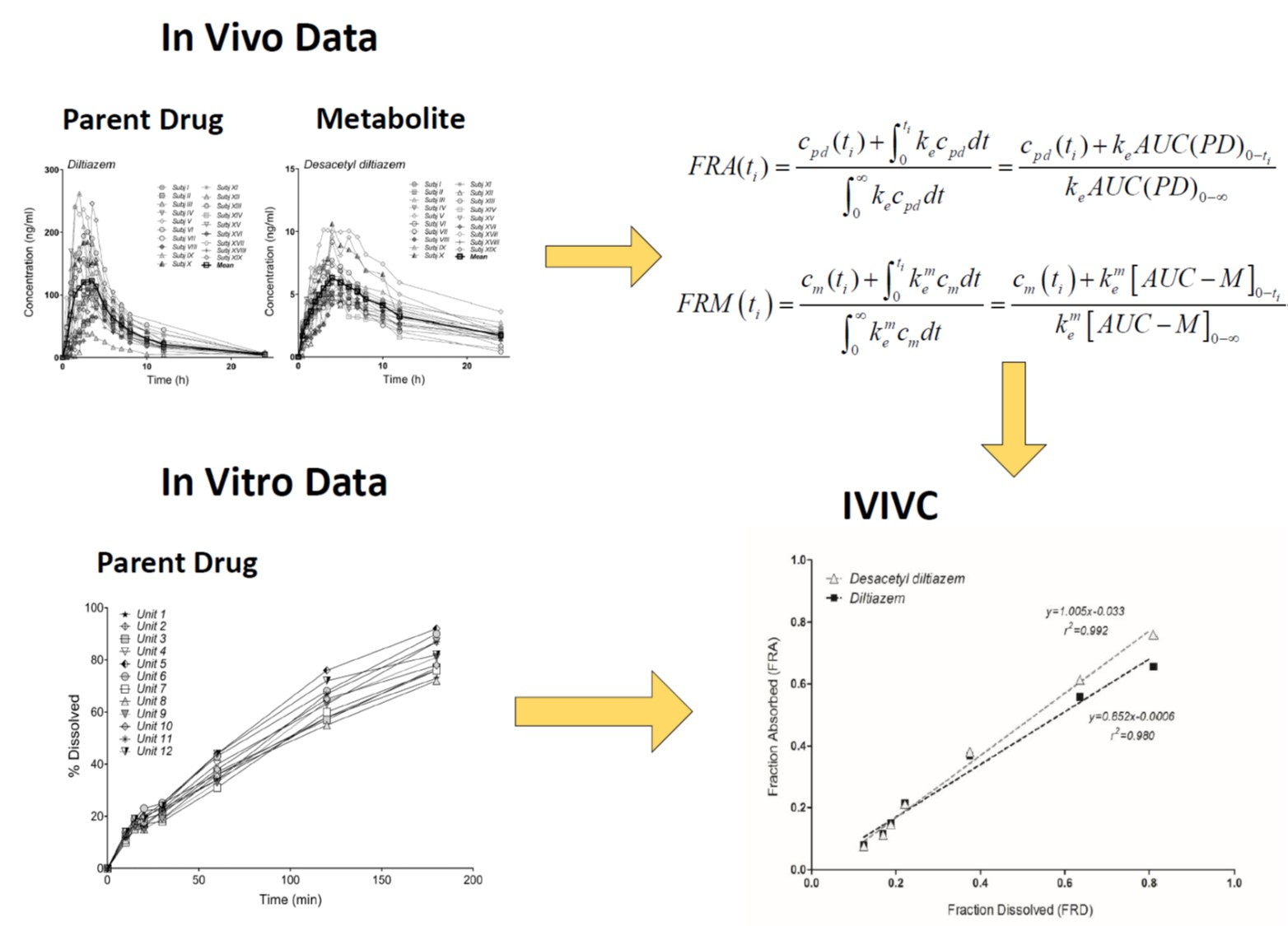
2.7. Treatment of In Vivo Data
2.8. Model-Independent Estimation of In Vivo Absorption/Dissolution by the Deconvolution of In Vivo Pharmacokinetics
3. Results
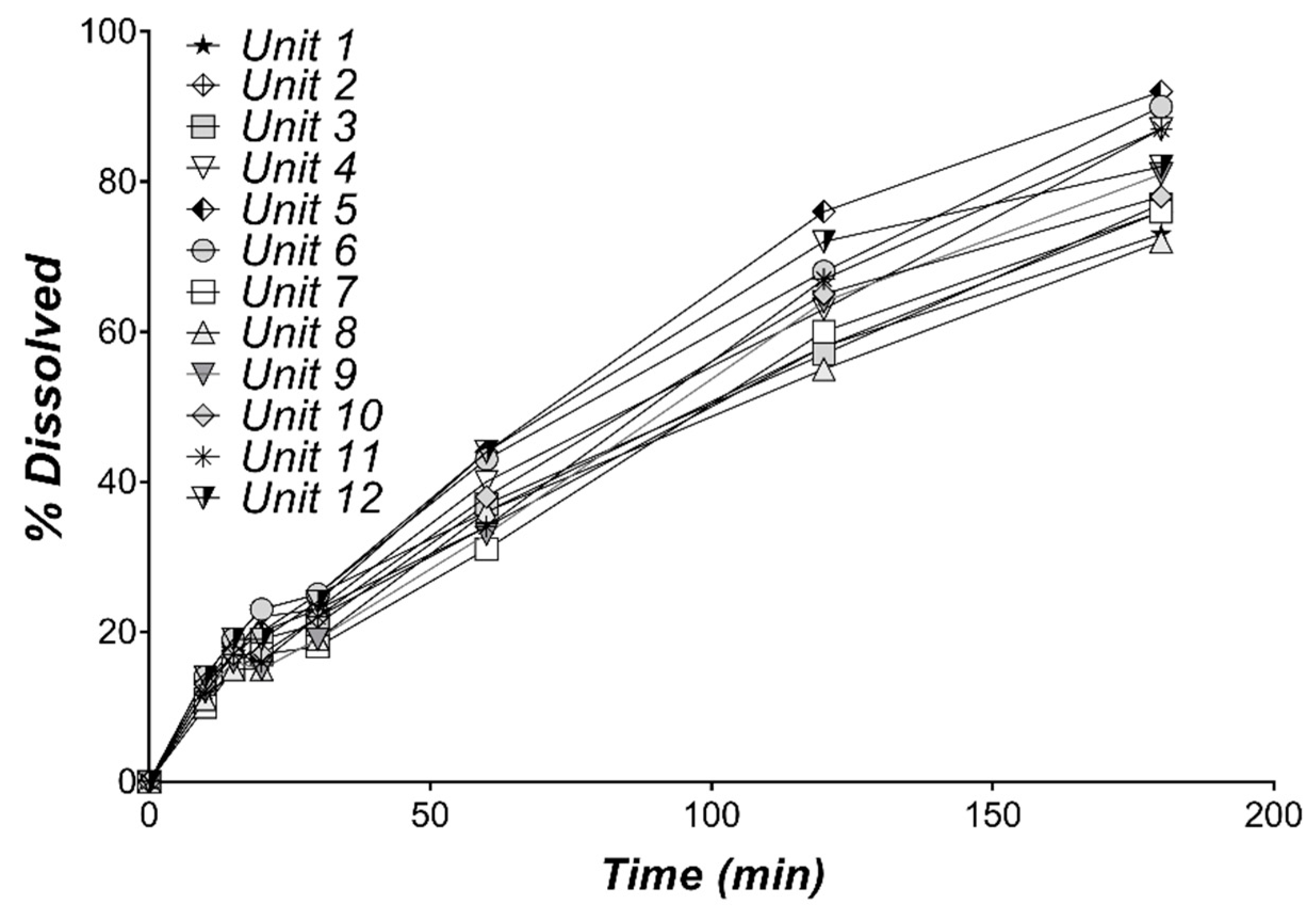
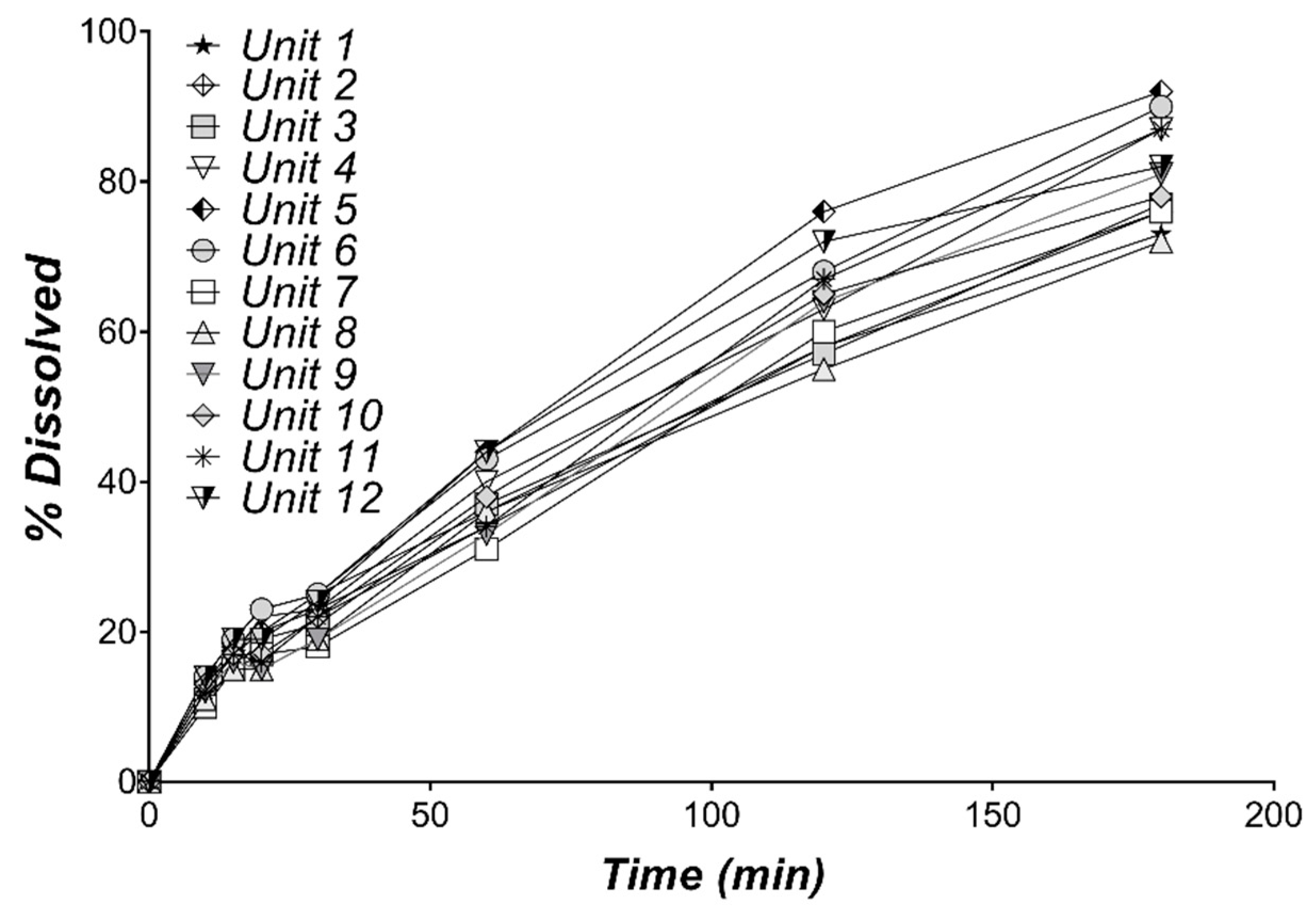
3.1. In Vitro Dissolution of Diltiazem
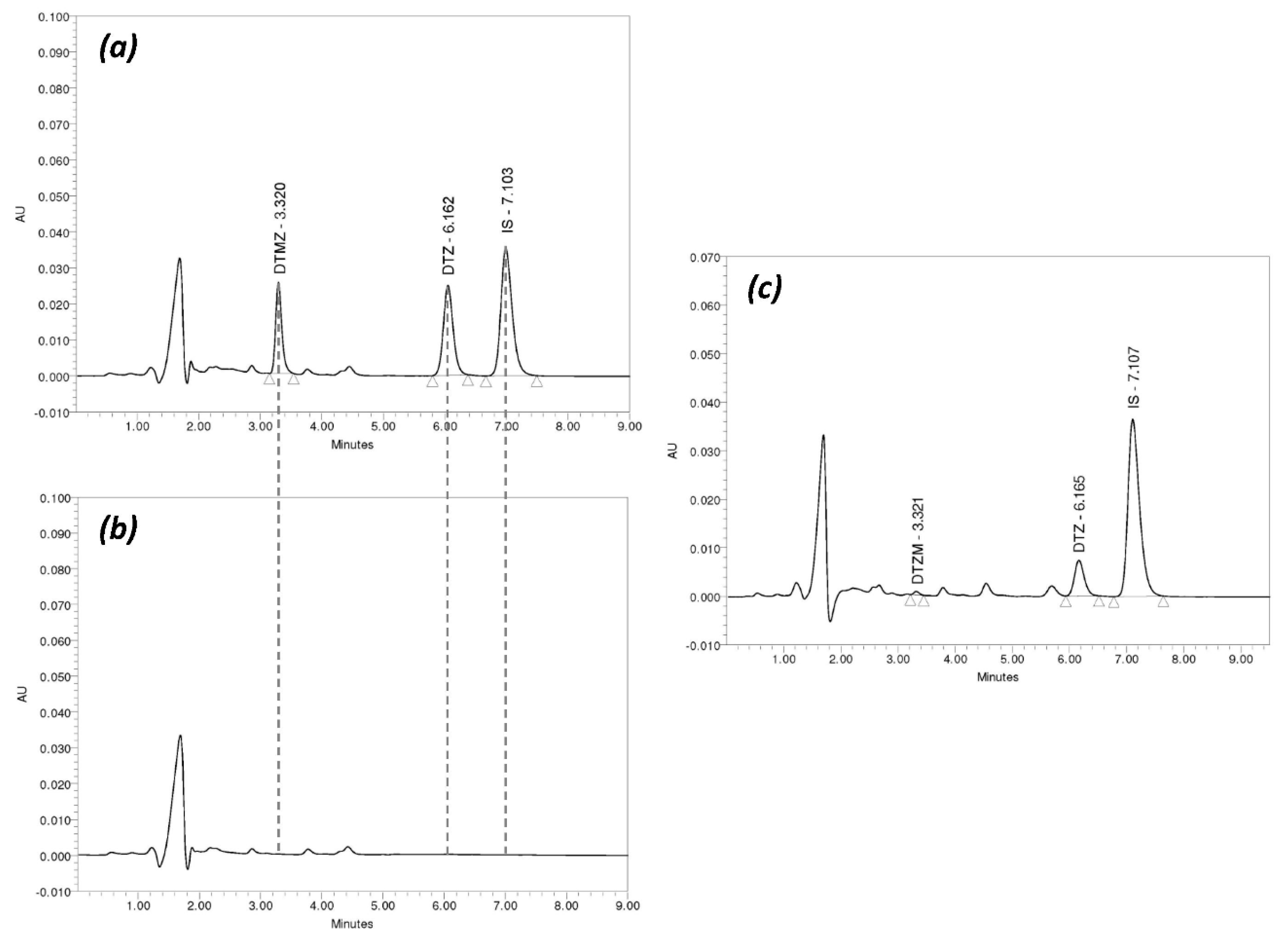
3.2. Chromatographic Method Validation
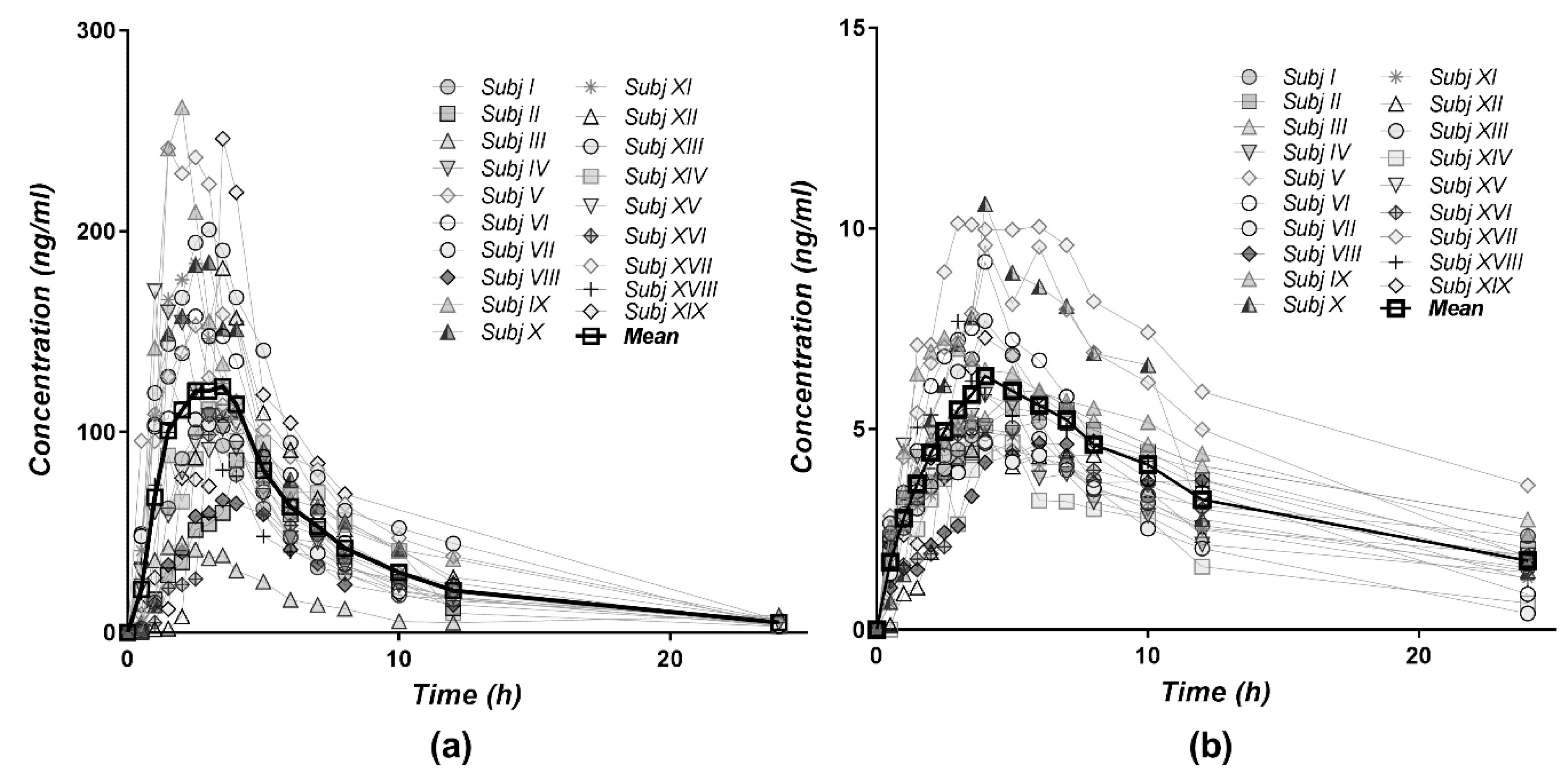
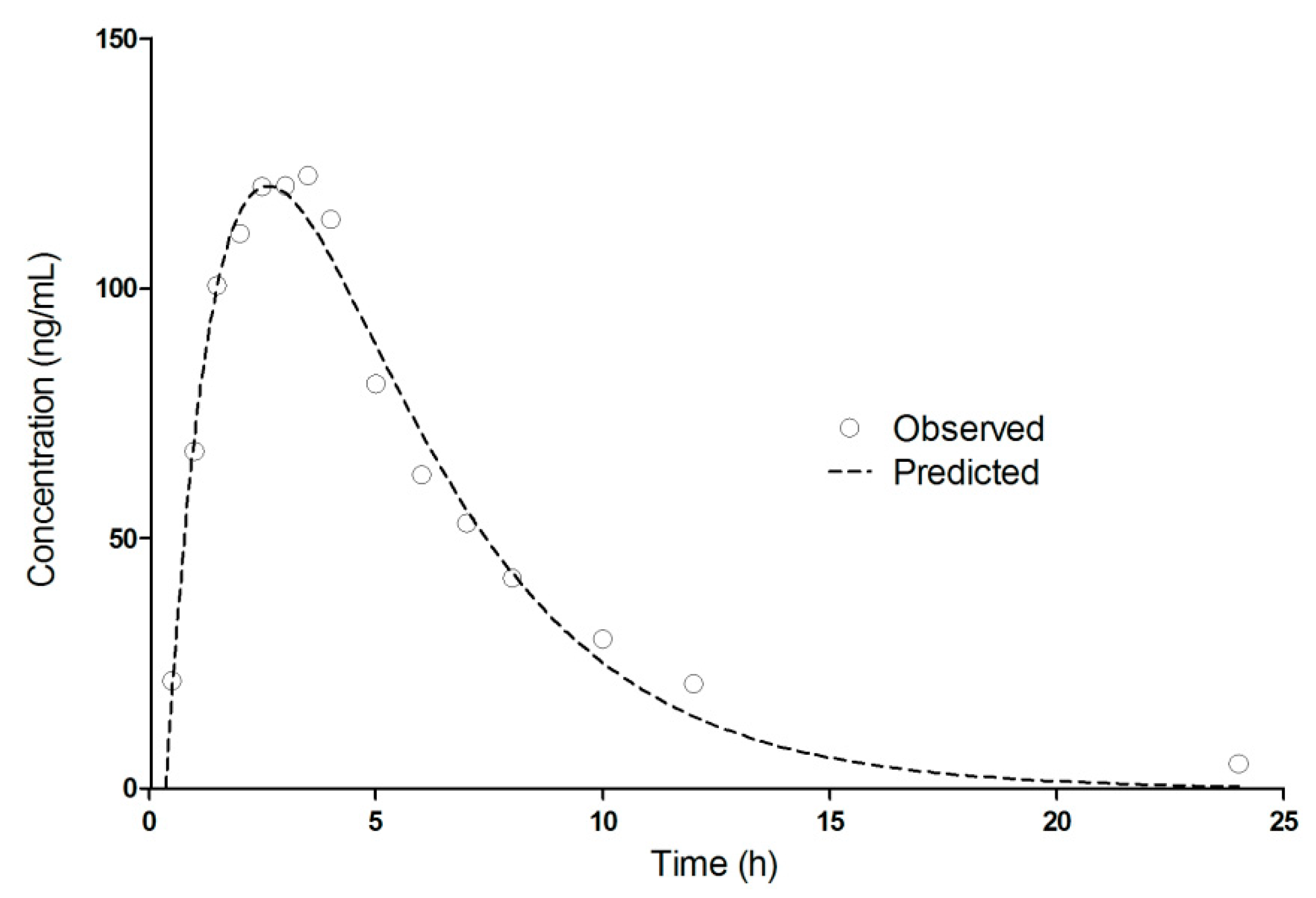
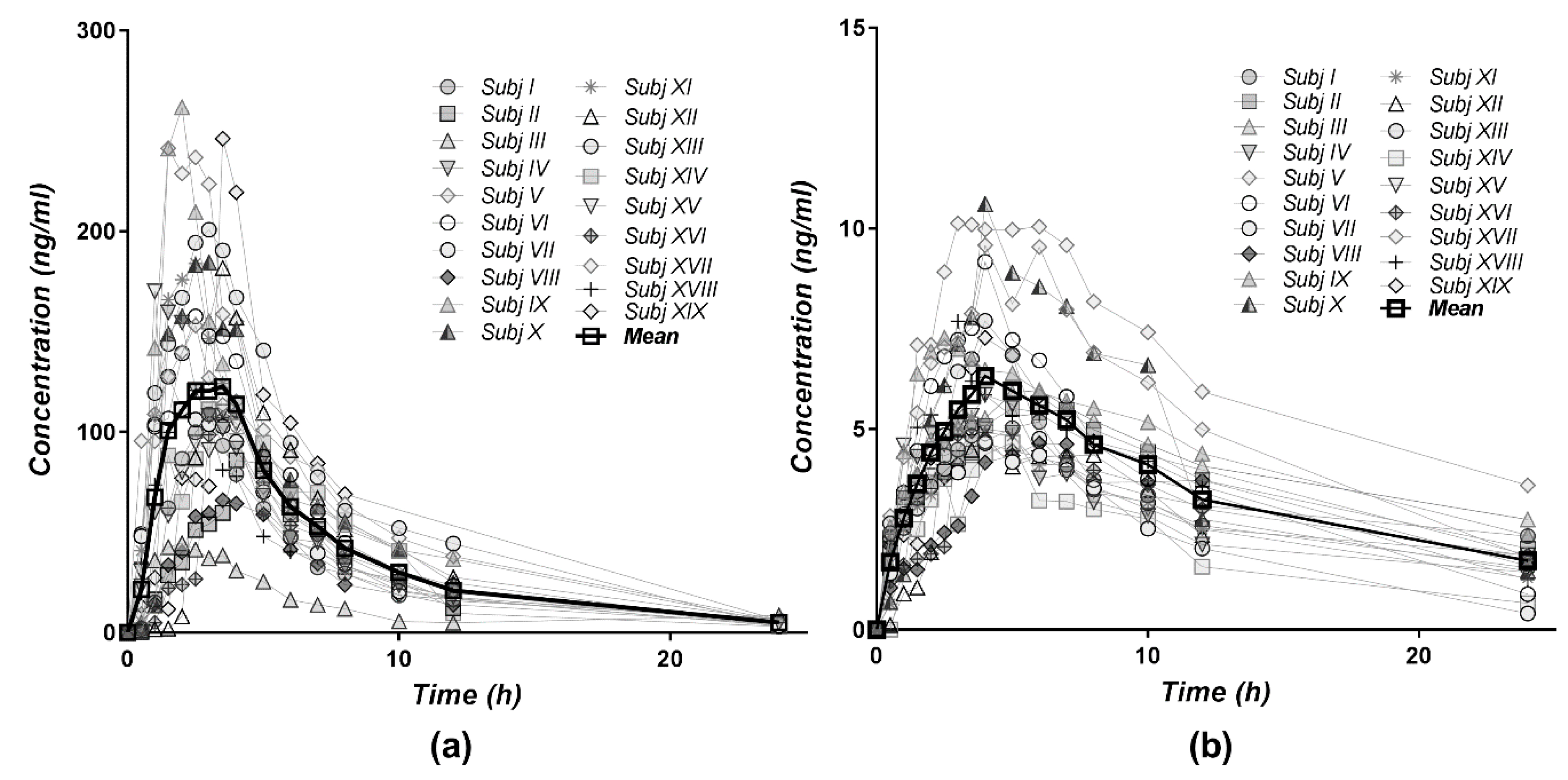
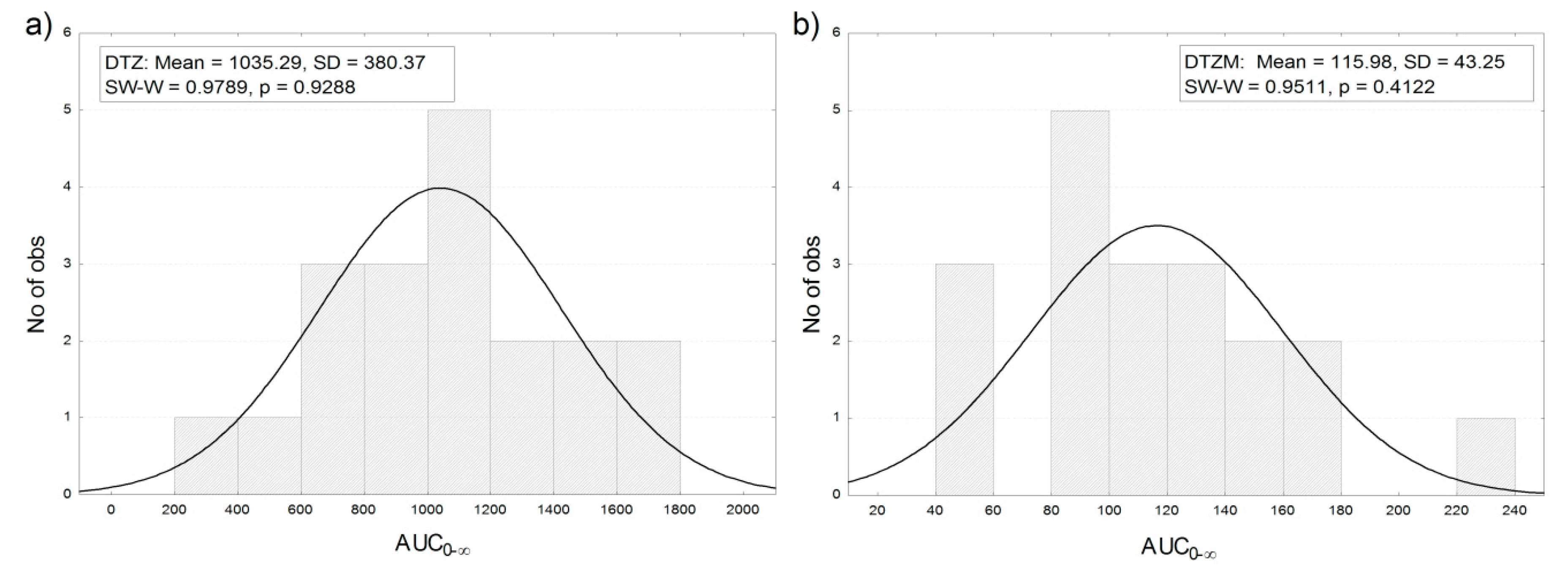
3.3. Pharmacokinetics of Diltiazem and Its Metabolite
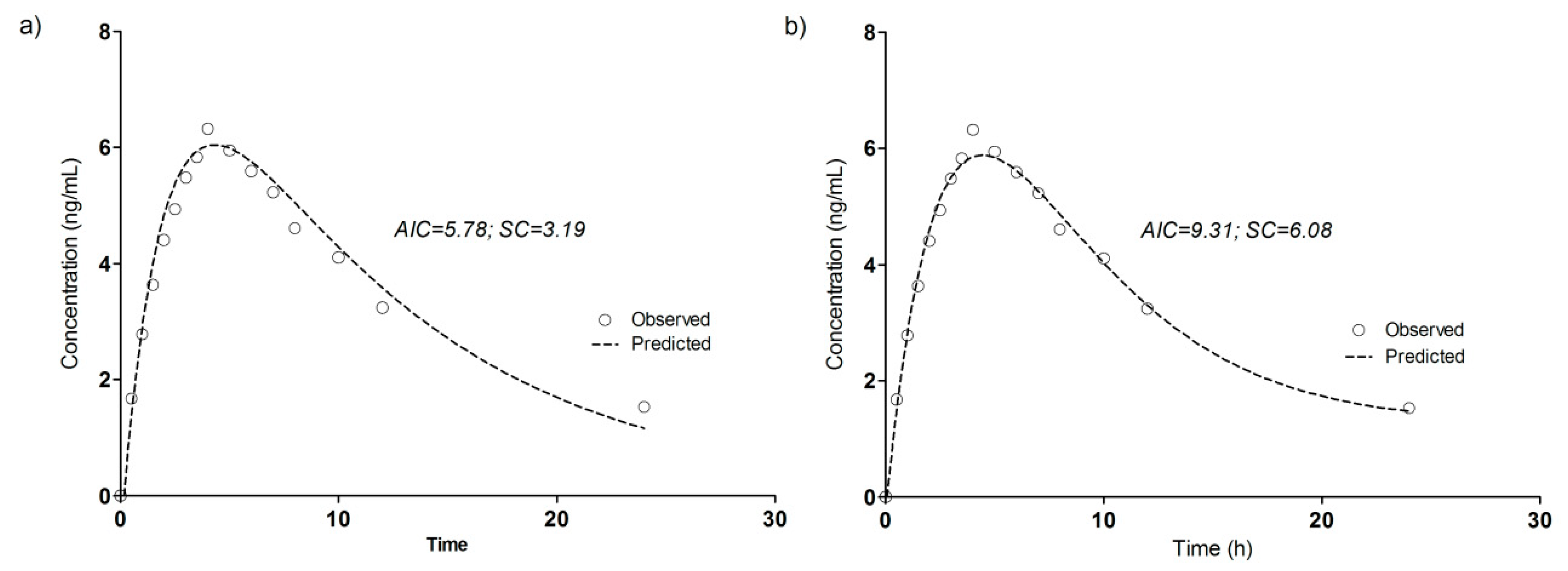
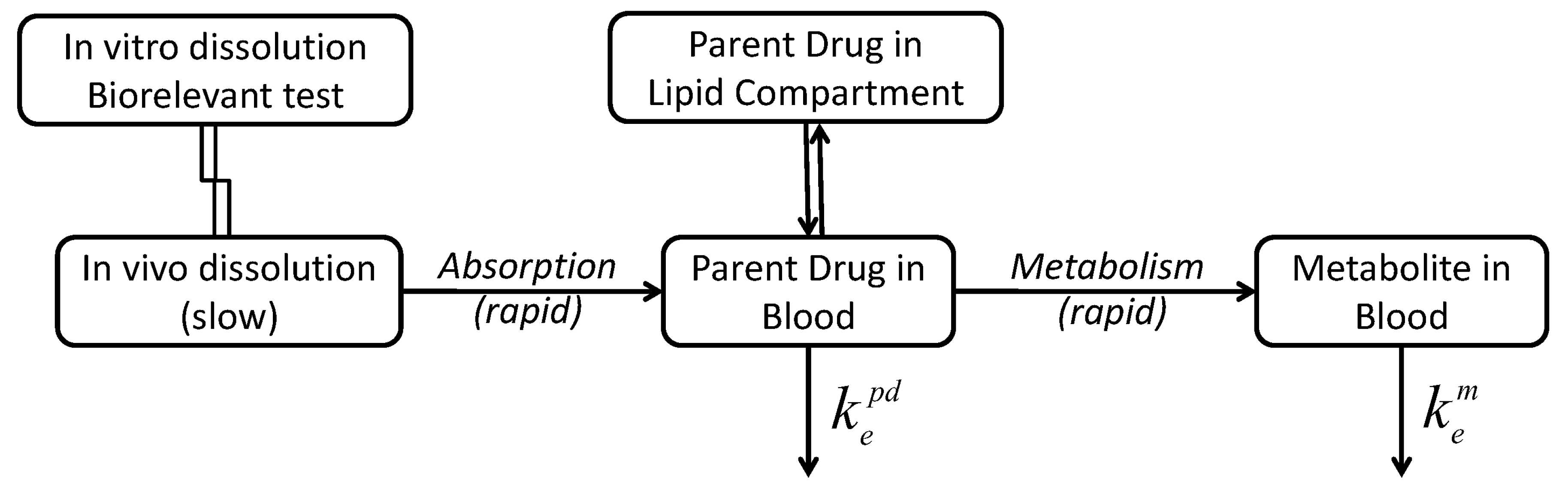
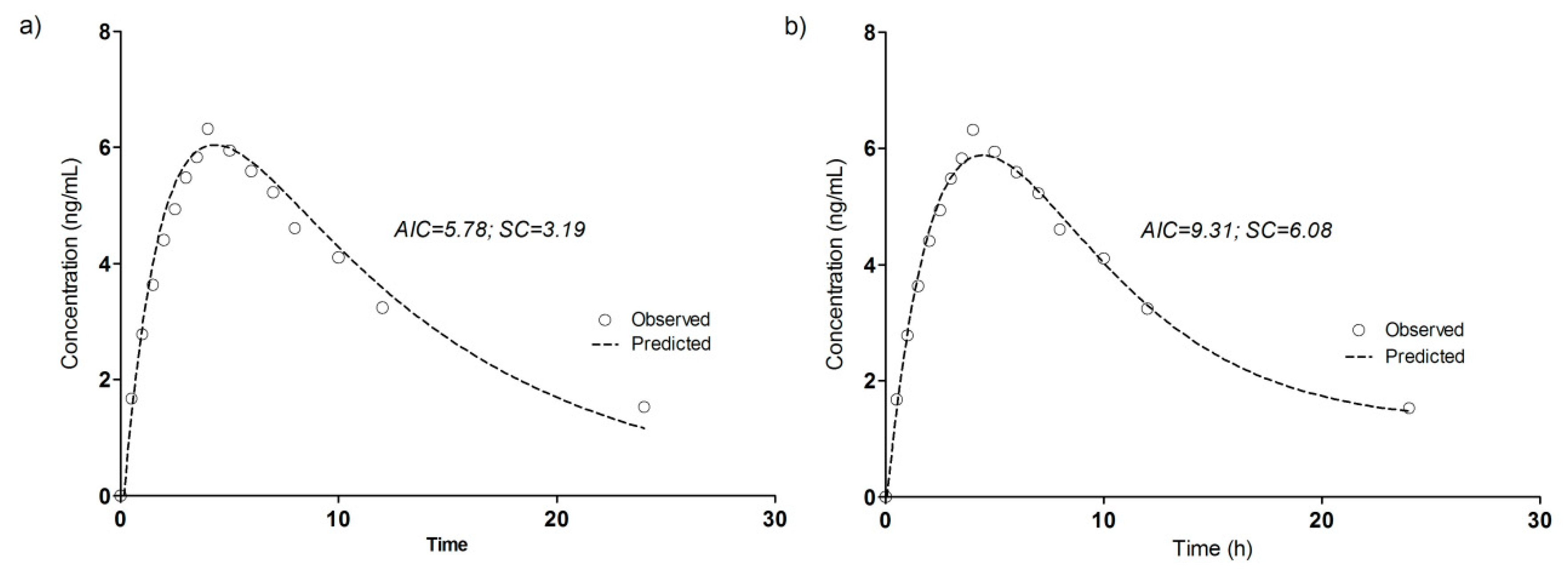
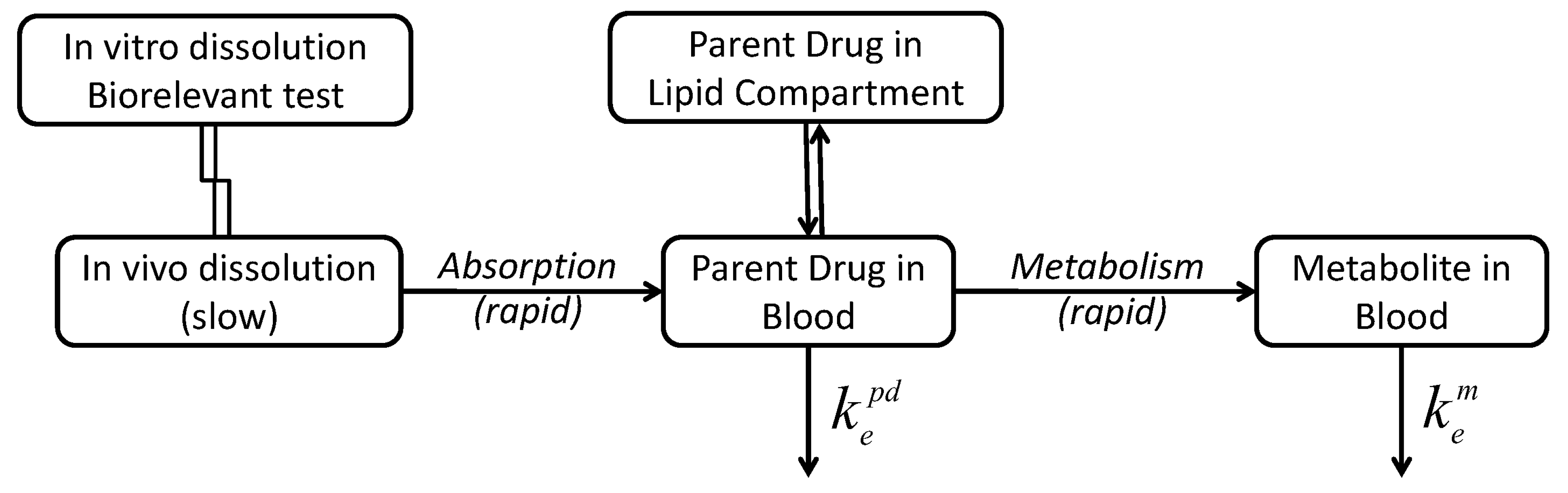
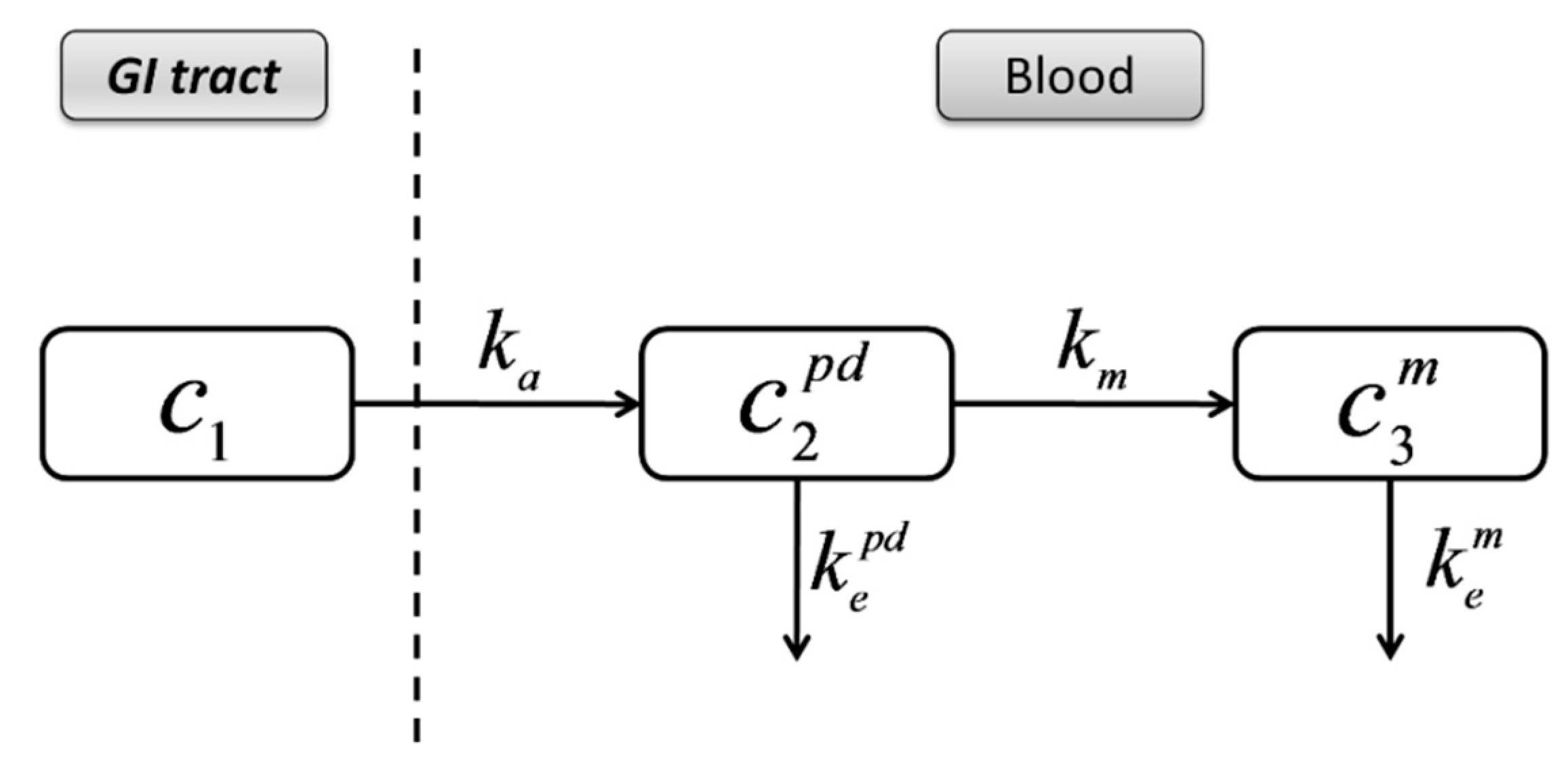
3.4. Compartmental Modeling of Diltiazem and Its Metabolite Pharmacokinetics
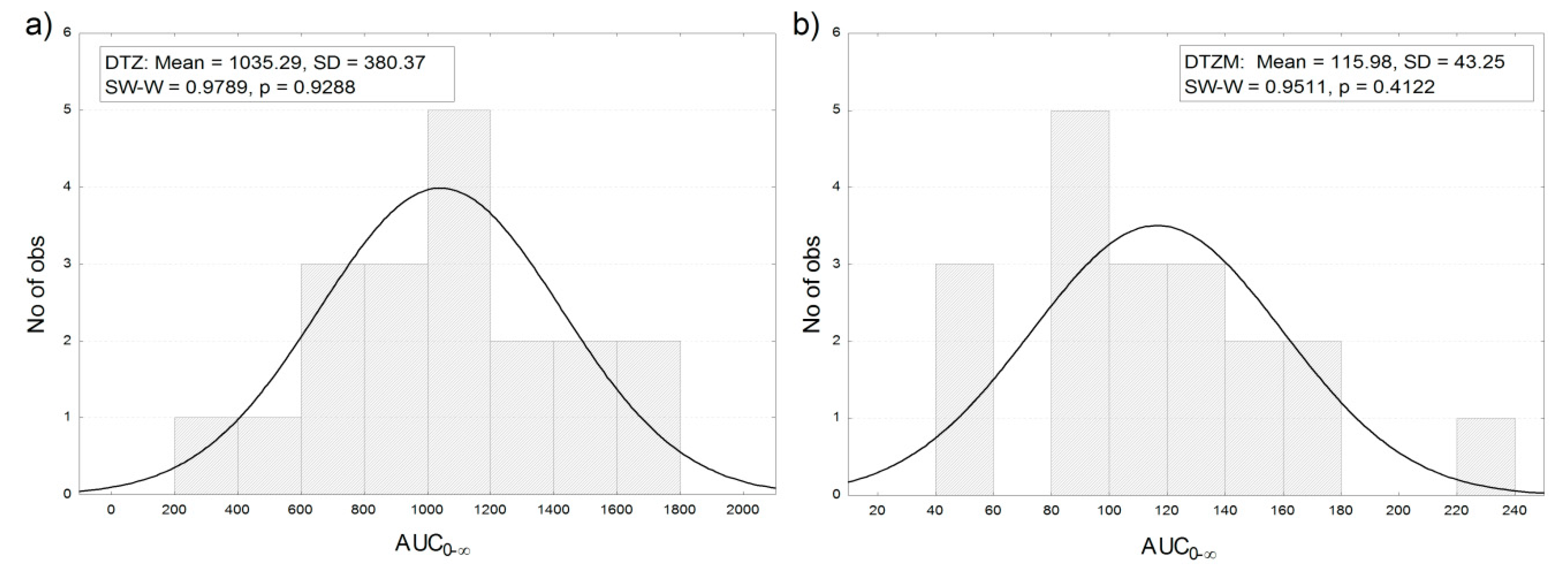
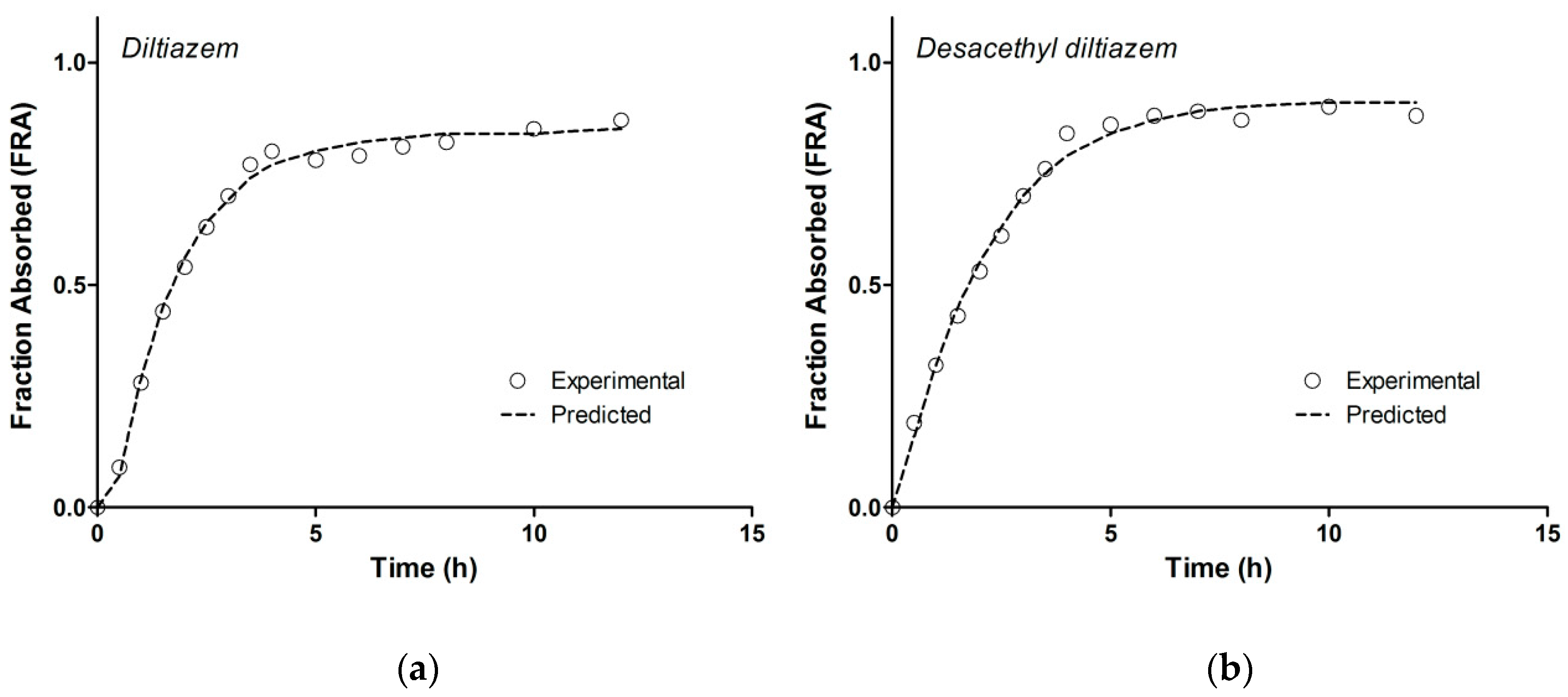
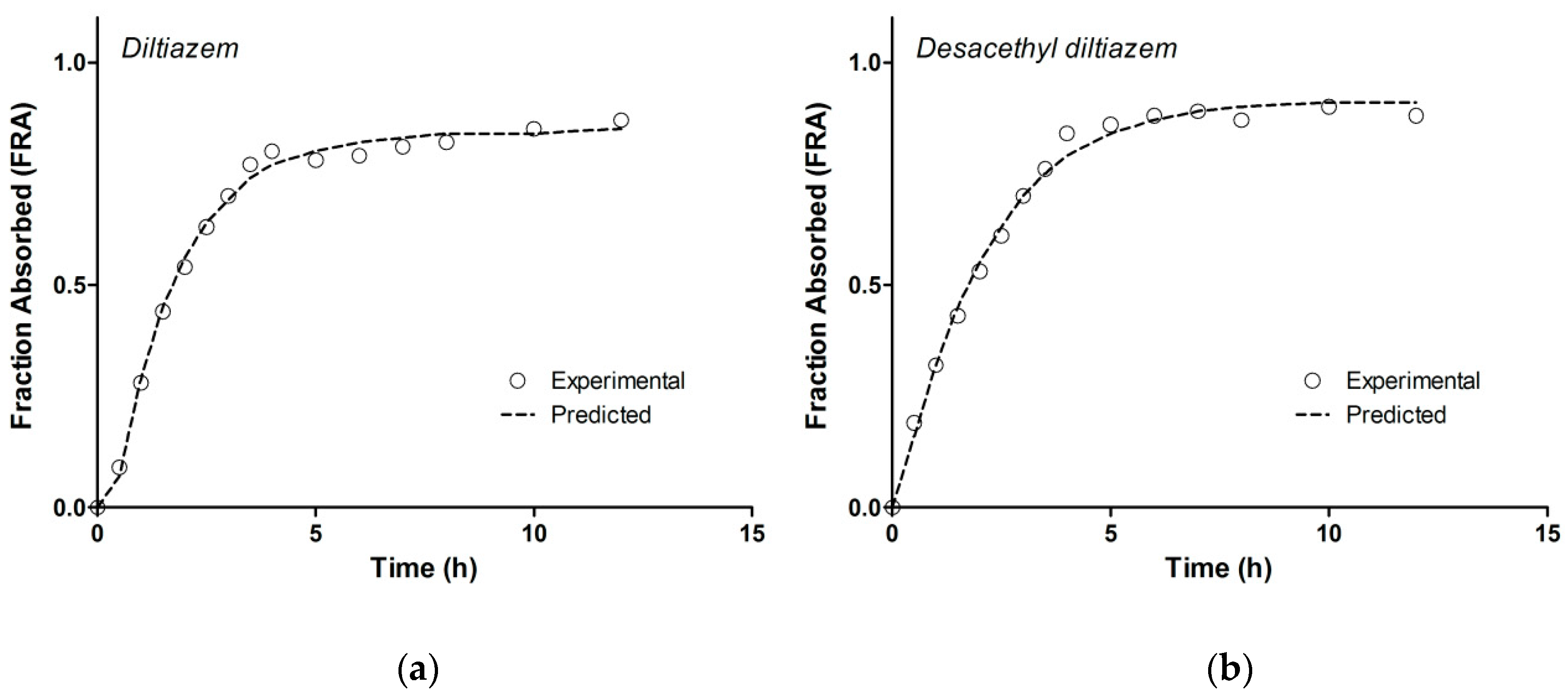
3.5. Model-Independent Estimation of In Vivo Absorption/Dissolution
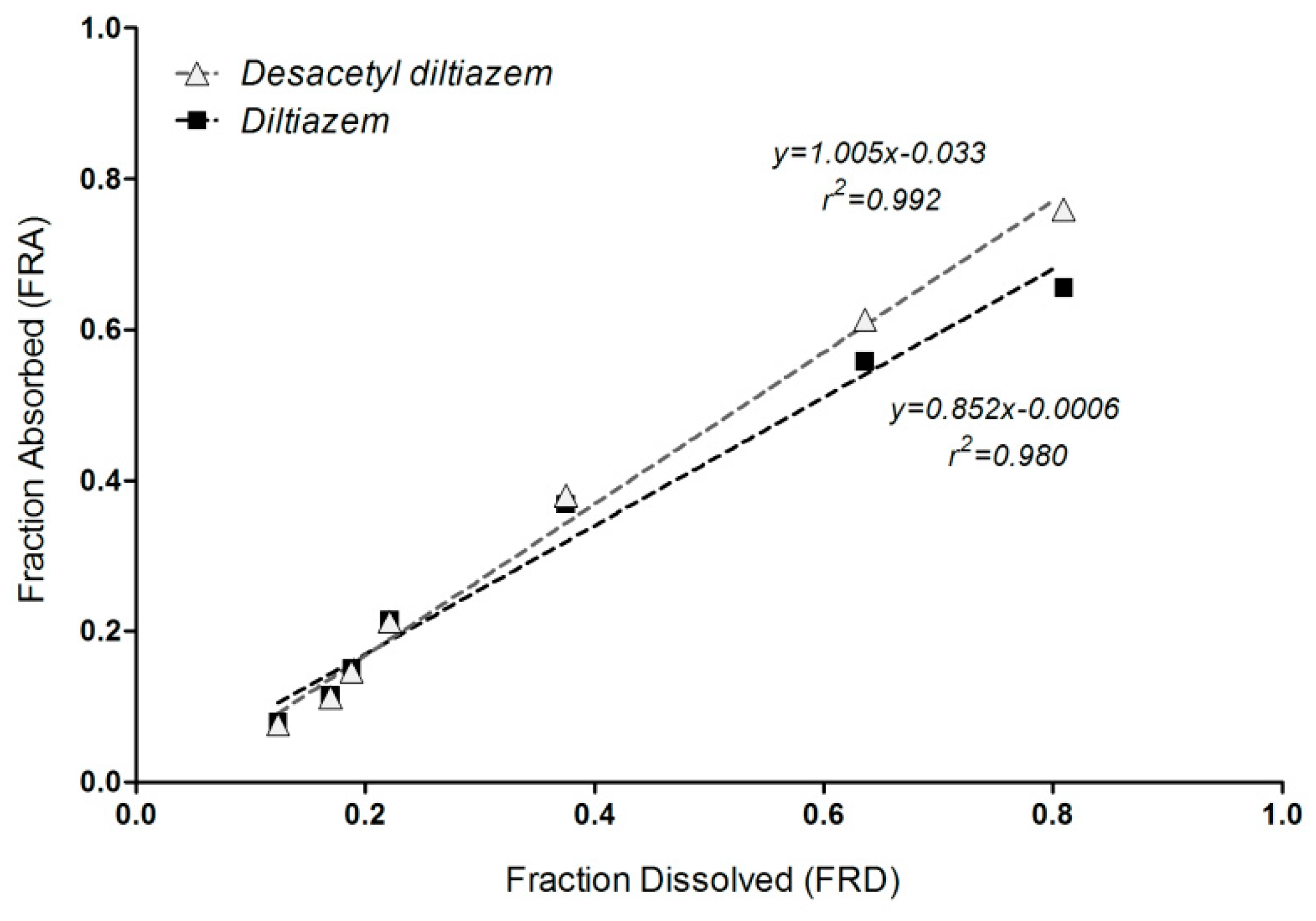
3.6. Correlation of Apparent Absorbed/Metabolized Fraction with In Vitro Dissolution of Diltiazem
4. Discussion
5. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Humbert, H.; Bosshardt, H.; Cabiac, M.-D.; Cabiac, M. In Vitro-in Vivo Correlation of a Modified-Release Oral Form of Ketotifen: In Vitro Dissolution Rate Specification. J. Pharm. Sci. 1994, 83, 131–136. [Google Scholar] [CrossRef] [PubMed]
- Eddington, N.D.; Marroum, P.; Uppoor, R.; Hussain, A.; Augsburger, L. Development and Internal Validation of an In Vitro-in Vivo Correlation for a Hydrophilic Metoprolol Tartrate Extended Release Tablet Formulation. Pharm. Res. 1998, 15, 466–473. [Google Scholar] [CrossRef]
- Mahayni, H.; Rekhi, G.; Uppoor, R.; Marroum, P.; Hussain, A.; Augsburger, L.; Eddington, N. Evaluation of “External” Predictability of an In Vitro–In Vivo Correlation for an Extended-Release Formulation Containing Metoprolol Tartrate. J. Pharm. Sci. 2000, 89, 1354–1361. [Google Scholar] [CrossRef]
- Takka, S.; Rajbhandari, S.; Sakr, A. Effect of anionic polymers on the release of propranolol hydrochloride from matrix tablets. Eur. J. Pharm. Biopharm. 2001, 52, 75–82. [Google Scholar] [CrossRef]
- Emami, J. In vitro-in vivo correlation: From theory to applications. J. Pharm. Pharm. Sci. 2006, 9, 169–189. [Google Scholar]
- Lake, O.; Olling, M.; Barends, D. In vitro/in vivo correlations of dissolution data of carbamazepine immediate release tablets with pharmacokinetic data obtained in healthy volunteers. Eur. J. Pharm. Biopharm. 1999, 48, 13–19. [Google Scholar] [CrossRef]
- Varshosaz, J.; Ghafghazi, T.; Raisi, A.; Falamarzian, M. Biopharmaceutical characterization of oral theophylline and aminophylline tablets. Quantitative correlation between dissolution and bioavailability studies. Eur. J. Pharm. Biopharm. 2000, 50, 301–306. [Google Scholar] [CrossRef]
- Rao, B.S.; Seshasayana, A.; Saradhi, S.P.; Kumar, N.R.; Narayan, C.P.; Murthy, K.R. Correlation of ‘in vitro’ release and ‘in vivo’ absorption characteristics of rifampicin from ethylcellulose coated nonpareil beads. Int. J. Pharm. 2001, 230, 1–9. [Google Scholar]
- Al-Behaisi, S.; Antal, I.; Morovján, G.; Szunyog, J.; Drabant, S.; Marton, S.; Klebovich, I. In vitro simulation of food effect on dissolution of deramciclane film-coated tablets and correlation with in vivo data in healthy volunteers. Eur. J. Pharm. Sci. 2002, 15, 157–162. [Google Scholar] [CrossRef]
- Mircioiu, C.; Mircioiu, I.; Voicu, V.; Miron, D. Dissolution-Bioequivalence Non-Correlations. Basic Clin. Pharmacol. Toxicol. 2005, 96, 262–264. [Google Scholar] [CrossRef]
- Meyer, M.C.; Straughn, A.B.; Mhatre, R.M.; Shah, V.P.; Williams, R.L.; Lesko, L.J. Lack of In Vivo/In Vitro Correlations for 50 mg and 250 mg Primidone Tablets. Pharm. Res. 1998, 15, 1085–1089. [Google Scholar] [CrossRef]
- Food and Drug Administration-Center for Drug Evaluation and Research (CDER). Guidance for Industry: Extended Release Oral Dosage Forms: Development, Evaluation, and Application of In Vitro/In Vivo Correlations. Available online: https://www.fda.gov/media/70939/download (accessed on 19 February 2019).
- Young, D. Significance of in Vitro in Vivo Correlation (ivivc). In International Bioequivalence Standards: A New Era; Amidon, G.L., Lesko, L.J., Midha, K.K., Shah, V.P., Hilfinger, J.M., Eds.; TSRL: Ann Arbor, MI, USA, 2006; pp. 41–48. [Google Scholar]
- USP 34-NF 29. The United States Pharmacopeia 32-the National Formulary 27; United States Pharmacopeial Convention Inc.: Rockville, MD, USA, 2011. [Google Scholar]
- Polli, J.E.; Crison, J.R.; Amidon, G.L. Novel approach to the analysis of in vitro-in vivo relationships. J. Pharm. Sci. 1996, 85, 753–760. [Google Scholar] [CrossRef]
- Dunne, A.; O’Hara, T.; DeVane, J. A new approach to modelling the relationship between in vitro and in vivo drug dissolution/absorption. Stat. Med. 1999, 18, 1865–1876. [Google Scholar] [CrossRef]
- Dunnex, A.; O’Hara, T.; DeVane, J.; Dunne, A. Level A in Vivo−in Vitro Correlation: Nonlinear Models and Statistical Methodology. J. Pharm. Sci. 1997, 86, 1245–1249. [Google Scholar] [CrossRef]
- Sirisuth, N.; Augsburger, L.L.; Eddington, N.D. Development and validation of a non-linear IVIVC model for a diltiazem extended release formulation. Biopharm. Drug Dispos. 2002, 23, 1–8. [Google Scholar] [CrossRef]
- Parojčić, J.; Ibric, S.; Djurić, Z.; Jovanović, M.; Corrigan, O.I. An investigation into the usefulness of generalized regression neural network analysis in the development of level A in vitro–in vivo correlation. Eur. J. Pharm. Sci. 2007, 30, 264–272. [Google Scholar] [CrossRef]
- Corrigan, O.I.; Devlin, Y.; Butler, J. Influence of dissolution medium buffer composition on ketoprofen release from ER products and in vitro–in vivo correlation. Int. J. Pharm. 2003, 254, 147–154. [Google Scholar] [CrossRef]
- Mendell-Harary, J.; Dowell, J.; Bigora, S.; Piscitelli, D.; Butler, J.; Farrell, C.; DeVane, J.G.; Young, D. Nonlinear in Vitro-in Vivo Correlations. In Results and Problems in Cell Differentiation; Springer Science and Business Media LLC: Berlin/Heidelberg, Germany, 1997; Volume 423, pp. 199–206. [Google Scholar]
- Sandulovici, R.; Prasacu, I.; Mircioiu, C.; Voicu, V.A.; Medvedovici, A.; Anuta, V. Mathematical and phenomenological criteria in selection of pharmacokinetic model for m1 metabolite of pentoxyphylline. FARMACIA 2009, 57, 235–246. [Google Scholar]
- Tvrdonova, M.; Dedik, L.; Mircioiu, C.; Miklovicova, D.; Ďurišová, M. Physiologically Motivated Time-Delay Model to Account for Mechanisms Underlying Enterohepatic Circulation of Piroxicam in Human Beings. Basic Clin. Pharmacol. Toxicol. 2009, 104, 35–42. [Google Scholar] [CrossRef]
- Chaffman, M.; Brogden, R.N.; Speight, T.M.; Avery, G.S. A review of its pharmacological properties and therapeutic efficacy. Drugs 1985, 29, 387–454. [Google Scholar] [CrossRef]
- Fagan, T.C. Diltiazem: Its place in the antihypertensive armamentarium. J. Cardiovasc. Pharmacol. 1991, 18, S26–S31. [Google Scholar] [CrossRef]
- Hermann, P.; Rodger, S.D.; Remones, G.; Thenot, J.P.; London, D.R.; Morselli, P.L. Pharmacokinetics of diltiazem after intravenous and oral administration. Eur. J. Clin. Pharmacol. 1983, 24, 349–352. [Google Scholar] [CrossRef]
- Caillé, G.; Boucher, S.; Spénard, J.; Lakhani, Z.; Russell, A.; Thiffault, J.; Grace, M.G. Diltiazem pharmacokinetics in elderly volunteers after single and multiple doses. Eur. J. Drug Metab. Pharmacokinet. 1991, 16, 75–80. [Google Scholar] [CrossRef]
- Yeung, P.K.F.; Montague, T.J.; Tsui, B.; McGregor, C. High-Performance Liquid Chromatographic Assay of Diltiazem and Six of Its Metabolites in Plasma: Application to a Pharmacokinetic Study in Healthy Volunteers. J. Pharm. Sci. 1989, 78, 592–597. [Google Scholar] [CrossRef]
- Food and Drug Administration-Center for Drug Evaluation and Research (CDER). Guidance for Industry: Bioanalytical Method Validation. Available online: https://www.fda.gov/regulatory-information/search-fda-guidance-documents/bioanalytical-method-validation-guidance-industry (accessed on 26 March 2019).
- Wagner, J.G.; Nelson, E. Kinetic Analysis of Blood Levels and Urinary Excretion in the Absorptive Phase after Single Doses of Drug. J. Pharm. Sci. 1964, 53, 1392–1403. [Google Scholar] [CrossRef]
- Kokate, A.; Li, X.; Williams, P.J.; Singh, P.; Jasti, B.R. In Silico Prediction of Drug Permeability Across Buccal Mucosa. Pharm. Res. 2009, 26, 1130–1139. [Google Scholar] [CrossRef]
- Chrenova, J.; Durisova, M.; Mircioui, C.; Dedik, L.; Mircioiu, C. Effect of gastric emptying and entero-hepatic circulation on bioequivalence assessment of ranitidine. Methods Find. Exp. Clin. Pharmacol. 2010, 32, 413. [Google Scholar] [CrossRef]
- Marchidanu, D.; Raducanu, N.; Miron, D.S.; Radulescu, F.S.; Anuta, V.; Mircioiu, I.; Prasacu, I. Comparative pharmacokinetics of rifampicin and 25-desacetyl rifampicin in healthy volunteers after single oral dose administration. FARMACIA 2013, 61, 398–410. [Google Scholar]
- Mircioiu, C.; Ionica, G.; Danilceac, A.; Miron, D.; Mircioiu, I.; Radulescu, F.S. Pharmacokinetic and mathematical outliers for drugs with active metabolites. Note i. Model independent analyses for pentoxifylline. FARMACIA 2010, 58, 264–278. [Google Scholar]
- Wu, C.Y.; Benet, L.Z. Predicting Drug disposition via application of BCS: Transport/absorption/elimination interplay and development of a biopharmaceutics. drug disposition classification SYSTEM. Pharm. Res. 2005, 22, 11–23. [Google Scholar] [CrossRef]
 ).
).


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Time (min) | Dissolved (%) | CV (%) |
|---|---|---|
| 10 | 12.42 | 10.56 |
| 15 | 17.00 | 9.04 |
| 20 | 18.67 | 15.88 |
| 30 | 22.17 | 11.19 |
| 60 | 37.67 | 10.46 |
| 120 | 63.58 | 9.66 |
| 180 | 80.92 | 8.29 |
| Sample Code | DTZ | DTZM | ||||||
|---|---|---|---|---|---|---|---|---|
| Nominal conc. (ng/mL) | Measured conc. (Mean ± SD, ng/mL) | RSD (%) | Bias (%) | Nominal conc. (ng/mL) | Measured conc. (Mean ± SD, ng/mL) | RSD (%) | Bias (%) | |
| Within-run | ||||||||
| LLOQ | 2.5 | 2.49 ± 0.14 | 5.71 | −0.56 | 1.25 | 1.26 ± 0.05 | 3.79 | 0.40 |
| QClow | 7.5 | 7.48 ± 0.29 | 3.89 | −0.25 | 3.75 | 3.72 ± 0.14 | 3.73 | −0.85 |
| QCmed | 150 | 148.83 ± 2.76 | 1.85 | −0.78 | 75 | 72.18 ± 1.65 | 2.29 | −3.76 |
| QChigh | 300 | 310.46 ± 3.70 | 1.19 | 3.49 | 150 | 146.26 ± 2.47 | 1.69 | −2.49 |
| Between runs | ||||||||
| LLOQ | 2.5 | 2.63 ± 0.17 | 6.43 | 5.04 | 1.25 | 1.32 ± 0.07 | 5.32 | 5.60 |
| QClow | 7.5 | 7.52 ± 0.31 | 4.15 | 0.24 | 3.75 | 3.74 ± 0.30 | 8.03 | −0.30 |
| QCmed | 150 | 146.22 ± 6.68 | 4.57 | −2.52 | 75 | 73.71 ± 3.42 | 4.64 | −1.72 |
| QChigh | 300 | 298.91 ± 12.52 | 4.19 | −0.36 | 150 | 149.37 ± 5.27 | 3.53 | −0.42 |
| Parameter | DTZ | DTZM | ||
|---|---|---|---|---|
| Mean | SD | Mean | SD | |
| Cmax (ng/mL) | 154 | 59.6 | 6.66 | 1.98 |
| Tmax (h) | 2.66 | 0.898 | 4 | 1.12 |
| ke (1/h) | 0.157 | 0.0237 | 0.074 | 0.0325 |
| t1/2 (h) | 4.51 | 0.655 | 11.2 | 5.25 |
| Parameter | DTZ | DTZM |
|---|---|---|
| ke (1/h) | 0.2861 | 0.0945 |
| ka (1/h) | 0.0655 | 0.492 |
| Tlag (h) | 0.36 | 0.162 |
| Cmax (ng/mL) | 120.5 | 6.04 |
| Tmax (h) | 2.51 | 4.20 |
| AUC0–∞ (ng/mL·h) | 800.9 | 94.66 |
| t1/2 (h) | 2.42 | 7.34 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mircioiu, C.; Anuta, V.; Mircioiu, I.; Nicolescu, A.; Fotaki, N. In Vitro–In Vivo Correlations Based on In Vitro Dissolution of Parent Drug Diltiazem and Pharmacokinetics of Its Metabolite. Pharmaceutics 2019, 11, 344. https://doi.org/10.3390/pharmaceutics11070344
Mircioiu C, Anuta V, Mircioiu I, Nicolescu A, Fotaki N. In Vitro–In Vivo Correlations Based on In Vitro Dissolution of Parent Drug Diltiazem and Pharmacokinetics of Its Metabolite. Pharmaceutics. 2019; 11(7):344. https://doi.org/10.3390/pharmaceutics11070344
Chicago/Turabian StyleMircioiu, Constantin, Valentina Anuta, Ion Mircioiu, Adrian Nicolescu, and Nikoletta Fotaki. 2019. "In Vitro–In Vivo Correlations Based on In Vitro Dissolution of Parent Drug Diltiazem and Pharmacokinetics of Its Metabolite" Pharmaceutics 11, no. 7: 344. https://doi.org/10.3390/pharmaceutics11070344
APA StyleMircioiu, C., Anuta, V., Mircioiu, I., Nicolescu, A., & Fotaki, N. (2019). In Vitro–In Vivo Correlations Based on In Vitro Dissolution of Parent Drug Diltiazem and Pharmacokinetics of Its Metabolite. Pharmaceutics, 11(7), 344. https://doi.org/10.3390/pharmaceutics11070344