
Plasmid-Based Reverse Genetics System Enabling One-Step Generation of Genotype 3 Hepatitis E Virus


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
2.1. Plasmids and Construction
2.2. In Vitro RNA Synthesis and Capping
2.3. Cell Culture and Transfection
2.4. Quantification of HEV RNA
2.5. The 5′ Rapid Amplification of cDNA Ends (5′RACE)
2.6. Detection of Genetic Marker
2.7. Western Blotting
2.8. Immunofluorescence
2.9. Northern Blotting
2.10. Virus Inoculation
3. Results
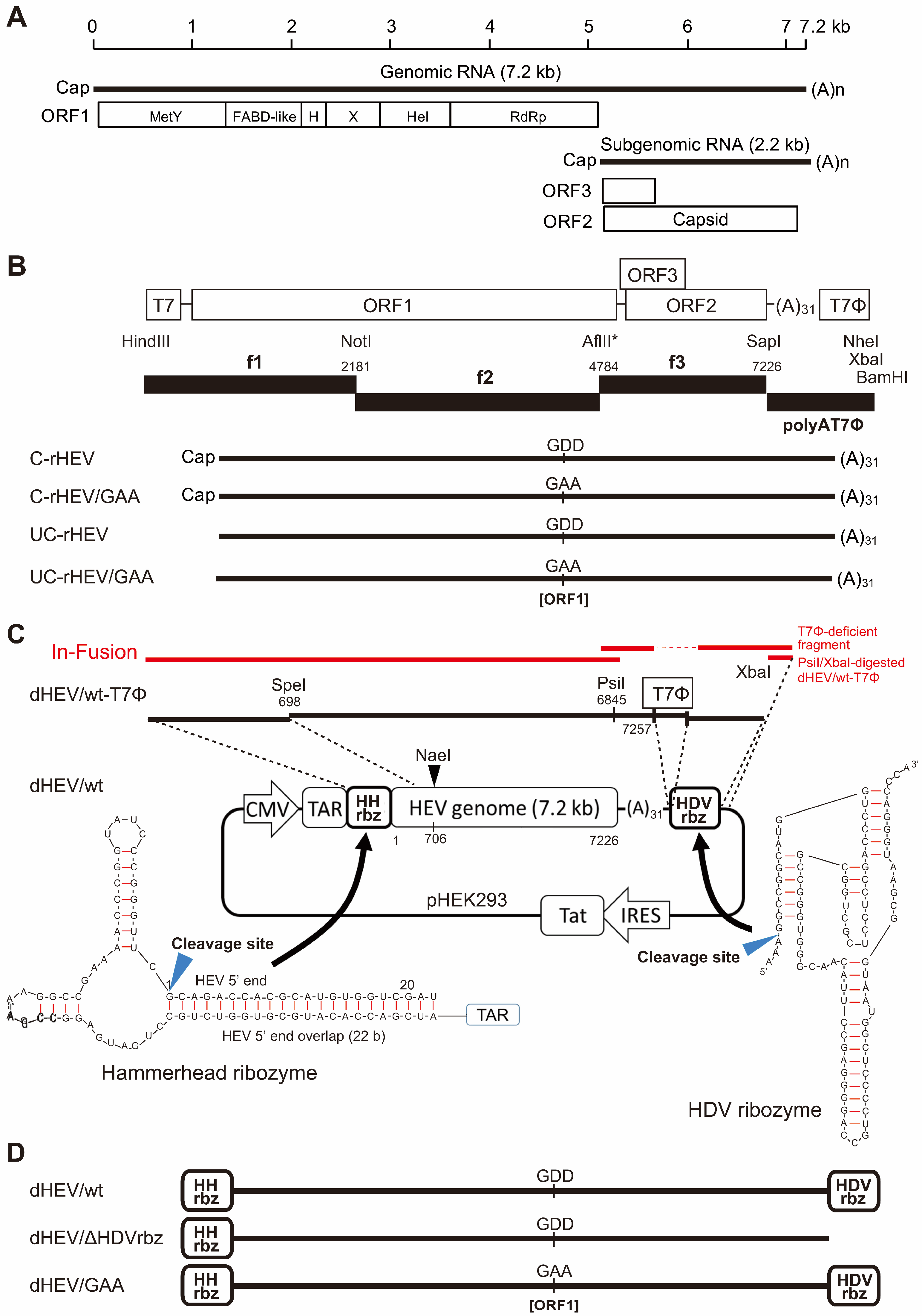
3.1. Construction of a Plasmid-Based HEV Expression Vector
3.2. Evaluation of Transfection Reagents
3.3. Comparison of HEV Production Efficiency Between the HEV Expression Plasmid Vector and In Vitro-Synthesized HEV RNA Genome
3.4. Delayed Capping of Plasmid-Derived HEV Genomic RNA in Cells
3.5. Retention of the Genetic Marker in Plasmid-Derived HEV Genome
3.6. Expression of Viral Proteins from a Plasmid-Based HEV Expression Vector
3.7. Replication of the Cellular HEV RNA Genome in HEV Expression Plasmid DNA-Transfected Cells
3.8. Infectivity of Progeny Virions from Plasmid-Based HEV
3.9. Effect of the Vaccinia Virus Capping Enzyme on HEV Replication
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
References
- Li, P.; Liu, J.; Li, Y.; Su, J.; Ma, Z.; Bramer, W.M.; Cao, W.; de Man, R.A.; Peppelenbosch, M.P.; Pan, Q. The Global Epidemiology of Hepatitis E Virus Infection: A Systematic Review and Meta-Analysis. Liver Int. 2020, 40, 1516–1528. [Google Scholar] [CrossRef] [PubMed]
- Horvatits, T.; Schulze zur Wiesch, J.; Lütgehetmann, M.; Lohse, A.W.; Pischke, S. The Clinical Perspective on Hepatitis E. Viruses 2019, 11, 617. [Google Scholar] [CrossRef] [PubMed]
- Emerson, S.U.; Purcell, R.H. Hepatitis E Virus. In Fields Virology, 6th ed.; Lippincott Williams & Wilkins: Philadelphia, PA, USA, 2013; pp. 2242–2258. [Google Scholar]
- Meng, X.-J. Zoonotic and Foodborne Transmission of Hepatitis E Virus. Semin. Liver Dis. 2013, 33, 41–49. [Google Scholar] [CrossRef]
- Ma, Z.; de Man, R.A.; Kamar, N.; Pan, Q. Chronic Hepatitis E: Advancing Research and Patient Care. J. Hepatol. 2022, 77, 1109–1123. [Google Scholar] [CrossRef] [PubMed]
- Damiris, K.; Aghaie Meybodi, M.; Niazi, M.; Pyrsopoulos, N. Hepatitis E in Immunocompromised Individuals. World J. Hepatol. 2022, 14, 482–494. [Google Scholar] [CrossRef]
- Pischke, S.; Hartl, J.; Pas, S.D.; Lohse, A.W.; Jacobs, B.C.; Van der Eijk, A.A. Hepatitis E Virus: Infection beyond the Liver? J. Hepatol. 2017, 66, 1082–1095. [Google Scholar] [CrossRef]
- Takahashi, M.; Hoshino, Y.; Tanaka, T.; Takahashi, H.; Nishizawa, T.; Okamoto, H. Production of Monoclonal Antibodies against Hepatitis E Virus Capsid Protein and Evaluation of Their Neutralizing Activity in a Cell Culture System. Arch. Virol. 2008, 153, 657–666. [Google Scholar] [CrossRef]
- Takahashi, M.; Tanaka, T.; Takahashi, H.; Hoshino, Y.; Nagashima, S.; Jirintai; Mizuo, H.; Yazaki, Y.; Takagi, T.; Azuma, M.; et al. Hepatitis E Virus (HEV) Strains in Serum Samples Can Replicate Efficiently in Cultured Cells despite the Coexistence of HEV Antibodies: Characterization of HEV Virions in Blood Circulation. J. Clin. Microbiol. 2010, 48, 1112–1125. [Google Scholar] [CrossRef]
- Yin, X.; Ambardekar, C.; Lu, Y.; Feng, Z. Distinct Entry Mechanisms for Nonenveloped and Quasi-Enveloped Hepatitis E Viruses. J. Virol. 2016, 90, 4232–4242. [Google Scholar] [CrossRef]
- Nagashima, S.; Takahashi, M.; Kobayashi, T.; Tanggis; Nishizawa, T.; Nishiyama, T.; Primadharsini, P.P.; Okamoto, H. Characterization of the Quasi-Enveloped Hepatitis E Virus Particles Released by the Cellular Exosomal Pathway. J. Virol. 2017, 91, e00822-17. [Google Scholar] [CrossRef]
- Purdy, M.A.; Drexler, J.F.; Meng, X.-J.; Norder, H.; Okamoto, H.; Van der Poel, W.H.M.; Reuter, G.; de Souza, W.M.; Ulrich, R.G.; Smith, D.B. ICTV Virus Taxonomy Profile: Hepeviridae 2022. J. Gen. Virol. 2022, 103, 001778. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Zhao, C.; Qi, Y.; Geng, Y. Hepatitis E Virus. Adv. Exp. Med. Biol. 2016, 948, 1–16. [Google Scholar] [CrossRef]
- Tam, A.W.; Smith, M.M.; Guerra, M.E.; Huang, C.C.; Bradley, D.W.; Fry, K.E.; Reyes, G.R. Hepatitis E Virus (HEV): Molecular Cloning and Sequencing of the Full-Length Viral Genome. Virology 1991, 185, 120–131. [Google Scholar] [CrossRef]
- Kabrane-Lazizi, Y.; Meng, X.J.; Purcell, R.H.; Emerson, S.U. Evidence That the Genomic RNA of Hepatitis E Virus Is Capped. J. Virol. 1999, 73, 8848–8850. [Google Scholar] [CrossRef]
- Koonin, E.V.; Gorbalenya, A.E.; Purdy, M.A.; Rozanov, M.N.; Reyes, G.R.; Bradley, D.W. Computer-Assisted Assignment of Functional Domains in the Nonstructural Polyprotein of Hepatitis E Virus: Delineation of an Additional Group of Positive-Strand RNA Plant and Animal Viruses. Proc. Natl. Acad. Sci. USA 1992, 89, 8259–8263. [Google Scholar] [CrossRef] [PubMed]
- Panda, S.K.; Varma, S.P.K. Hepatitis E: Molecular Virology and Pathogenesis. J. Clin. Exp. Hepatol. 2013, 3, 114–124. [Google Scholar] [CrossRef] [PubMed]
- Fieulaine, S.; Tubiana, T.; Bressanelli, S. De novo modelling of HEV replication polyprotein: Five-domain breakdown and involvement of flexibility in functional regulation. Virology 2023, 578, 128–140. [Google Scholar] [CrossRef]
- Yin, X.; Ying, D.; Lhomme, S.; Tang, Z.; Walker, C.M.; Xia, N.; Zheng, Z.; Feng, Z. Origin, Antigenicity, and Function of a Secreted Form of ORF2 in Hepatitis E Virus Infection. Proc. Natl. Acad. Sci. USA 2018, 115, 4773–4778. [Google Scholar] [CrossRef]
- Montpellier, C.; Wychowski, C.; Sayed, I.M.; Meunier, J.-C.; Saliou, J.-M.; Ankavay, M.; Bull, A.; Pillez, A.; Abravanel, F.; Helle, F.; et al. Hepatitis E Virus Lifecycle and Identification of 3 Forms of the ORF2 Capsid Protein. Gastroenterology 2018, 154, 211–223.e8. [Google Scholar] [CrossRef]
- Smith, D.B.; Izopet, J.; Nicot, F.; Simmonds, P.; Jameel, S.; Meng, X.-J.; Norder, H.; Okamoto, H.; van der Poel, W.H.M.; Reuter, G.; et al. Update: Proposed Reference Sequences for Subtypes of Hepatitis E Virus (Species Orthohepevirus A). J. Gen. Virol. 2020, 101, 692–698. [Google Scholar] [CrossRef]
- Raji, Y.E.; Toung, O.P.; Taib, N.M.; Sekawi, Z. Bin Hepatitis E Virus: An Emerging Enigmatic and Underestimated Pathogen. Saudi J. Biol. Sci. 2022, 29, 499–512. [Google Scholar] [CrossRef]
- Lee, G.-H.; Tan, B.-H.; Teo, E.C.-Y.; Lim, S.-G.; Dan, Y.-Y.; Wee, A.; Aw, P.P.K.; Zhu, Y.; Hibberd, M.L.; Tan, C.-K.; et al. Chronic Infection with Camelid Hepatitis E Virus in a Liver Transplant Recipient Who Regularly Consumes Camel Meat and Milk. Gastroenterology 2016, 150, 355–357.e3. [Google Scholar] [CrossRef] [PubMed]
- Primadharsini, P.P.; Nagashima, S.; Okamoto, H. Mechanism of Cross-Species Transmission, Adaptive Evolution and Pathogenesis of Hepatitis E Virus. Viruses 2021, 13, 909. [Google Scholar] [CrossRef] [PubMed]
- Alonso, D.; Mondragón, A. Mechanisms of Catalytic RNA Molecules. Biochem. Soc. Trans. 2021, 49, 1529–1535. [Google Scholar] [CrossRef] [PubMed]
- Peng, H.; Latifi, B.; Müller, S.; Lupták, A.; Chen, I.A. Self-Cleaving Ribozymes: Substrate Specificity and Synthetic Biology Applications. RSC Chem. Biol. 2021, 2, 1370–1383. [Google Scholar] [CrossRef]
- Hammann, C.; Luptak, A.; Perreault, J.; de la Peña, M. The Ubiquitous Hammerhead Ribozyme. RNA 2012, 18, 871–885. [Google Scholar] [CrossRef]
- Criglar, J.M.; Crawford, S.E.; Estes, M.K. Plasmid-Based Reverse Genetics for Probing Phosphorylation-Dependent Viroplasm Formation in Rotaviruses. Virus Res. 2021, 291, 198193. [Google Scholar] [CrossRef]
- Álvarez, Á.L.; Arboleya, A.; Abade Dos Santos, F.A.; García-Manso, A.; Nicieza, I.; Dalton, K.P.; Parra, F.; Martín-Alonso, J.M. Highs and Lows in Calicivirus Reverse Genetics. Viruses 2024, 16, 866. [Google Scholar] [CrossRef]
- Zhang, Q.; Zhu, S.; Zhang, X.; Su, L.; Ni, J.; Zhang, Y.; Fang, L. Recent Insights into Reverse Genetics of Norovirus. Virus Res. 2023, 325, 199046. [Google Scholar] [CrossRef]
- Sosnovtsev, S.; Sosnovtseva, S.; Green, K.Y. Recovery of Feline Calicivirus from Plasmid DNA Containing a Full-Length Copy of the Genome; European Society for Veterinary Virology and Central Veterinary Laboratory: Reading, UK, 1997; pp. 125–130. [Google Scholar]
- Willenbrink, W.; Neubert, W.J. Long-Term Replication of Sendai Virus Defective Interfering Particle Nucleocapsids in Stable Helper Cell Lines. J. Virol. 1994, 68, 8413–8417. [Google Scholar] [CrossRef]
- Radecke, F.; Spielhofer, P.; Schneider, H.; Kaelin, K.; Huber, M.; Dötsch, C.; Christiansen, G.; Billeter, M.A. Rescue of Measles Viruses from Cloned DNA. EMBO J. 1995, 14, 5773–5784. [Google Scholar] [CrossRef] [PubMed]
- Kanai, Y.; Komoto, S.; Kawagishi, T.; Nouda, R.; Nagasawa, N.; Onishi, M.; Matsuura, Y.; Taniguchi, K.; Kobayashi, T. Entirely Plasmid-Based Reverse Genetics System for Rotaviruses. Proc. Natl. Acad. Sci. USA 2017, 114, 2349–2354. [Google Scholar] [CrossRef] [PubMed]
- Hamajima, R.; Lusiany, T.; Minami, S.; Nouda, R.; Nurdin, J.A.; Yamasaki, M.; Kobayashi, N.; Kanai, Y.; Kobayashi, T. A Reverse Genetics System for Human Rotavirus G2P[4]. J. Gen. Virol. 2022, 103, 001816. [Google Scholar] [CrossRef]
- Philip, A.A.; Dai, J.; Katen, S.P.; Patton, J.T. Simplified Reverse Genetics Method to Recover Recombinant Rotaviruses Expressing Reporter Proteins. J. Vis. Exp. 2020, 158, e61039. [Google Scholar] [CrossRef]
- Oka, T.; Takagi, H.; Tohya, Y. Development of a Novel Single Step Reverse Genetics System for Feline Calicivirus. J. Virol. Methods 2014, 207, 178–181. [Google Scholar] [CrossRef]
- Cheng, J.; Tang, A.; Chen, J.; Zhang, D.; Meng, C.; Li, C.; Wei, H.; Liu, G. A CDNA-Based Reverse Genetics System for Feline Calicivirus Identifies the 3′ Untranslated Region as an Essential Element for Viral Replication. Arch. Virol. 2023, 168, 33. [Google Scholar] [CrossRef]
- Taube, S.; Wobus, C.E. A Novel Reverse Genetics System for Human Norovirus. Trends Microbiol. 2014, 22, 604–606. [Google Scholar] [CrossRef]
- Wang, X.; Zhang, D.; Tang, A.; Zhang, M.; Zhu, S.; Zhu, Y.; Li, B.; Meng, C.; Li, C.; Zhu, J.; et al. Establishment of a Reverse Genetics System for Virulent Systemic Feline Calicivirus Using Circular Polymerase Extension Reaction. J. Virol. Methods 2024, 330, 115031. [Google Scholar] [CrossRef]
- Pretorius, J.M.; Huismans, H.; Theron, J. Establishment of an Entirely Plasmid-Based Reverse Genetics System for Bluetongue Virus. Virology 2015, 486, 71–77. [Google Scholar] [CrossRef]
- Scholz, J.; Falkenhagen, A.; Bock, C.-T.; Johne, R. Reverse Genetics Approaches for Hepatitis E Virus and Related Viruses. Curr. Opin. Virol. 2020, 44, 121–128. [Google Scholar] [CrossRef]
- Scholz, J.; Bächlein, C.; Gadicherla, A.K.; Falkenhagen, A.; Tausch, S.H.; Johne, R. Establishment of a Plasmid-Based Reverse Genetics System for the Cell Culture-Adapted Hepatitis E Virus Genotype 3c Strain 47832c. Pathogens 2020, 9, 157. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, T.; Takahashi, M.; Kusano, E.; Okamoto, H. Development and Evaluation of an Efficient Cell-Culture System for Hepatitis E Virus. J. Gen. Virol. 2007, 88, 903–911. [Google Scholar] [CrossRef]
- Lorenzo, F.R.; Tanaka, T.; Takahashi, H.; Ichiyama, K.; Hoshino, Y.; Yamada, K.; Inoue, J.; Takahashi, M.; Okamoto, H. Mutational Events during the Primary Propagation and Consecutive Passages of Hepatitis E Virus Strain JE03-1760F in Cell Culture. Virus Res. 2008, 137, 86–96. [Google Scholar] [CrossRef]
- Yamada, K.; Takahashi, M.; Hoshino, Y.; Takahashi, H.; Ichiyama, K.; Tanaka, T.; Okamoto, H. Construction of an Infectious CDNA Clone of Hepatitis E Virus Strain JE03-1760F That Can Propagate Efficiently in Cultured Cells. J. Gen. Virol. 2009, 90, 457–462. [Google Scholar] [CrossRef] [PubMed]
- Nagashima, S.; Kobayashi, T.; Tanaka, T.; Tanggis; Jirintai, S.; Takahashi, M.; Nishizawa, T.; Okamoto, H. Analysis of adaptive mutations selected during the consecutive passages of hepatitis E virus produced from an infectious cDNA clone. Virus Res. 2016, 223, 170–180. [Google Scholar] [CrossRef]
- Been, M.D.; Wickham, G.S. Self-Cleaving Ribozymes of Hepatitis Delta Virus RNA. Eur. J. Biochem. 1997, 247, 741–753. [Google Scholar] [CrossRef]
- Herold, J.; Andino, R. Poliovirus Requires a Precise 5′ End for Efficient Positive-Strand RNA Synthesis. J. Virol. 2000, 74, 6394–6400. [Google Scholar] [CrossRef] [PubMed]
- Davis, M.W.; Jorgensen, E.M. ApE, A Plasmid Editor: A Freely Available DNA Manipulation and Visualization Program. Front. Bioinform. 2022, 2, 818619. [Google Scholar] [CrossRef]
- Takahashi, M.; Yamada, K.; Hoshino, Y.; Takahashi, H.; Ichiyama, K.; Tanaka, T.; Okamoto, H. Monoclonal Antibodies Raised against the ORF3 Protein of Hepatitis E Virus (HEV) Can Capture HEV Particles in Culture Supernatant and Serum but Not Those in Feces. Arch. Virol. 2008, 153, 1703–1713. [Google Scholar] [CrossRef]
- Primadharsini, P.P.; Takahashi, M.; Nishizawa, T.; Sato, Y.; Nagashima, S.; Murata, K.; Okamoto, H. The Full-Genome Analysis and Generation of an Infectious cDNA Clone of a Genotype 6 Hepatitis E Virus Variant Obtained from a Japanese Wild Boar: In Vitro Cultivation in Human Cell Lines. Viruses 2024, 16, 842. [Google Scholar] [CrossRef]
- Zhang, W.; Ami, Y.; Suzaki, Y.; Doan, Y.H.; Takeda, N.; Muramatsu, M.; Li, T.-C. Generation of a Bactrian Camel Hepatitis E Virus by a Reverse Genetics System. J. Gen. Virol. 2021, 102, 001618. [Google Scholar] [CrossRef] [PubMed]
- Tanggis; Kobayashi, T.; Takahashi, M.; Jirintai, S.; Nishizawa, T.; Nagashima, S.; Nishiyama, T.; Kunita, S.; Hayama, E.; Tanaka, T.; et al. An Analysis of Two Open Reading Frames (ORF3 and ORF4) of Rat Hepatitis E Virus Genome Using Its Infectious CDNA Clones with Mutations in ORF3 or ORF4. Virus Res. 2018, 249, 16–30. [Google Scholar] [CrossRef]
- Huang, F.F.; Pierson, F.W.; Toth, T.E.; Meng, X.J. Construction and characterization of infectious cDNA clones of a chicken strain of hepatitis E virus (HEV), avian HEV. J Gen Virol. 2005, 86 Pt 9, 2585–2593. [Google Scholar] [CrossRef]
- Cai, H.-L.; Huang, Y.-W. Reverse Genetics Systems for SARS-CoV-2: Development and Applications. Virol. Sin. 2023, 38, 837–850. [Google Scholar] [CrossRef] [PubMed]
- Arias, A.; Bailey, D.; Chaudhry, Y.; Goodfellow, I. Development of a Reverse-Genetics System for Murine Norovirus 3: Long-Term Persistence Occurs in the Caecum and Colon. J. Gen. Virol. 2012, 93, 1432–1441. [Google Scholar] [CrossRef]
- Katayama, K.; Murakami, K.; Sharp, T.M.; Guix, S.; Oka, T.; Takai-Todaka, R.; Nakanishi, A.; Crawford, S.E.; Atmar, R.L.; Estes, M.K. Plasmid-Based Human Norovirus Reverse Genetics System Produces Reporter-Tagged Progeny Virus Containing Infectious Genomic RNA. Proc. Natl. Acad. Sci. USA 2014, 111, E4043–E4052. [Google Scholar] [CrossRef]
- Kobayashi, S.; Yoshii, K.; Hirano, M.; Muto, M.; Kariwa, H. A Novel Reverse Genetics System for Production of Infectious West Nile Virus Using Homologous Recombination in Mammalian Cells. J. Virol. Methods 2017, 240, 14–20. [Google Scholar] [CrossRef] [PubMed]
- Zheng, H.; Zheng, X.; Tong, W.; Liu, F.; Liang, C.; Wang, T.; Gao, F.; Li, L.; Shan, T.; Li, G.; et al. A Simple Method for Developing an Infectious CDNA Clone of Japanese Encephalitis Virus. Virus Genes 2017, 53, 4–14. [Google Scholar] [CrossRef]
- Beall, A.; Yount, B.; Lin, C.-M.; Hou, Y.; Wang, Q.; Saif, L.; Baric, R. Characterization of a Pathogenic Full-Length CDNA Clone and Transmission Model for Porcine Epidemic Diarrhea Virus Strain PC22A. MBio 2016, 7, e01451-15. [Google Scholar] [CrossRef]
- Chapellier, B.; Tange, S.; Tasaki, H.; Yoshida, K.; Zhou, Y.; Sakon, N.; Katayama, K.; Nakanishi, A. Examination of a Plasmid-Based Reverse Genetics System for Human Astrovirus. Microbiol. Immunol. 2015, 59, 586–596. [Google Scholar] [CrossRef]
- Minami, S.; Nouda, R.; Hirai, K.; Chen, Z.; Kotaki, T.; Kanai, Y.; Kobayashi, T. Establishment of Reverse Genetics Systems for Colorado Tick Fever Virus. PLoS Pathog. 2025, 21, e1012921. [Google Scholar] [CrossRef] [PubMed]
- Nouda, R.; Minami, S.; Kanai, Y.; Kawagishi, T.; Nurdin, J.A.; Yamasaki, M.; Kuwata, R.; Shimoda, H.; Maeda, K.; Kobayashi, T. Development of an Entirely Plasmid-Based Reverse Genetics System for 12-Segmented Double-Stranded RNA Viruses. Proc. Natl. Acad. Sci. USA 2021, 118, e2105334118. [Google Scholar] [CrossRef] [PubMed]
- Chi, Y.-I.; Martick, M.; Lares, M.; Kim, R.; Scott, W.G.; Kim, S.-H. Capturing Hammerhead Ribozyme Structures in Action by Modulating General Base Catalysis. PLoS Biol. 2008, 6, e234. [Google Scholar] [CrossRef] [PubMed]

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kobayashi, T.; Nishiyama, T.; Yamada, K.; Murata, K.; Okamoto, H. Plasmid-Based Reverse Genetics System Enabling One-Step Generation of Genotype 3 Hepatitis E Virus. Viruses 2025, 17, 669. https://doi.org/10.3390/v17050669
Kobayashi T, Nishiyama T, Yamada K, Murata K, Okamoto H. Plasmid-Based Reverse Genetics System Enabling One-Step Generation of Genotype 3 Hepatitis E Virus. Viruses. 2025; 17(5):669. https://doi.org/10.3390/v17050669
Chicago/Turabian StyleKobayashi, Tominari, Takashi Nishiyama, Kentaro Yamada, Kazumoto Murata, and Hiroaki Okamoto. 2025. "Plasmid-Based Reverse Genetics System Enabling One-Step Generation of Genotype 3 Hepatitis E Virus" Viruses 17, no. 5: 669. https://doi.org/10.3390/v17050669
APA StyleKobayashi, T., Nishiyama, T., Yamada, K., Murata, K., & Okamoto, H. (2025). Plasmid-Based Reverse Genetics System Enabling One-Step Generation of Genotype 3 Hepatitis E Virus. Viruses, 17(5), 669. https://doi.org/10.3390/v17050669