Glycine Cleavage System and cAMP Receptor Protein Co-Regulate CRISPR/cas3 Expression to Resist Bacteriophage


 ,
, 
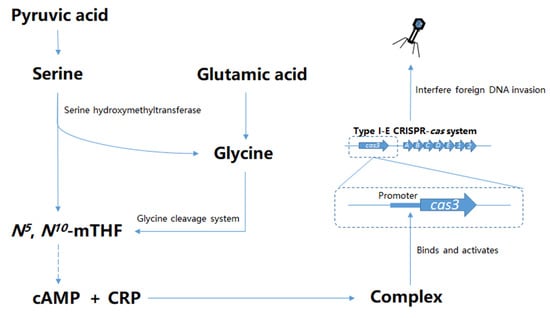
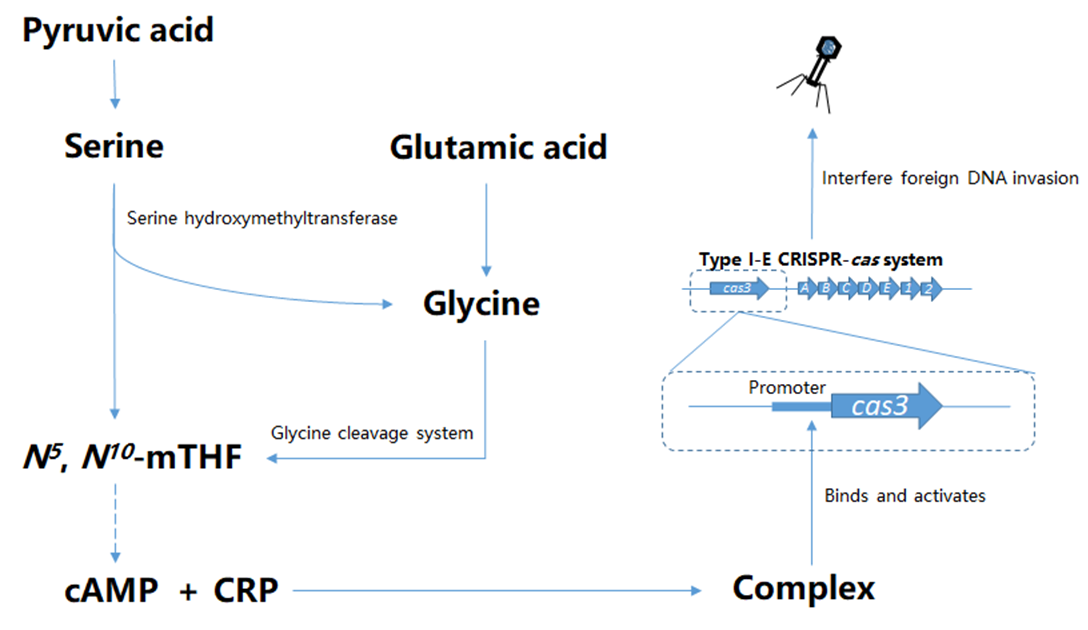
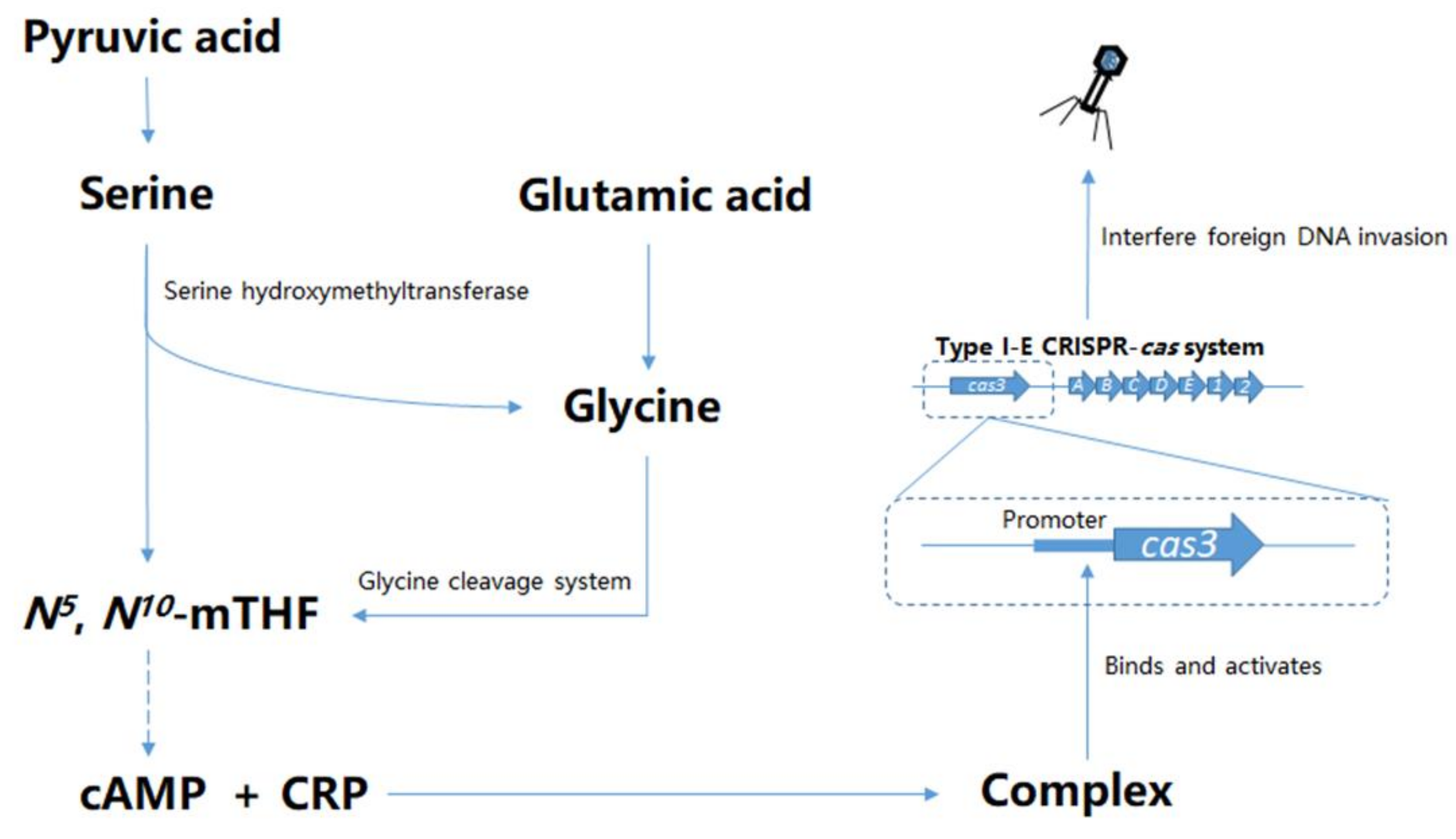
Abstract
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
2.1. Strains, Plasmids, and Growth Conditions
2.2. β-Galactosidase Assays
2.3. Construction and Identification of Transposon Mutants
2.4. DNA Pull-Down Assays
2.5. Western Blotting
2.6. Electrophoretic Mobility Shift Assays
2.7. DNase I Footprinting Assay
2.8. Lytic Infection Efficiency Assays
2.9. Plasmid Transformation Assays
3. Results
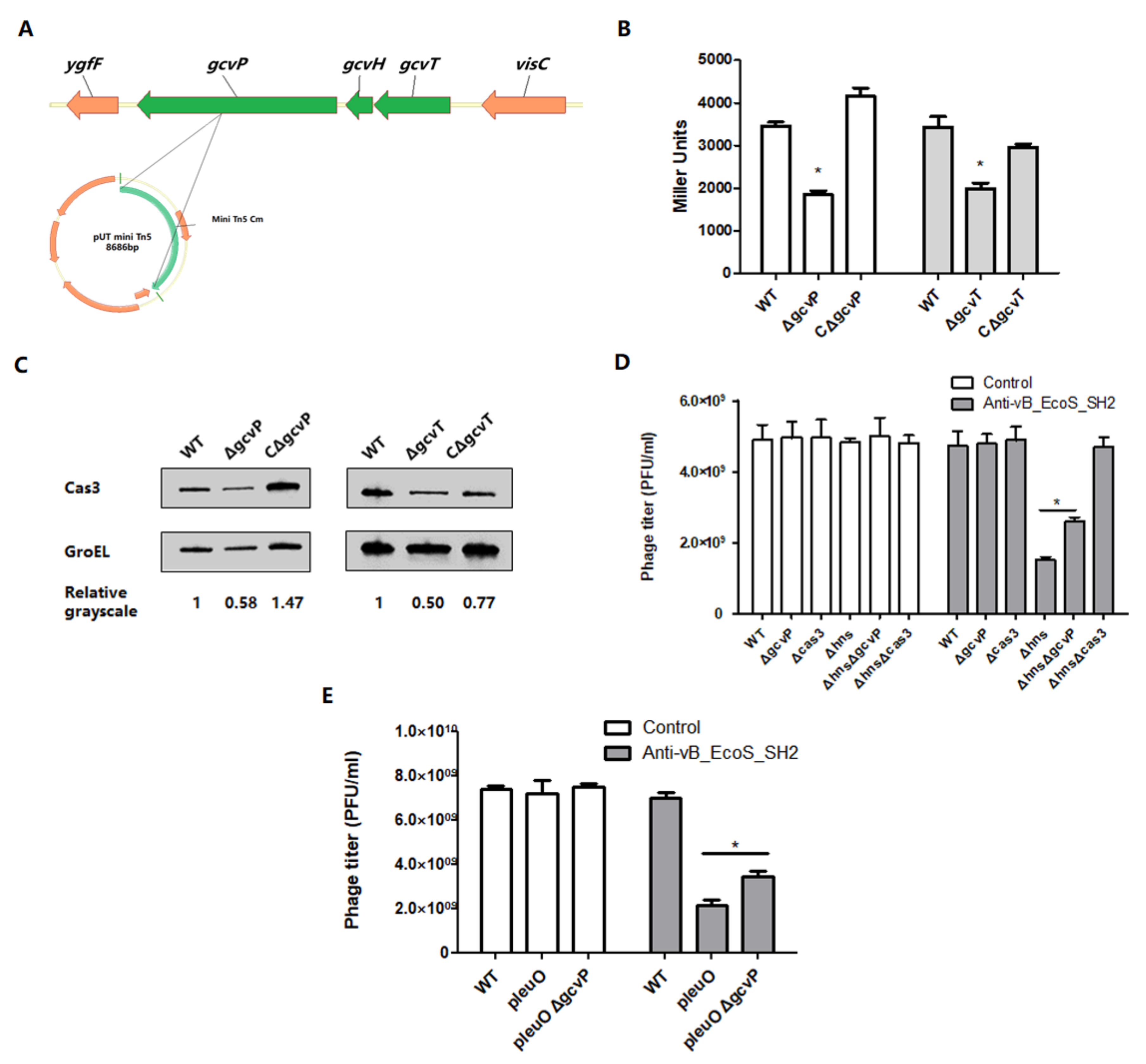
3.1. Mutation of gcvP or gcvT Decreases cas3 Expression to Affect Phage Infection
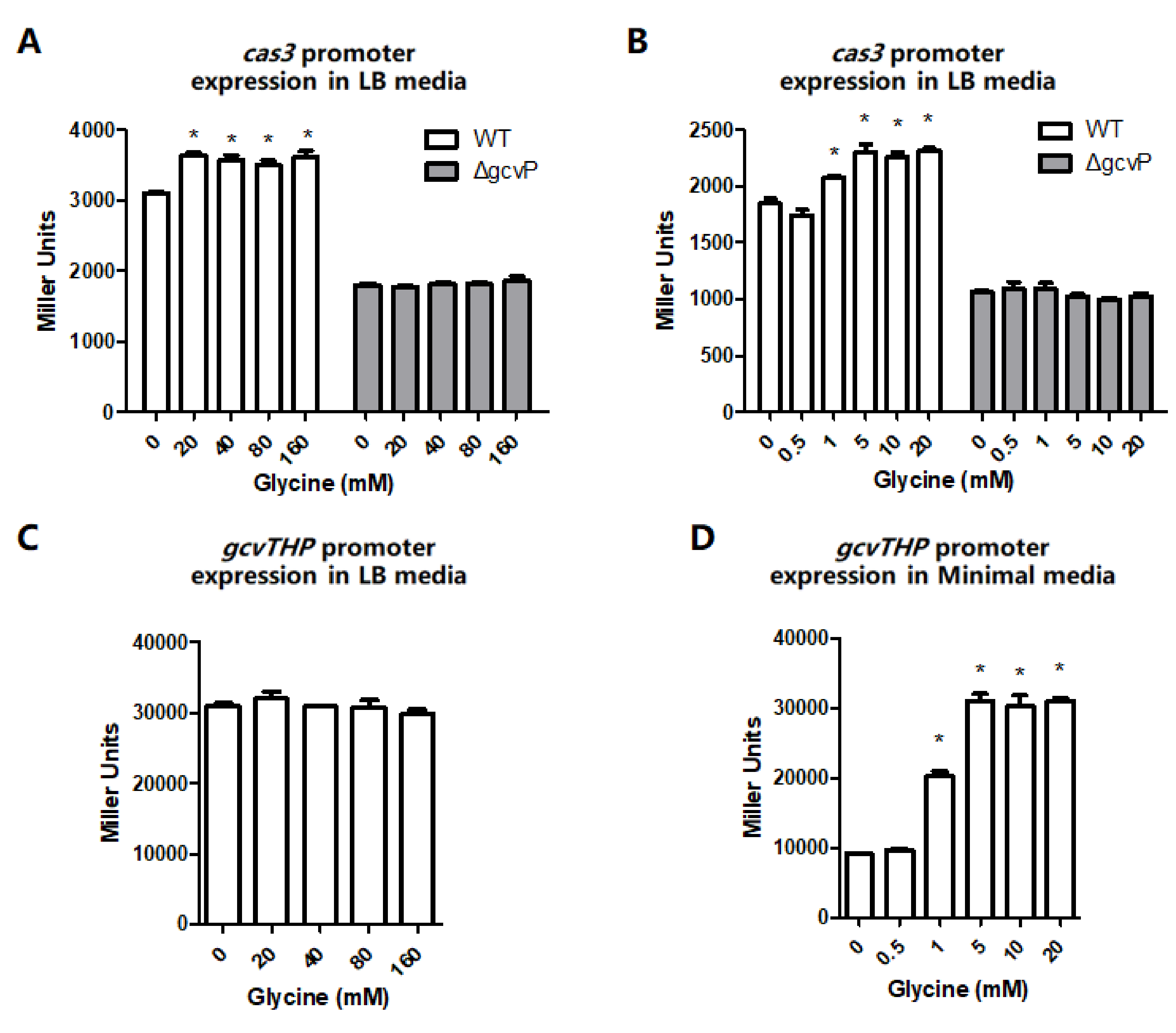
3.2. The cas3 and gcvTHP Promoter Activities are Promoted by Glycine
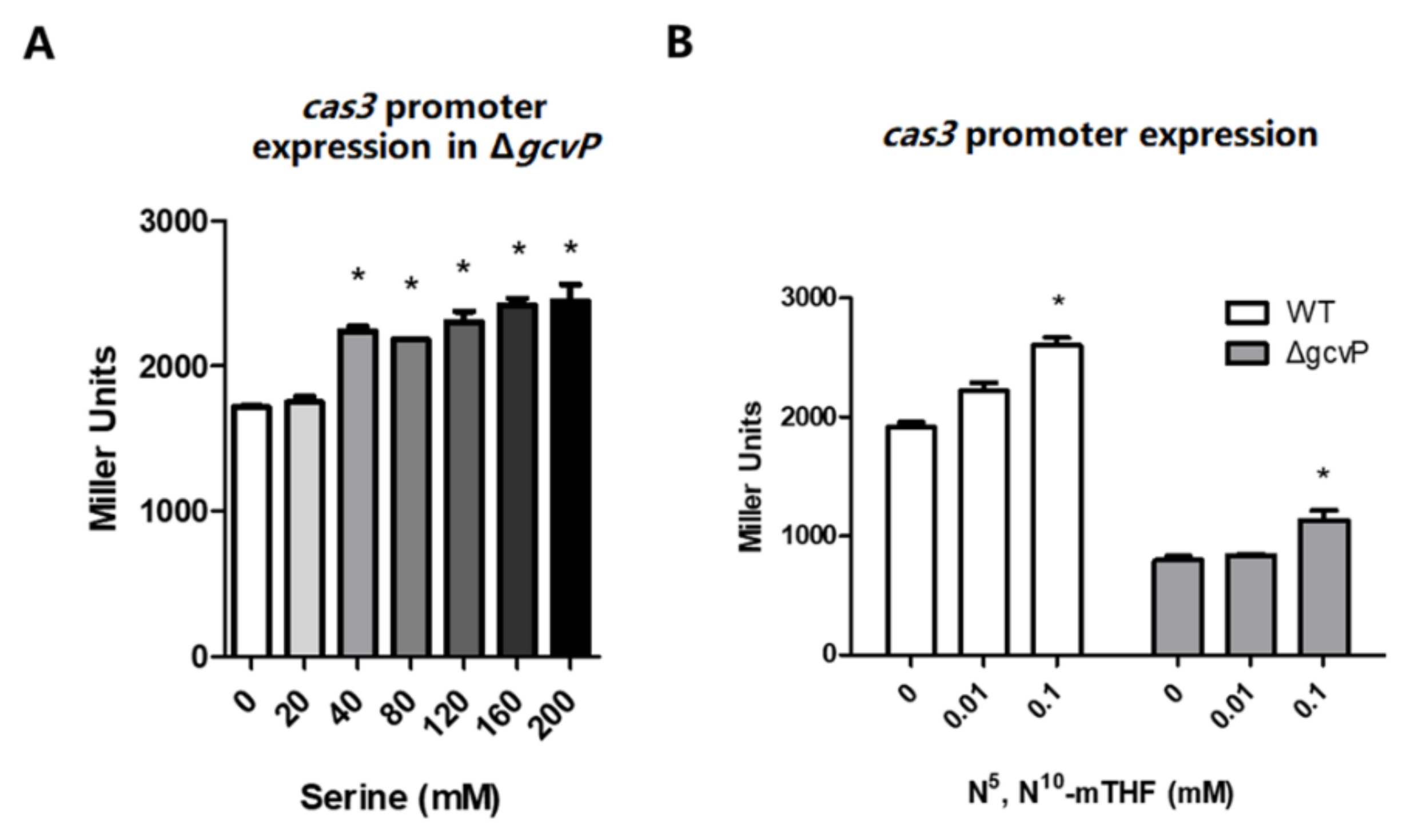
3.3. N5, N10-Methylene Tetrahydrofolate Promotes cas3 Promoter Activity
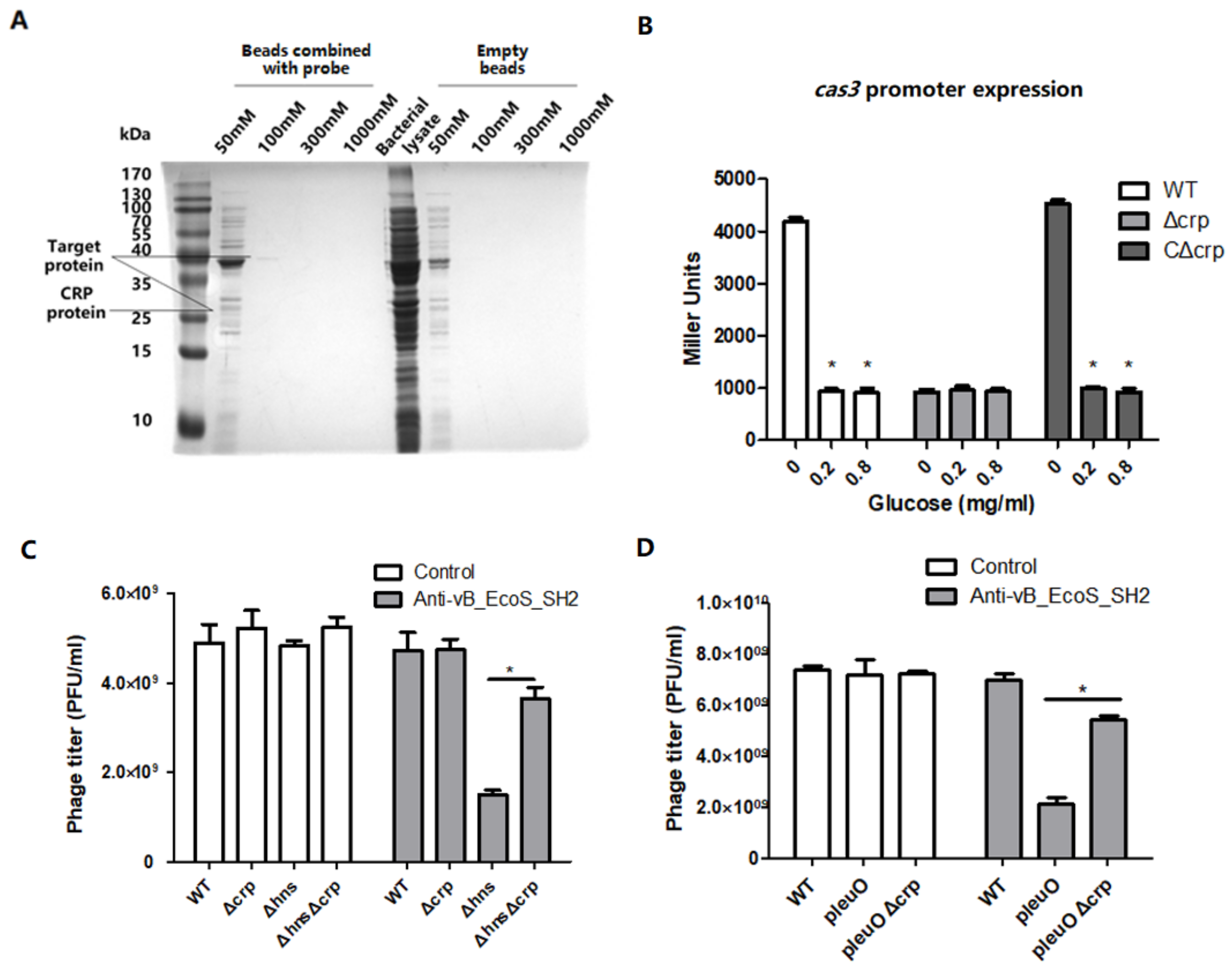
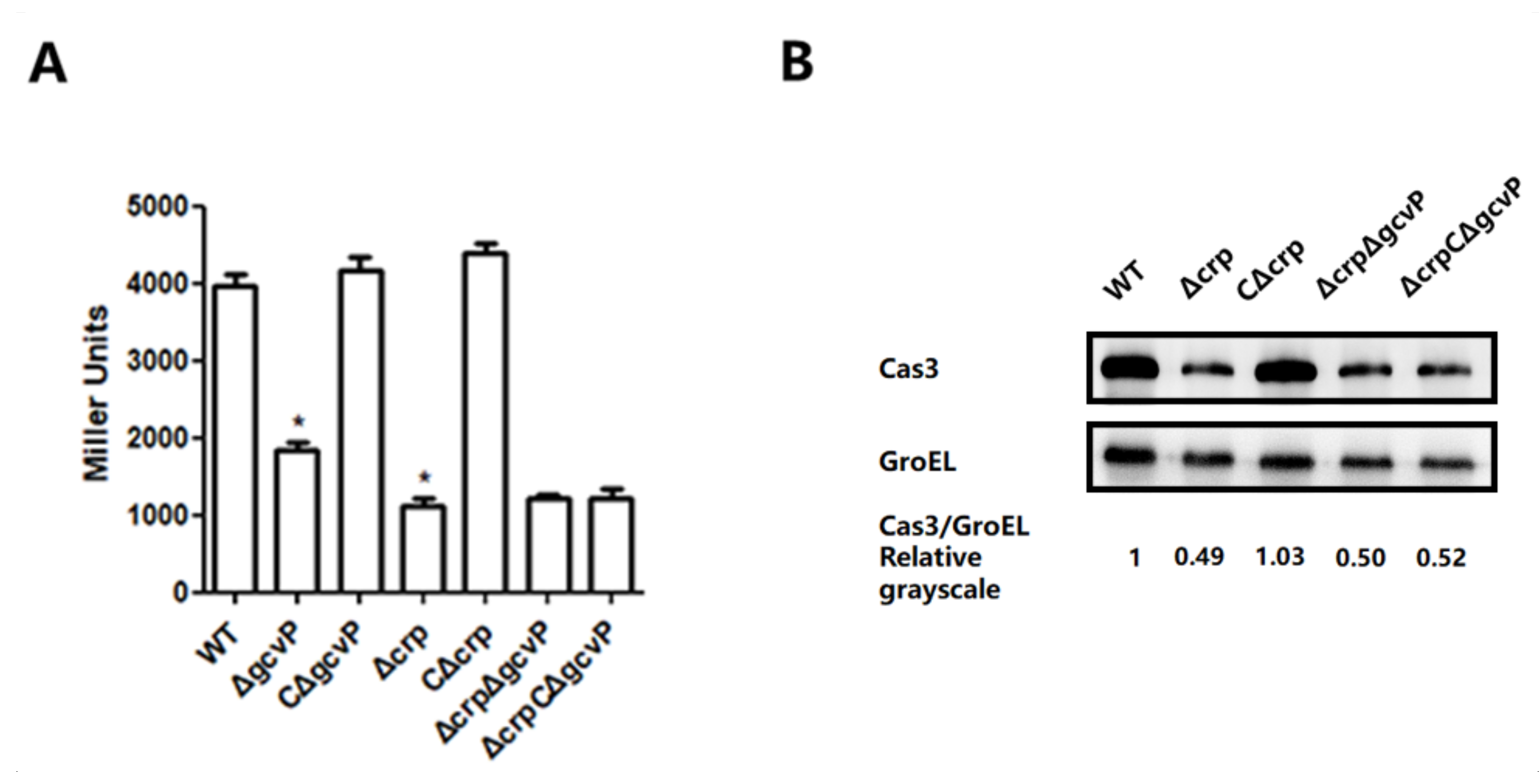
3.4. CRP Positively Regulates the Transcription of cas3 Gene to Affect Phage Infection
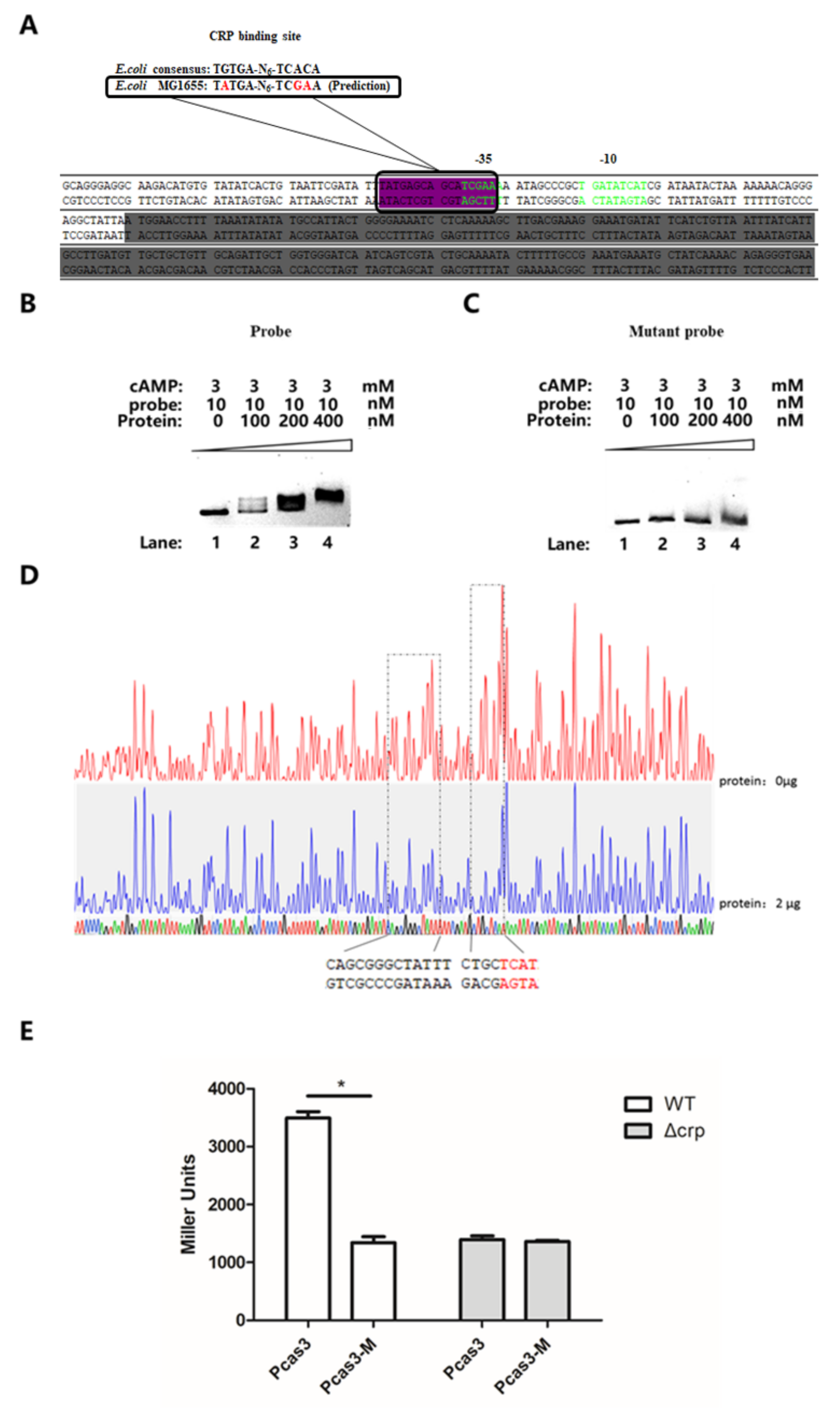
3.5. CRP Activates cas3 Expression by Binding to Transcriptional Initiation Area
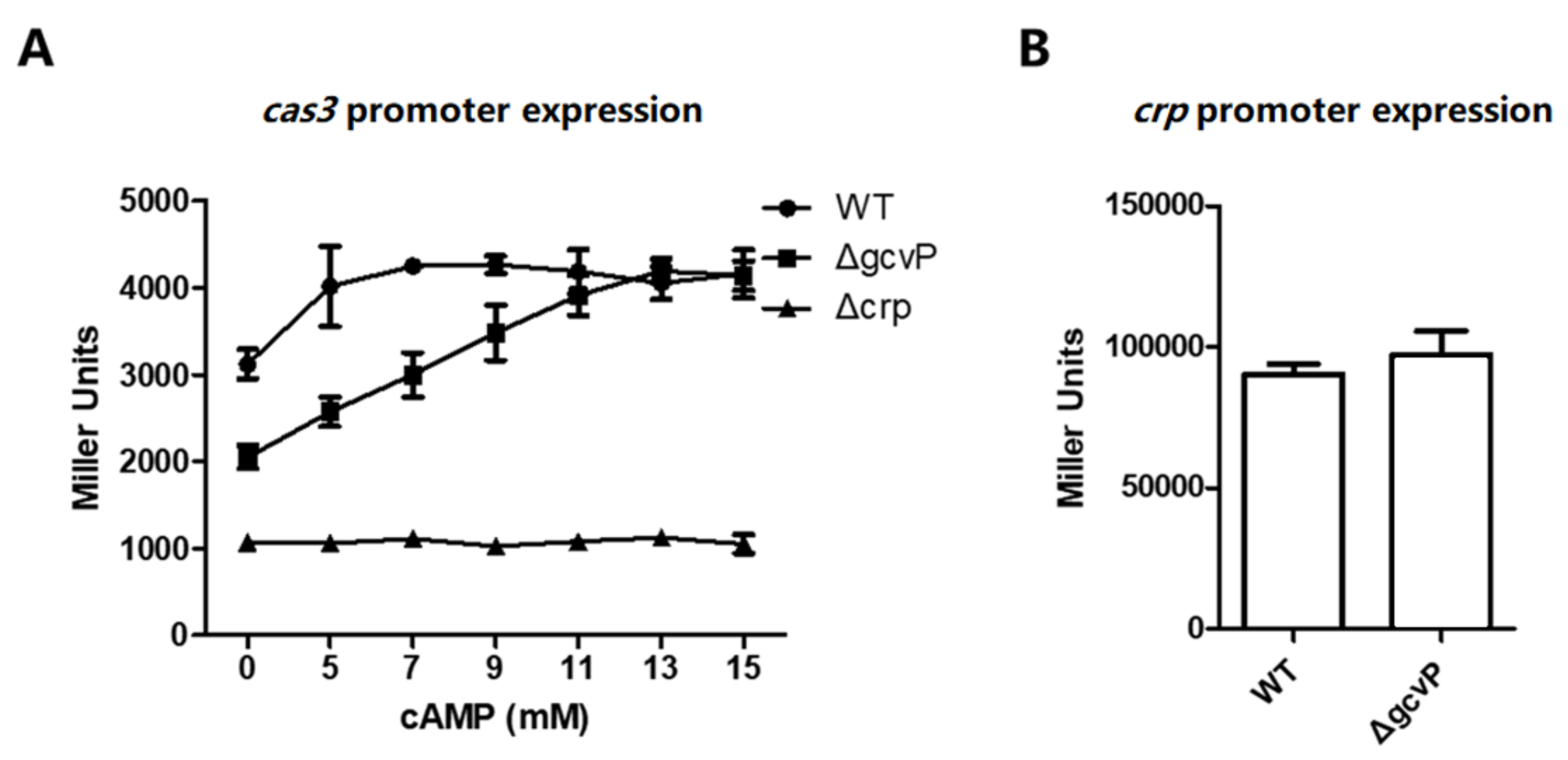
3.6. The Effect of GCS on cas3 Expression is Mediated by CRP
3.7. The Sufficient Concentration of cAMP Restores cas3 Promoter Activity Reduced by the Mutation of GCS
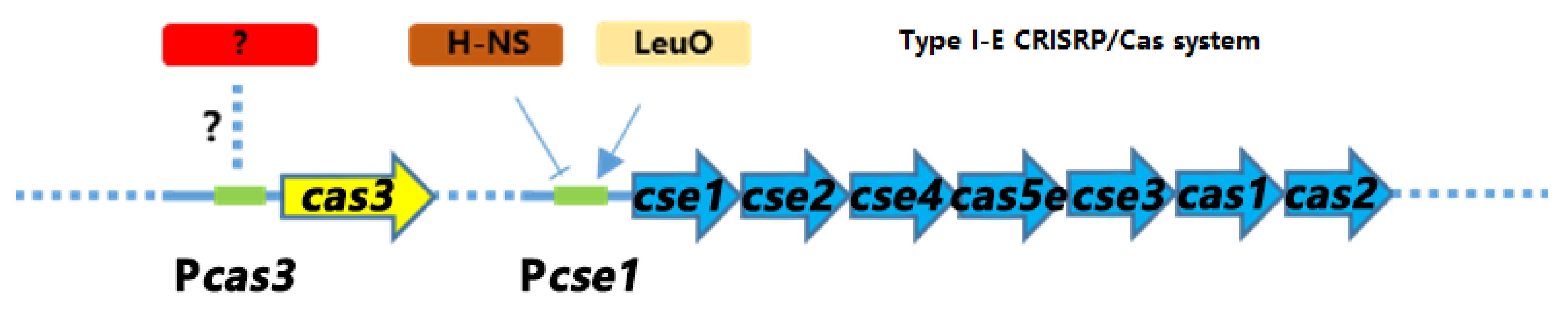
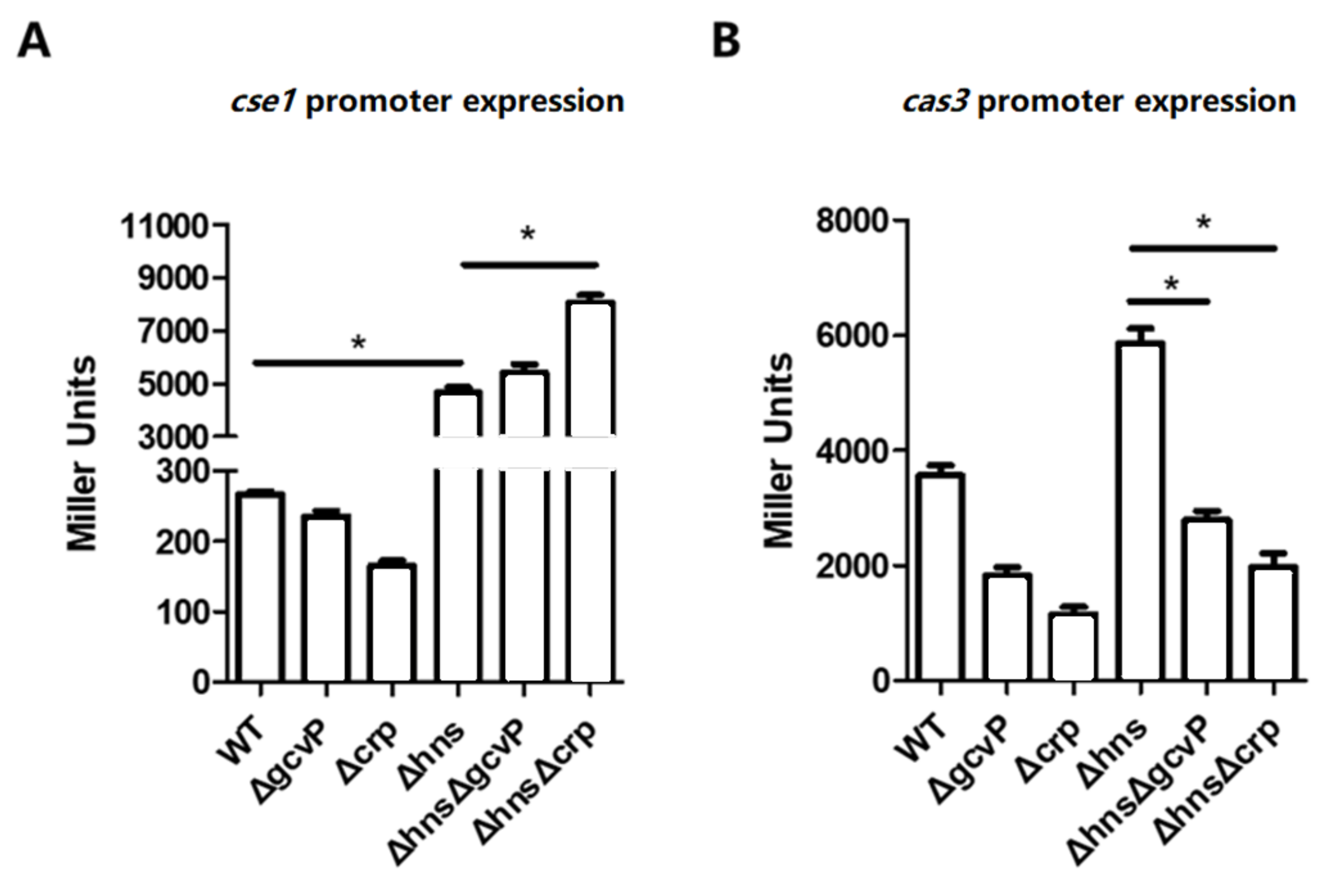
3.8. The Activation of cse1 by H-NS Has No Effect on the Regulation of cas3 by GCS and CRP
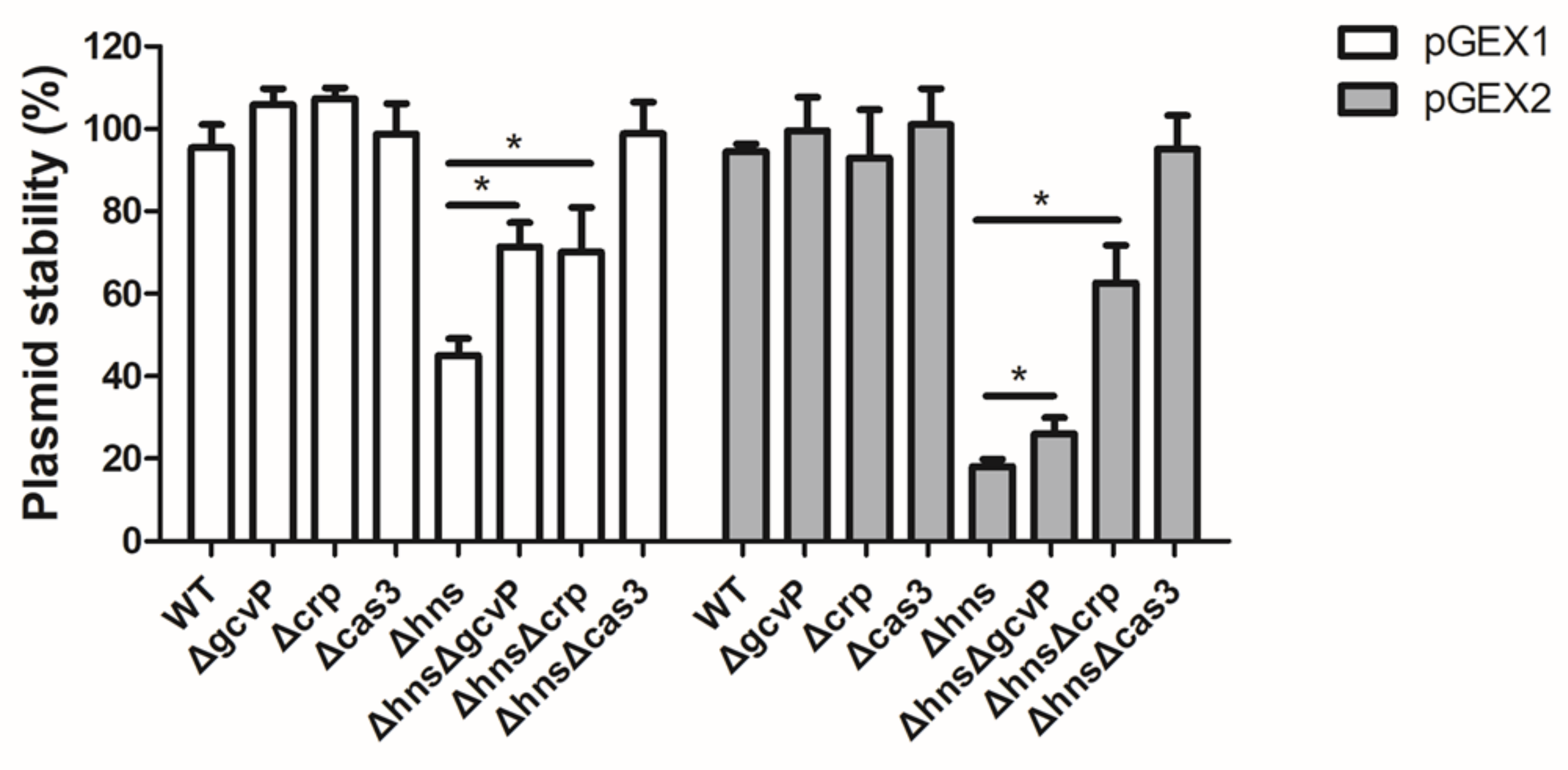
3.9. Mutation of gcvP or crp Increases the Stability of Foreign Plasmids
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Bergh, O.; Børsheim, K.Y.; Bratbak, G.; Heldal, M. High abundance of viruses found in aquatic environments. Nature 1989, 340, 467–468. [Google Scholar] [CrossRef] [PubMed]
- Bickle, T.A.; Krüger, D.H. Biology of DNA restriction. Microbiol. Rev. 1993, 57, 434–450. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Rifat, D.; Wright, N.T.; Varney, K.M.; Weber, D.J.; Black, L.W. Restriction endonuclease inhibitor IPI* of bacteriophage T4: A novel structure for a dedicated target. J. Mol. Biol. 2008, 375, 720–734. [Google Scholar] [CrossRef] [PubMed]
- Labrie, S.J.; Samson, J.E.; Moineau, S. Bacteriophage resistance mechanisms. Nat. Rev. Microbiol. 2010, 8, 317–327. [Google Scholar] [CrossRef]
- Sorek, R.; Lawrence, C.M.; Wiedenheft, B. CRISPR-Mediated Adaptive Immune Systems in Bacteria and Archaea. Annu. Rev. Biochem. 2013, 82, 237–266. [Google Scholar] [CrossRef]
- Liao, H.K.; Gu, Y.; Diaz, A.; Marlett, J.; Takahashi, Y.; Li, M.; Suzuki, K.; Xu, R.; Hishida, T.; Chang, C.J.; et al. Use of the CRISPR/Cas9 system as an intracellular defense against HIV-1 infection in human cells. Nat. Commun. 2015, 6, 6413. [Google Scholar] [CrossRef]
- van der Oost, J.; Jore, M.M.; Westra, E.R.; Lundgren, M.; Brouns, S.J.J. CRISPR-based adaptive and heritable immunity in prokaryotes. Trends Biochem. Sci. 2009, 34, 401–407. [Google Scholar] [CrossRef]
- Yin, S.; Jensen, M.A.; Bai, J.; Debroy, C.; Barrangou, R.; Dudley, E.G. The evolutionary divergence of Shiga toxin-producing Escherichia coli is reflected in clustered regularly interspaced short palindromic repeat (CRISPR) spacer composition. Appl. Environ. Microbiol. 2013, 79, 5710–5720. [Google Scholar] [CrossRef]
- Jansen, R.; Embden, J.D.; Gaastra, W.; Schouls, L.M. Identification of genes that are associated with DNA repeats in prokaryotes. Mol. Microbiol. 2002, 43, 1565–1575. [Google Scholar] [CrossRef]
- Brouns, S.J.J.; Jore, M.M.; Lundgren, M.; Westra, E.R.; Slijkhuis, R.J.H.; Snijders, A.P.L.; Dickman, M.J.; Makarova, K.S.; Koonin, E.V.; van der Oost, J. Small CRISPR RNAs guide antiviral defense in prokaryotes. Science 2008, 321, 960–964. [Google Scholar] [CrossRef]
- Westra, E.R.; van Erp, P.B.G.; Kunne, T.; Wong, S.P.; Staals, R.H.J.; Seegers, C.L.C.; Bollen, S.; Jore, M.M.; Semenova, E.; Severinov, K.; et al. CRISPR Immunity Relies on the Consecutive Binding and Degradation of Negatively Supercoiled Invader DNA by Cascade and Cas3. Mol. Cell 2012, 46, 595–605. [Google Scholar] [CrossRef] [PubMed]
- Westra, E.R.; van Houte, S.; Oyesiku-Blakemore, S.; Makin, B.; Broniewski, J.M.; Best, A.; Bondy-Denomy, J.; Davidson, A.; Boots, M.; Buckling, A. Parasite Exposure Drives Selective Evolution of Constitutive versus Inducible Defense. Curr. Biol. 2015, 25, 1043–1049. [Google Scholar] [CrossRef] [PubMed]
- Tan, D.M.; Lo Svenningsen, S.; Middelboe, M. Quorum Sensing Determines the Choice of Antiphage Defense Strategy in Vibrio anguillarum. Mbio 2015, 6. [Google Scholar] [CrossRef] [PubMed]
- Semenova, E.; Severinov, K. Come Together: CRISPR-Cas Immunity Senses the Quorum. Mol. Cell 2016, 64, 1013–1015. [Google Scholar] [CrossRef] [PubMed]
- Westra, E.R.; Pul, U.; Heidrich, N.; Jore, M.M.; Lundgren, M.; Stratmann, T.; Wurm, R.; Raine, A.; Mescher, M.; van Heereveld, L.; et al. H-NS-mediated repression of CRISPR-based immunity in Escherichia coli K12 can be relieved by the transcription activator LeuO. Mol. Microbiol. 2010, 77, 1380–1393. [Google Scholar] [CrossRef]
- Patterson, A.G.; Chang, J.T.; Taylor, C.; Fineran, P.C. Regulation of the Type I-F CRISPR-Cas system by CRP-cAMP and GalM controls spacer acquisition and interference. Nucleic Acids Res. 2015, 43, 6038–6048. [Google Scholar] [CrossRef]
- Koonin, E.V.; Makarova, K.S.; Zhang, F. Diversity, classification and evolution of CRISPR-Cas systems. Curr. Opin. Microbiol. 2017, 37, 67–78. [Google Scholar] [CrossRef]
- Salcedo, E.; Sims, P.F.G.; Hyde, J.E. A glycine-cleavage complex as part of the folate one-carbon metabolism of Plasmodium falciparum. Trends Parasitol. 2005, 21, 406–411. [Google Scholar] [CrossRef][Green Version]
- Kolb, A.; Busby, S.; Buc, H.; Garges, S.; Adhya, S. Transcriptional regulation by cAMP and its receptor protein. Annu. Rev. Biochem. 1993, 62, 749–795. [Google Scholar] [CrossRef]
- Zhang, X.; Bremer, H. Control of the Escherichia coli rrnB P1 promoter strength by ppGpp. J. Biol. Chem. 1995, 270, 11181–11189. [Google Scholar] [CrossRef]
- Jutras, B.L.; Verma, A.; Stevenson, B. Identification of novel DNA-binding proteins using DNA-affinity chromatography/pull down. Curr. Protoc. Microbiol. 2012. [Google Scholar] [CrossRef]
- Wang, Z.F.; Kong, L.C.; Liu, Y.; Fu, Q.; Cui, Z.L.; Wang, J.; Ma, J.J.; Wang, H.A.; Yan, Y.X.; Sun, J.H. A Phage Lysin Fused to a Cell-Penetrating Peptide Kills Intracellular Methicillin-Resistant Staphylococcus aureus in Keratinocytes and Has Potential as a Treatment for Skin Infections in Mice. Appl. Env. Microb. 2018, 84. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Cen, X.F.; Zhao, G.P.; Wang, J. Characterization of a new GlnR binding box in the promoter of amtB in Streptomyces coelicolor inferred a PhoP/GlnR competitive binding mechanism for transcriptional regulation of amtB. J. Bacteriol. 2012, 194, 5237–5244. [Google Scholar] [CrossRef] [PubMed]
- Yang, C.D.; Chen, Y.H.; Huang, H.Y.; Huang, H.D.; Tseng, C.P. CRP represses the CRISPR/Cas system in Escherichia coli: Evidence that endogenous CRISPR spacers impede phage P1 replication. Mol. Microbiol. 2014, 92, 1072–1091. [Google Scholar] [CrossRef]
- Fu, Q.; Li, S.Y.; Wang, Z.F.; Shan, W.Y.; Ma, J.J.; Cheng, Y.Q.; Wang, H.G.; Yan, Y.X.; Sun, J.H. H-NS Mutation-Mediated CRISPR-Cas Activation inhibits Phage Release and Toxin Production of Escherichia coli Stx2 Phage Lysogen. Front. Microbiol. 2017, 8, 652. [Google Scholar] [CrossRef] [PubMed]
- Kikuchi, G.; Motokawa, Y.; Yoshida, T.; Hiraga, K. Glycine cleavage system: Reaction mechanism, physiological significance, and hyperglycinemia. P Jpn. Acad. B Phys. 2008, 84, 246–263. [Google Scholar] [CrossRef]
- Semenova, E.; Jore, M.M.; Datsenko, K.A.; Semenova, A.; Westra, E.R.; Wanner, B.; van der Oost, J.; Brouns, S.J.J.; Severinov, K. Interference by clustered regularly interspaced short palindromic repeat (CRISPR) RNA is governed by a seed sequence. Proc. Natl. Acad. Sci. USA 2011, 108, 10098–10103. [Google Scholar] [CrossRef]
- Meedel, T.H.; Pizer, L.I. Regulation of one-carbon biosynthesis and utilization in Escherichia coli. J. Bacteriol. 1974, 118, 905. [Google Scholar] [CrossRef]
- Yoshida, T.; Kikuchi, G. Comparative Study on Major Pathways of Glycine and Serine Catabolism in Vertebrate Livers. J. Biochem. 1972, 72, 1503–1516. [Google Scholar] [CrossRef]
- Kikuchi, G. The glycine cleavage system: Composition, reaction mechanism, and physiological significance. Mol. Cell. Biochem. 1973, 1, 169–187. [Google Scholar] [CrossRef]
- Lawson, C.L.; Swigon, D.; Murakami, K.S.; Darst, S.A.; Berman, H.M.; Ebright, R.H. Catabolite activator protein: DNA binding and transcription activation. Curr. Opin. Struc. Biol. 2004, 14, 10–20. [Google Scholar] [CrossRef] [PubMed]
- Busby, S.; Ebright, R.H. Transcription activation by catabolite activator protein (CAP). J. Mol. Biol. 1999, 293, 199–213. [Google Scholar] [CrossRef] [PubMed]
- Pul, U.; Wurm, R.; Arslan, Z.; Geissen, R.; Hofmann, N.; Wagner, R. Identification and characterization of E-coli CRISPR-cas promoters and their silencing by H-NS. Mol. Microbiol. 2010, 75, 1495–1512. [Google Scholar] [CrossRef] [PubMed]
- Brown, M.J.; Russo, B.C.; O’Dee, D.M.; Schmitt, D.M.; Nau, G.J. The contribution of the glycine cleavage system to the pathogenesis of Francisella tularensis. Microbes Infect. 2014, 16, 300–309. [Google Scholar] [CrossRef]
- Lorio, J.C.; Kim, W.S.; Krishnan, A.H.; Krishnan, H.B. Disruption of the Glycine Cleavage System Enables Sinorhizobium fredii USDA257 To Form Nitrogen-Fixing Nodules on Agronomically Improved North American Soybean Cultivars. Appl. Env. Microb. 2010, 76, 4185–4193. [Google Scholar] [CrossRef]
- Zheng, D.L.; Constantinidou, C.; Hobman, J.L.; Minchin, S.D. Identification of the CRP regulon using in vitro and in vivo transcriptional profiling. Nucleic Acids Res. 2004, 32, 5874–5893. [Google Scholar] [CrossRef]
- Gosset, G.; Zhang, Z.; Nayyar, S.; Cuevas, W.A.; Saier, M.H. Transcriptome Analysis of Crp-Dependent Catabolite Control of Gene Expression in Escherichia coli. J. Bacteriol. 2004, 186, 3516–3524. [Google Scholar] [CrossRef]
- Grainger, D.C.; Hurd, D.; Harrison, M.; Holdstock, J.; Busby, S.J.W. Studies of the distribution of Escherichia coli cAMP-receptor protein and RNA polymerase along the E-coli chromosome. Proc. Natl. Acad. Sci. USA 2005, 102, 17693–17698. [Google Scholar] [CrossRef]
- Tan, K.; Moreno-Hagelsieb, G.; Collado-Vides, J.; Stormo, G.D. A comparative genomics approach to prediction of new members of regulons. Genome Res. 2001, 11, 566–584. [Google Scholar] [CrossRef]
- Shinkai, A.; Kira, S.; Nakagawa, N.; Kashihara, A.; Kuramitsu, S.; Yokoyama, S. Transcription activation mediated by a cyclic AMP receptor protein from Thermus thermophilus HB8. J. Bacteriol. 2007, 189, 3891–3901. [Google Scholar] [CrossRef]
- Hoyland-Kroghsbo, N.M.; Paczkowski, J.; Mukherjee, S.; Broniewski, J.; Westra, E.; Bondy-Denomy, J.; Bassler, B.L. Quorum sensing controls the Pseudomonas aeruginosa CRISPR-Cas adaptive immune system. Proc. Natl. Acad. Sci. USA 2017, 114, 131–135. [Google Scholar] [CrossRef] [PubMed]
- Buettner, M.J.; Spitz, E.; Rickenberg, H.V. Cyclic adenosine 3’,5’-monophosphate in Escherichia coli. J. Bacteriol. 1973, 114, 1068–1073. [Google Scholar] [CrossRef] [PubMed]
- Maiuolo, J.; Oppedisano, F.; Gratteri, S.; Muscoli, C.; Mollace, V. Regulation of uric acid metabolism and excretion. Int. J. Cardiol. 2016, 213, 8–14. [Google Scholar] [CrossRef] [PubMed]
- Pedley, A.M.; Benkovic, S.J. A New View into the Regulation of Purine Metabolism: The Purinosome. Trends Biochem. Sci. 2017, 42, 141–154. [Google Scholar] [CrossRef]
- Steegborn, C. Structure, mechanism, and regulation of soluble adenylyl cyclases-similarities and differences to transmembrane adenylyl cyclases. Biochim. Biophys Acta. 2014, 1842, 2535–2547. [Google Scholar] [CrossRef]
- Kao, K.C.; Tran, L.M.; Liao, J.C. A global regulatory role of gluconeogenic genes in Escherichia coli revealed by transcriptome network analysis. J. Biol. Chem. 2005, 280, 36079–36087. [Google Scholar] [CrossRef]
- Vazquez, A.; Markert, E.K.; Oltvai, Z.N. Serine biosynthesis with one carbon catabolism and the glycine cleavage system represents a novel pathway for ATP generation. PLoS ONE 2011, 6, e25881. [Google Scholar] [CrossRef]
- Pougach, K.; Semenova, E.; Bogdanova, E.; Datsenko, K.A.; Djordjevic, M.; Wanner, B.L.; Severinov, K. Transcription, processing and function of CRISPR cassettes in Escherichia coli. Mol. Microbiol. 2010, 77, 1367–1379. [Google Scholar] [CrossRef]
- Tedeschi, P.M.; Markert, E.K.; Gounder, M.; Lin, H.; Dvorzhinski, D.; Dolfi, S.C.; Chan, L.L.; Qiu, J.; DiPaola, R.S.; Hirshfield, K.M.; et al. Contribution of serine, folate and glycine metabolism to the ATP, NADPH and purine requirements of cancer cells. Cell Death Dis. 2013, 4, e877. [Google Scholar] [CrossRef]











© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yang, D.; Wang, Z.; Ma, J.; Fu, Q.; Wu, L.; Wang, H.; Wang, S.; Yan, Y.; Sun, J. Glycine Cleavage System and cAMP Receptor Protein Co-Regulate CRISPR/cas3 Expression to Resist Bacteriophage. Viruses 2020, 12, 90. https://doi.org/10.3390/v12010090
Yang D, Wang Z, Ma J, Fu Q, Wu L, Wang H, Wang S, Yan Y, Sun J. Glycine Cleavage System and cAMP Receptor Protein Co-Regulate CRISPR/cas3 Expression to Resist Bacteriophage. Viruses. 2020; 12(1):90. https://doi.org/10.3390/v12010090
Chicago/Turabian StyleYang, Denghui, Zhaofei Wang, Jingjiao Ma, Qiang Fu, Lifei Wu, Hengan Wang, Shaohui Wang, Yaxian Yan, and Jianhe Sun. 2020. "Glycine Cleavage System and cAMP Receptor Protein Co-Regulate CRISPR/cas3 Expression to Resist Bacteriophage" Viruses 12, no. 1: 90. https://doi.org/10.3390/v12010090
APA StyleYang, D., Wang, Z., Ma, J., Fu, Q., Wu, L., Wang, H., Wang, S., Yan, Y., & Sun, J. (2020). Glycine Cleavage System and cAMP Receptor Protein Co-Regulate CRISPR/cas3 Expression to Resist Bacteriophage. Viruses, 12(1), 90. https://doi.org/10.3390/v12010090