Interferon-Independent Upregulation of Interferon-Stimulated Genes during Human Cytomegalovirus Infection is Dependent on IRF3 Expression



Abstract
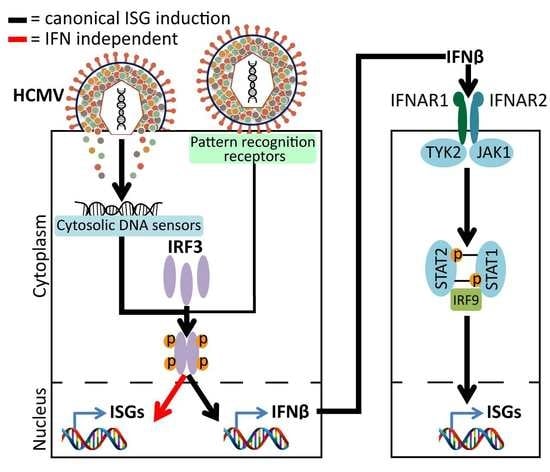
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
2.1. Cell Culture, Viral Infection and Treatment of Cells with Conditioned Supernatants
2.2. Quantitative Reverse Transcription Polymerase Chain Reaction (qRT-PCR)
2.3. Western Blot
2.4. CRISPR/Cas-9
3. Results
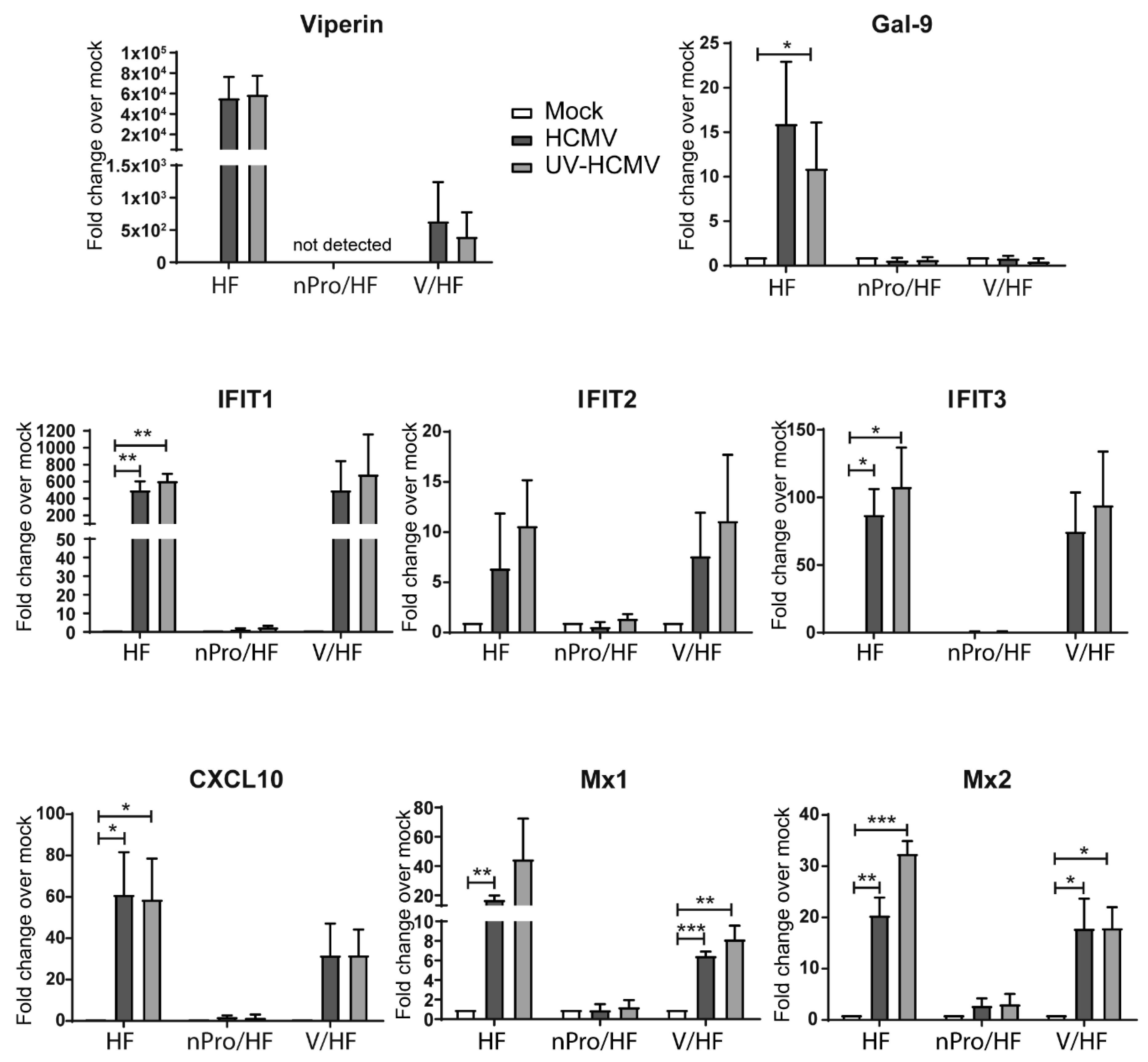
3.1. Transcript Upregulation of Interferon (IFN)-Independent Interferon-Stimulated Genes (ISGs) Following Infection with Human Cytomegalovirus (HCMV) and Ultraviolet-Irradiated HCMV (UV-HCMV) in IRF3-Deficient Human Foreskin Fibroblasts (HFs)
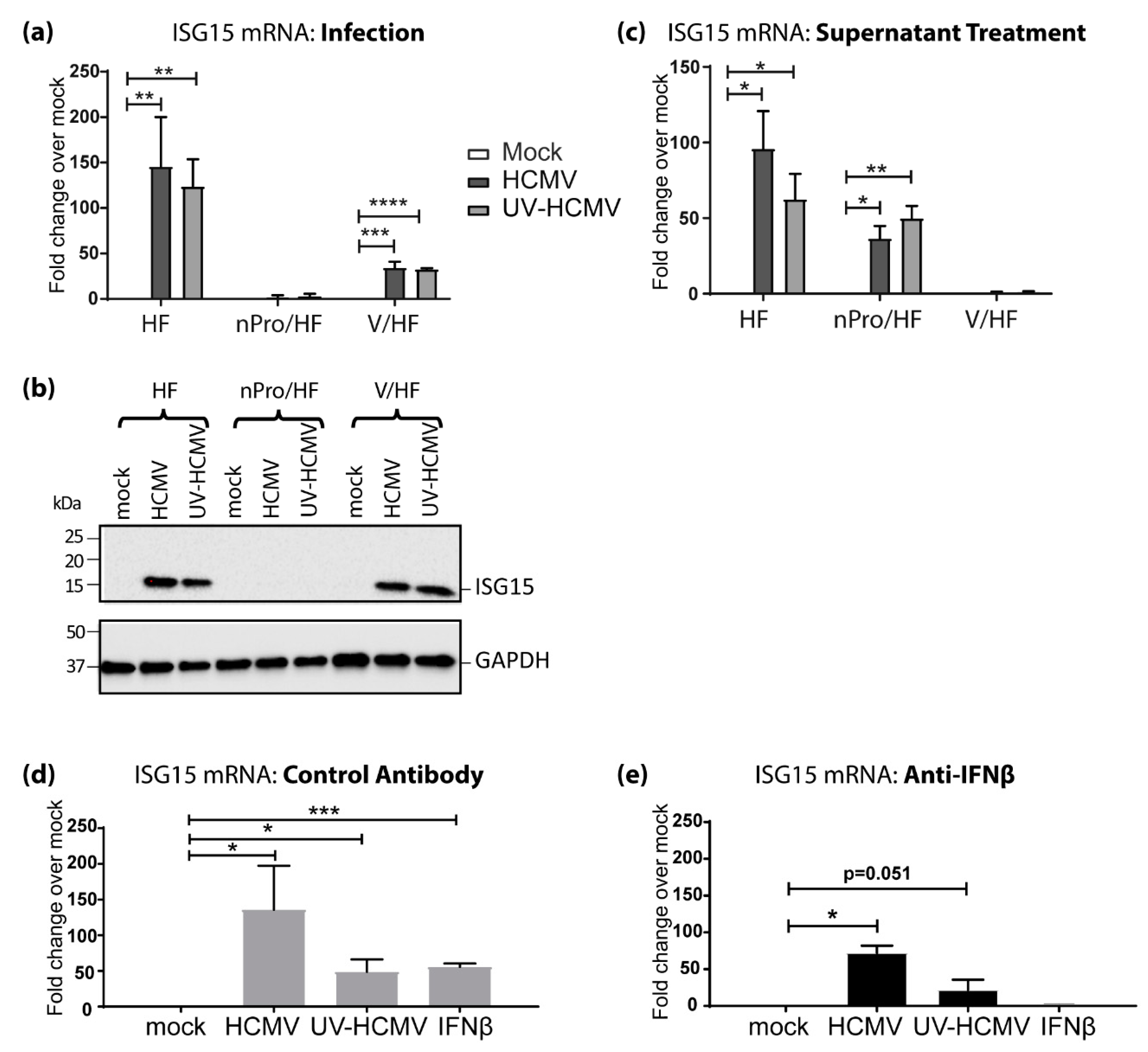
3.2. IFN-Independent, IRF3-Dependent Induction of ISG15 Expression Contributes Significantly to Its Upregulation by HCMV Infection
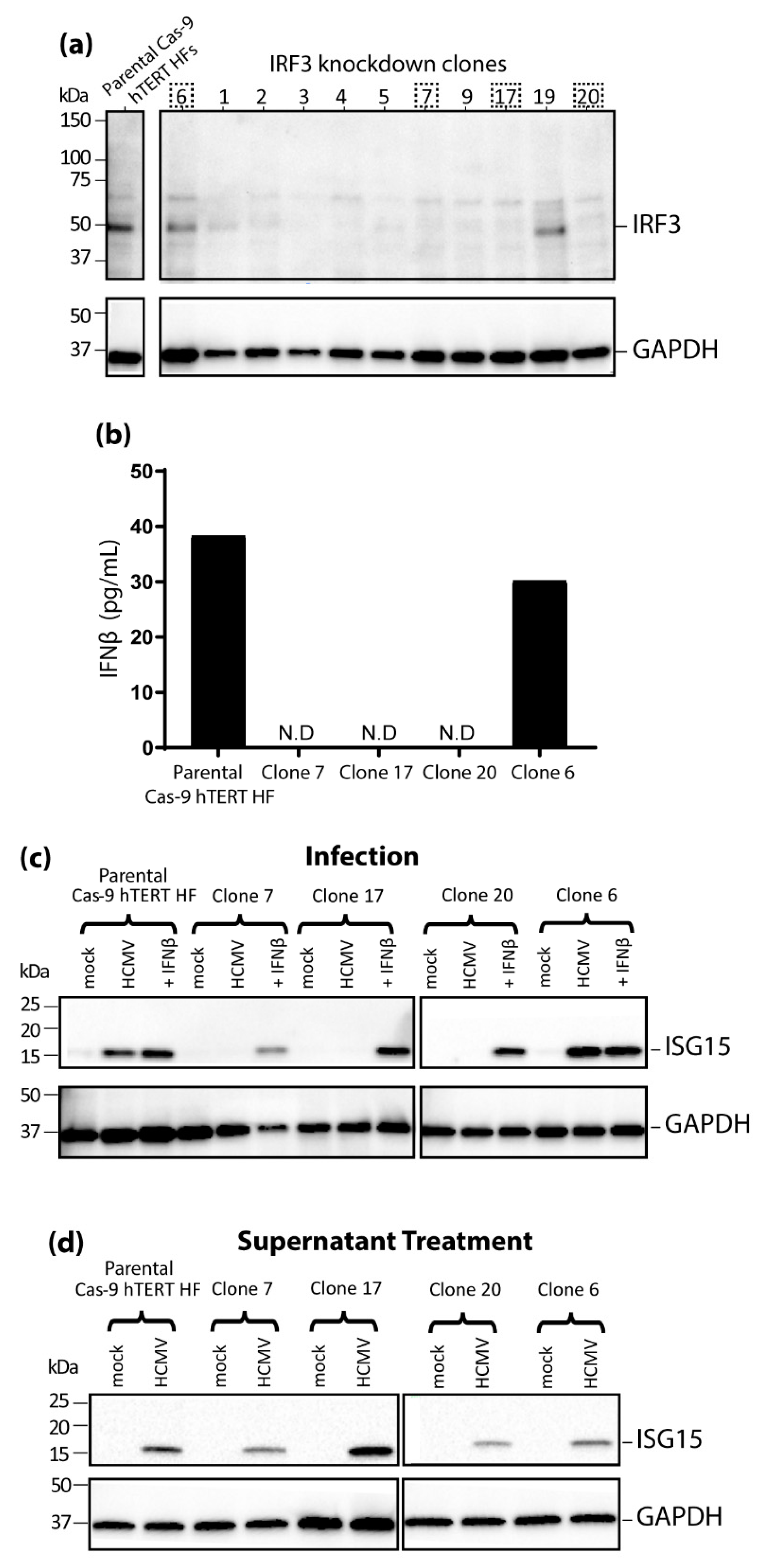
3.3. IRF3 Knockout (KO) by CRISPR/Cas-9 Inhibits ISG15 Protein Expression during HCMV Infection Mirroring the Phenotype Seen in nPro/HFs
4. Discussion
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Seale, H.; MacIntyre, C.R.; Gidding, H.F.; Backhouse, J.L.; Dwyer, D.E.; Gilbert, L. National Serosurvey of Cytomegalovirus in Australia. Clin. Vaccine Immunol. 2006, 13, 1181–1184. [Google Scholar] [CrossRef] [PubMed]
- Lachmann, R.; Loenenbach, A.; Waterboer, T.; Brenner, N.; Pawlita, M.; Michel, A.; Thamm, M.; Poethko-Müller, C.; Wichmann, O.; Wiese-Posselt, M. Cytomegalovirus (CMV) seroprevalence in the adult population of Germany. PLoS ONE 2018, 13, e0200267. [Google Scholar] [CrossRef] [PubMed]
- Staras, S.A.S.; Dollard, S.C.; Radford, K.W.; Flanders, W.D.; Pass, R.F.; Cannon, M.J. Seroprevalence of Cytomegalovirus Infection in the United States, 1988–1994. Clin. Infect. Dis. 2006, 43, 1143–1151. [Google Scholar] [CrossRef] [PubMed]
- Mujtaba, G.; Shaukat, S.; Angez, M.; Alam, M.M.; Hasan, F.; Zahoor Zaidi, S.S.; Shah, A.A. Seroprevalence of Human Cytomegalovirus (HCMV) infection in pregnant women and outcomes of pregnancies with active infection. JPMA J. Pak. Med. Assoc. 2016, 66, 1009–1014. [Google Scholar]
- Correa, C.B.; Kourí, V.; Verdasquera, D.; Martínez, P.A.; Alvarez, A.; Alemán, Y.; Pérez, L.; Viera, J.; González, R.; Pérez, E.; et al. HCMV seroprevalence and associated risk factors in pregnant women, Havana City, 2007 to 2008. Prenat. Diagn. 2010, 30, 888–892. [Google Scholar] [CrossRef] [PubMed]
- Cannon, M.J.; Schmid, D.S.; Hyde, T.B. Review of cytomegalovirus seroprevalence and demographic characteristics associated with infection. Rev. Med. Virol. 2010, 20, 202–213. [Google Scholar] [CrossRef] [PubMed]
- Griffiths, P.; Baraniak, I.; Reeves, M. The pathogenesis of human cytomegalovirus. J. Pathol. 2015, 235, 288–297. [Google Scholar] [CrossRef]
- McMullan, B.J.; Palasanthiran, P.; Jones, C.L.A.; Hall, B.P.M.; Robertson, P.W.; Howard, J.; Rawlinson, W.D. Congenital cytomegalovirus–time to diagnosis, management and clinical sequelae in Australia: Opportunities for earlier identification. Med. J. Aust. 2011, 194, 625–629. [Google Scholar]
- Cannon, M.J.; Griffiths, P.D.; Aston, V.; Rawlinson, W.D. Universal newborn screening for congenital CMV infection: What is the evidence of potential benefit? Rev. Med. Virol. 2014, 24, 291–307. [Google Scholar] [CrossRef]
- Cannon, M.J. Congenital cytomegalovirus (CMV) epidemiology and awareness. J. Clin. Virol. Off. Publ. Pan Am. Soc. Clin. Virol. 2009, 46 (Suppl. 4), S6–S10. [Google Scholar] [CrossRef]
- Cheeran, M.C.J.; Lokensgard, J.R.; Schleiss, M.R. Neuropathogenesis of congenital cytomegalovirus infection: Disease mechanisms and prospects for intervention. Clin. Microbiol. Rev. 2009, 22, 99–126. [Google Scholar] [CrossRef] [PubMed]
- Cannon, M.J.; Westbrook, K.; Levis, D.; Schleiss, M.R.; Thackeray, R.; Pass, R.F. Awareness of and behaviors related to child-to-mother transmission of cytomegalovirus. Prev. Med. 2012, 54, 351–357. [Google Scholar] [CrossRef] [PubMed]
- Nakamura, K.; Eizuru, Y.; Minamishima, Y. Effect of natural human interferon-beta on the replication of human cytomegalovirus. J. Med. Virol. 1988, 26, 363–373. [Google Scholar] [CrossRef]
- Delannoy, A.S.; Hober, D.; Bouzidi, A.; Wattre, P. Role of interferon alpha (IFN-alpha) and interferon gamma (IFN-gamma) in the control of the infection of monocyte-like cells with human cytomegalovirus (HCMV). Microbiol. Immunol. 1999, 43, 1087–1096. [Google Scholar] [CrossRef] [PubMed]
- Sainz, B.; LaMarca, H.; Garry, R.; Morris, C. Synergistic inhibition of human cytomegalovirus replication by interferon-alpha/beta and interferon-gamma. Virol. J. 2005, 2, 14. [Google Scholar] [CrossRef] [PubMed]
- McSharry, B.P.; Forbes, S.K.; Avdic, S.; Randall, R.E.; Wilkinson, G.W.; Abendroth, A.; Slobedman, B. Abrogation of the interferon response promotes more efficient human cytomegalovirus replication. J. Virol. 2015, 89, 1479–1483. [Google Scholar] [CrossRef] [PubMed]
- Compton, T.; Kurt-Jones, E.A.; Boehme, K.W.; Belko, J.; Latz, E.; Golenbock, D.T.; Finberg, R.W. Human cytomegalovirus activates inflammatory cytokine responses via CD14 and Toll-like receptor 2. J. Virol. 2003, 77, 4588–4596. [Google Scholar] [CrossRef] [PubMed]
- Boehme, K.W.; Guerrero, M.; Compton, T. Human cytomegalovirus envelope glycoproteins B and H are necessary for TLR2 activation in permissive cells. J. Immunol. (Baltimore, Md.: 1950) 2006, 177, 7094–7102. [Google Scholar] [CrossRef]
- Au, W.C.; Moore, P.A.; Lowther, W.; Juang, Y.T.; Pitha, P.M. Identification of a member of the interferon regulatory factor family that binds to the interferon-stimulated response element and activates expression of interferon-induced genes. Proc. Natl. Acad. Sci. USA 1995, 92, 11657–11661. [Google Scholar] [CrossRef]
- DeFilippis, V.R.; Robinson, B.; Keck, T.M.; Hansen, S.G.; Nelson, J.A.; Früh, K.J. Interferon Regulatory Factor 3 Is Necessary for Induction of Antiviral Genes during Human Cytomegalovirus Infection. J. Virol. 2006, 80, 1032–1037. [Google Scholar] [CrossRef]
- Marie, I.; Durbin, J.E.; Levy, D.E. Differential viral induction of distinct interferon-alpha genes by positive feedback through interferon regulatory factor-7. Embo J. 1998, 17, 6660–6669. [Google Scholar] [CrossRef]
- Yurochko, A.D.; Hwang, E.S.; Rasmussen, L.; Keay, S.; Pereira, L.; Huang, E.S. The human cytomegalovirus UL55 (gB) and UL75 (gH) glycoprotein ligands initiate the rapid activation of Sp1 and NF-kappaB during infection. J. Virol. 1997, 71, 5051–5059. [Google Scholar]
- Sun, L.; Wu, J.; Du, F.; Chen, X.; Chen, Z.J. Cyclic GMP-AMP synthase is a cytosolic DNA sensor that activates the type I interferon pathway. Science (New York, N.Y.) 2013, 339, 786–791. [Google Scholar] [CrossRef]
- Gariano, G.R.; Dell’Oste, V.; Bronzini, M.; Gatti, D.; Luganini, A.; De Andrea, M.; Gribaudo, G.; Gariglio, M.; Landolfo, S. The intracellular DNA sensor IFI16 gene acts as restriction factor for human cytomegalovirus replication. PLoS Pathog. 2012, 8, e1002498. [Google Scholar] [CrossRef]
- DeFilippis, V.R.; Alvarado, D.; Sali, T.; Rothenburg, S.; Fruh, K. Human cytomegalovirus induces the interferon response via the DNA sensor ZBP1. J. Virol. 2010, 84, 585–598. [Google Scholar] [CrossRef]
- Liu, S.; Cai, X.; Wu, J.; Cong, Q.; Chen, X.; Li, T.; Du, F.; Ren, J.; Wu, Y.T.; Grishin, N.V.; et al. Phosphorylation of innate immune adaptor proteins MAVS, STING, and TRIF induces IRF3 activation. Science (New York, N.Y.) 2015, 347, aaa2630. [Google Scholar] [CrossRef]
- Bianco, C.; Mohr, I. Restriction of Human Cytomegalovirus Replication by ISG15, a Host Effector Regulated by cGAS-STING Double-Stranded-DNA Sensing. J. Virol. 2017, 91, e02483-16. [Google Scholar] [CrossRef]
- Fu, X.Y. A transcription factor with SH2 and SH3 domains is directly activated by an interferon alpha-induced cytoplasmic protein tyrosine kinase(s). Cell 1992, 70, 323–335. [Google Scholar] [CrossRef]
- Schoggins, J.W.; Wilson, S.J.; Panis, M.; Murphy, M.Y.; Jones, C.T.; Bieniasz, P.; Rice, C.M. A diverse range of gene products are effectors of the type I interferon antiviral response. Nature 2011, 472, 481–485. [Google Scholar] [CrossRef]
- Isaacson, M.K.; Juckem, L.K.; Compton, T. Virus Entry and Innate Immune Activation. In Human Cytomegalovirus; Shenk, T.E., Stinski, M.F., Eds.; Springer: Berlin/Heidelberg, Germany, 2008; pp. 85–100. [Google Scholar] [CrossRef]
- Nicholl, M.J.; Robinson, L.H.; Preston, C.M. Activation of cellular interferon-responsive genes after infection of human cells with herpes simplex virus type 1. J. Gen. Virol. 2000, 81, 2215–2218. [Google Scholar] [CrossRef]
- Zhu, H.; Cong, J.-P.; Shenk, T. Use of differential display analysis to assess the effect of human cytomegalovirus infection on the accumulation of cellular RNAs: Induction of interferon-responsive RNAs. Proc. Natl. Acad. Sci. USA 1997, 94, 13985–13990. [Google Scholar] [CrossRef] [PubMed]
- Preston, C.M.; Harman, A.N.; Nicholl, M.J. Activation of interferon response factor-3 in human cells infected with herpes simplex virus type 1 or human cytomegalovirus. J. Virol. 2001, 75, 8909–8916. [Google Scholar] [CrossRef] [PubMed]
- Browne, E.P.; Wing, B.; Coleman, D.; Shenk, T. Altered Cellular mRNA Levels in Human Cytomegalovirus-Infected Fibroblasts: Viral Block to the Accumulation of Antiviral mRNAs. J. Virol. 2001, 75, 12319–12330. [Google Scholar] [CrossRef] [PubMed]
- Yoneyama, M.; Suhara, W.; Fukuhara, Y.; Fukuda, M.; Nishida, E.; Fujita, T. Direct triggering of the type I interferon system by virus infection: Activation of a transcription factor complex containing IRF-3 and CBP/p300. EMBO J. 1998, 17, 1087–1095. [Google Scholar] [CrossRef] [PubMed]
- WATHELET, M.G.; BERR, P.M.; HUEZ, G.A. Regulation of gene expression by cytokines and virus in human cells lacking the type-I interferon locus. Eur. J. Biochem. 1992, 206, 901–910. [Google Scholar] [CrossRef] [PubMed]
- Guo, J.; Peters, K.L.; Sen, G.C. Induction of the human protein P56 by interferon, double-stranded RNA, or virus infection. Virology 2000, 267, 209–219. [Google Scholar] [CrossRef] [PubMed]
- McSharry, B.P.; Burgert, H.-G.; Owen, D.P.; Stanton, R.J.; Prod’homme, V.; Sester, M.; Koebernick, K.; Groh, V.; Spies, T.; Cox, S.; et al. Adenovirus E3/19K Promotes Evasion of NK Cell Recognition by Intracellular Sequestration of the NKG2D Ligands Major Histocompatibility Complex Class I Chain-Related Proteins A and B. J. Virol. 2008, 82, 4585–4594. [Google Scholar] [CrossRef] [PubMed]
- McSharry, B.P.; Forbes, S.K.; Cao, J.Z.; Avdic, S.; Machala, E.A.; Gottlieb, D.J.; Abendroth, A.; Slobedman, B. Human cytomegalovirus upregulates expression of the lectin galectin 9 via induction of beta interferon. J. Virol. 2014, 88, 10990–10994. [Google Scholar] [CrossRef] [PubMed]
- Stanton, R.J.; Baluchova, K.; Dargan, D.J.; Cunningham, C.; Sheehy, O.; Seirafian, S.; McSharry, B.P.; Neale, M.L.; Davies, J.A.; Tomasec, P.; et al. Reconstruction of the complete human cytomegalovirus genome in a BAC reveals RL13 to be a potent inhibitor of replication. J. Clin. Investig. 2010, 120, 3191–3208. [Google Scholar] [CrossRef]
- Gibson, W. Structure and formation of the cytomegalovirus virion. Curr. Topics Microbiol. Immunol. 2008, 325, 187–204. [Google Scholar]
- Sanjana, N.E.; Shalem, O.; Zhang, F. Improved vectors and genome-wide libraries for CRISPR screening. Nat. Methods 2014, 11, 783. [Google Scholar] [CrossRef] [PubMed]
- Sali, T.M.; Pryke, K.M.; Abraham, J.; Liu, A.; Archer, I.; Broeckel, R.; Staverosky, J.A.; Smith, J.L.; Al-Shammari, A.; Amsler, L.; et al. Characterization of a Novel Human-Specific STING Agonist that Elicits Antiviral Activity Against Emerging Alphaviruses. PLoS Pathog. 2015, 11, e1005324. [Google Scholar] [CrossRef] [PubMed]
- Hilton, L.; Moganeradj, K.; Zhang, G.; Chen, Y.-H.; Randall, R.E.; McCauley, J.W.; Goodbourn, S. The NPro Product of Bovine Viral Diarrhea Virus Inhibits DNA Binding by Interferon Regulatory Factor 3 and Targets It for Proteasomal Degradation. J. Virol. 2006, 80, 11723–11732. [Google Scholar] [CrossRef] [PubMed]
- Andrejeva, J.; Young, D.F.; Goodbourn, S.; Randall, R.E. Degradation of STAT1 and STAT2 by the V Proteins of Simian Virus 5 and Human Parainfluenza Virus Type 2, Respectively: Consequences for Virus Replication in the Presence of Alpha/Beta and Gamma Interferons. J. Virol. 2002, 76, 2159–2167. [Google Scholar] [CrossRef] [PubMed]
- Chin, K.C.; Cresswell, P. Viperin (cig5), an IFN-inducible antiviral protein directly induced by human cytomegalovirus. Proc. Natl. Acad. Sci. USA 2001, 98, 15125–15130. [Google Scholar] [CrossRef] [PubMed]
- Boehme, K.W.; Singh, J.; Perry, S.T.; Compton, T. Human cytomegalovirus elicits a coordinated cellular antiviral response via envelope glycoprotein B. J. Virol. 2004, 78, 1202–1211. [Google Scholar] [CrossRef] [PubMed]
- Stirnweiss, A.; Ksienzyk, A.; Klages, K.; Rand, U.; Grashoff, M.; Hauser, H.; Kroger, A. IFN regulatory factor-1 bypasses IFN-mediated antiviral effects through viperin gene induction. J. Immunol. (Baltimore, Md.: 1950) 2010, 184, 5179–5185. [Google Scholar] [CrossRef]
- Ashley, C.L.; Glass, M.S.; Abendroth, A.; McSharry, B.P.; Slobedman, B. Nuclear domain 10 components upregulated via interferon during human cytomegalovirus infection potently regulate viral infection. J. Gen. Virol. 2017, 98, 1795–1805. [Google Scholar] [CrossRef]
- Grandvaux, N.; Servant, M.J.; tenOever, B.; Sen, G.C.; Balachandran, S.; Barber, G.N.; Lin, R.; Hiscott, J. Transcriptional Profiling of Interferon Regulatory Factor 3 Target Genes: Direct Involvement in the Regulation of Interferon-Stimulated Genes. J. Virol. 2002, 76, 5532–5539. [Google Scholar] [CrossRef]
- Rusinova, I.; Forster, S.; Yu, S.; Kannan, A.; Masse, M.; Cumming, H.; Chapman, R.; Hertzog, P.J. INTERFEROME v2.0: An updated database of annotated interferon-regulated genes. Nucleic Acids Res. 2013, 41, D1040–D1046. [Google Scholar] [CrossRef]
- Kim, Y.J.; Kim, E.T.; Kim, Y.-E.; Lee, M.K.; Kwon, K.M.; Kim, K.I.; Stamminger, T.; Ahn, J.-H. Consecutive Inhibition of ISG15 Expression and ISGylation by Cytomegalovirus Regulators. PLoS Pathog. 2016, 12, e1005850. [Google Scholar] [CrossRef] [PubMed]
- Lee, M.K.; Kim, Y.J.; Kim, Y.-E.; Han, T.-H.; Milbradt, J.; Marschall, M.; Ahn, J.-H. Transmembrane protein pUL50 of human cytomegalovirus inhibits ISGylation by downregulating UBE1L. J. Virol. 2018. [Google Scholar] [CrossRef] [PubMed]
- Zimmermann, C.; Büscher, N.; Krauter, S.; Krämer, N.; Wolfrum, U.; Sehn, E.; Tenzer, S.; Plachter, B. The abundant tegument protein pUL25 of human cytomegalovirus prevents proteasomal degradation of pUL26 and supports its suppression of ISGylation. J. Virol. 2018. [Google Scholar] [CrossRef] [PubMed]
- Collins, S.E.; Noyce, R.S.; Mossman, K.L. Innate cellular response to virus particle entry requires IRF3 but not virus replication. J. Virol. 2004, 78, 1706–1717. [Google Scholar] [CrossRef] [PubMed]
- Baca, L.M.; Genis, P.; Kalvakolanu, D.; Sen, G.; Meltzer, M.S.; Zhou, A.; Silverman, R. Regulation of interferon-α-inducible cellular genes in human immunodeficiency virus-infected monocytes. J. Leukocyte Biol. 1994, 55, 299–309. [Google Scholar] [CrossRef] [PubMed]
- Seo, J.-Y.; Cresswell, P. Viperin Regulates Cellular Lipid Metabolism during Human Cytomegalovirus Infection. PLoS Pathog. 2013, 9, e1003497. [Google Scholar] [CrossRef] [PubMed]
- Helbig, K.J.; Beard, M.R. The Role of Viperin in the Innate Antiviral Response. J. Mol. Biol. 2014, 426, 1210–1219. [Google Scholar] [CrossRef]
- Seo, J.-Y.; Yaneva, R.; Cresswell, P. Viperin: A Multifunctional, Interferon-Inducible Protein that Regulates Virus Replication. Cell Host Microbe 2011, 10, 534–539. [Google Scholar] [CrossRef]



© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ashley, C.L.; Abendroth, A.; McSharry, B.P.; Slobedman, B. Interferon-Independent Upregulation of Interferon-Stimulated Genes during Human Cytomegalovirus Infection is Dependent on IRF3 Expression. Viruses 2019, 11, 246. https://doi.org/10.3390/v11030246
Ashley CL, Abendroth A, McSharry BP, Slobedman B. Interferon-Independent Upregulation of Interferon-Stimulated Genes during Human Cytomegalovirus Infection is Dependent on IRF3 Expression. Viruses. 2019; 11(3):246. https://doi.org/10.3390/v11030246
Chicago/Turabian StyleAshley, Caroline L., Allison Abendroth, Brian P. McSharry, and Barry Slobedman. 2019. "Interferon-Independent Upregulation of Interferon-Stimulated Genes during Human Cytomegalovirus Infection is Dependent on IRF3 Expression" Viruses 11, no. 3: 246. https://doi.org/10.3390/v11030246
APA StyleAshley, C. L., Abendroth, A., McSharry, B. P., & Slobedman, B. (2019). Interferon-Independent Upregulation of Interferon-Stimulated Genes during Human Cytomegalovirus Infection is Dependent on IRF3 Expression. Viruses, 11(3), 246. https://doi.org/10.3390/v11030246