From a Movement-Deficient Grapevine Fanleaf Virus to the Identification of a New Viral Determinant of Nematode Transmission


 , ,
, , 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
2.1. Virus Strains and cDNA Clones
2.2. Cloning of 2CCP Mutations into GFLV-EGFP Infectious Clones
2.3. Capsid Structure Representation and Analyses
2.4. In Vitro Transcription and Inoculation
2.5. Fluorescence Visualization and Data Processing
2.6. Virus-Like Particle Production in Plants
2.7. Double-Antibody Sandwich-Enzyme-Linked Immunosorbent Assay (DAS-ELISA)
2.8. Immunocapture Reverse Transcription Polymerase Chain Reaction (IC-RT-PCR) and Sequencing
2.9. Electron Microscopy
2.10. Western Blot Analysis
2.11. Nematode Transmission Tests
3. Results
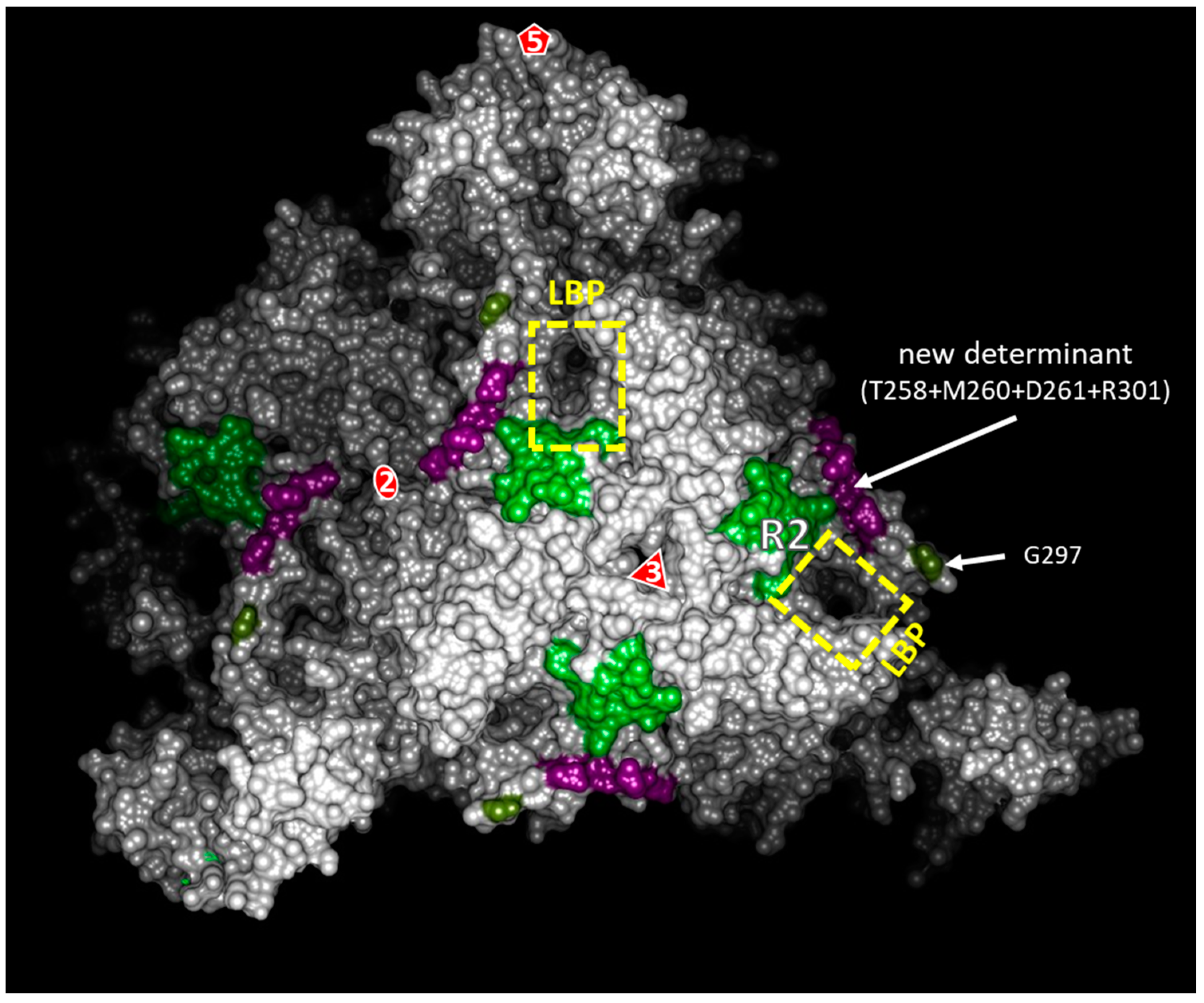
3.1. Structural Environments of Regions R3, R4, and R5 of the GFLV 2CCP
3.2. A GFLV Encoding EGFP Allows Short Distance Movement Visualization in Planta
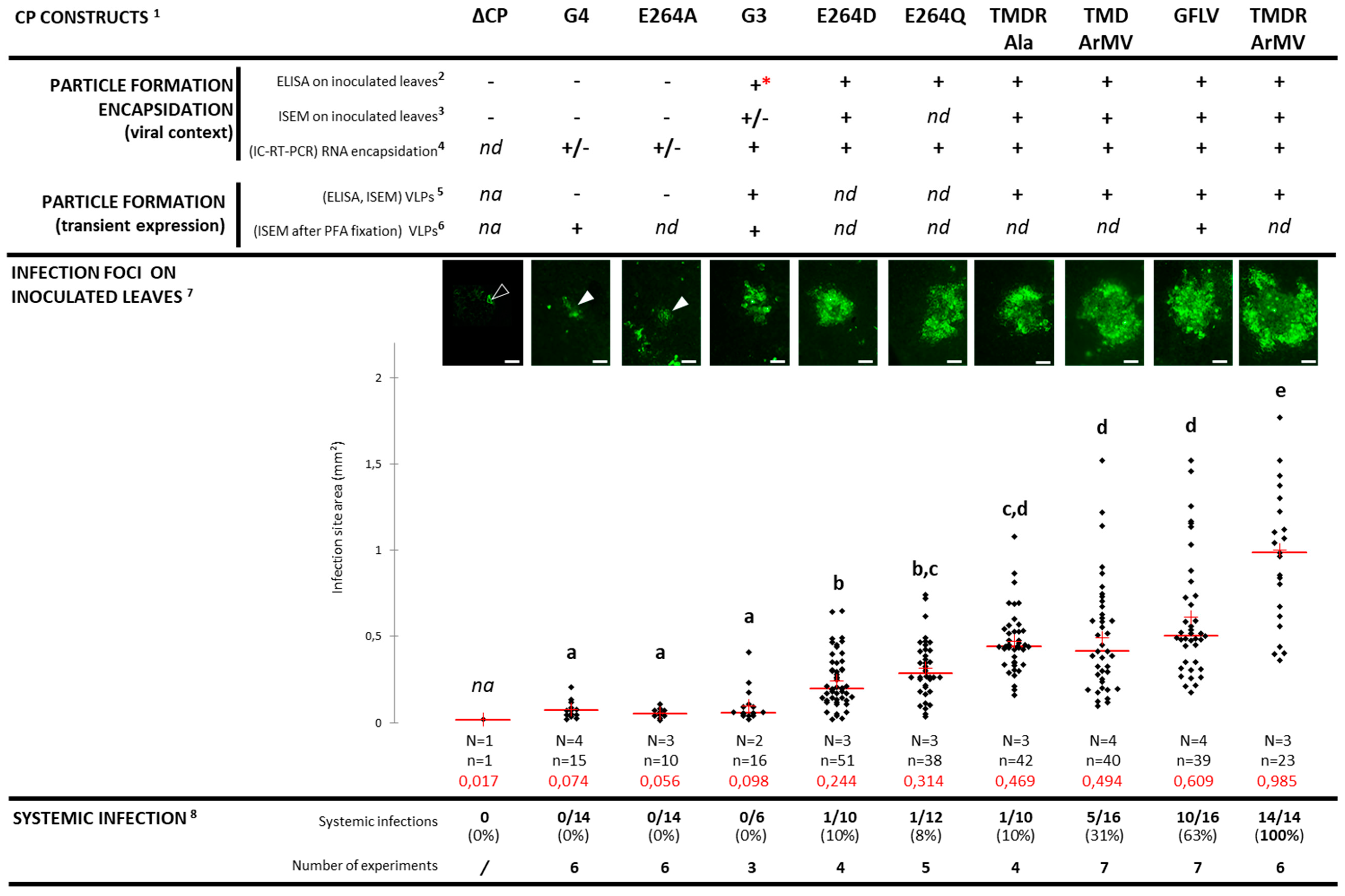
3.3. Cell-to-Cell Movement of Chimeric Viruses G3-EGFP and G4-EGFP Is Impaired But Not Abolished
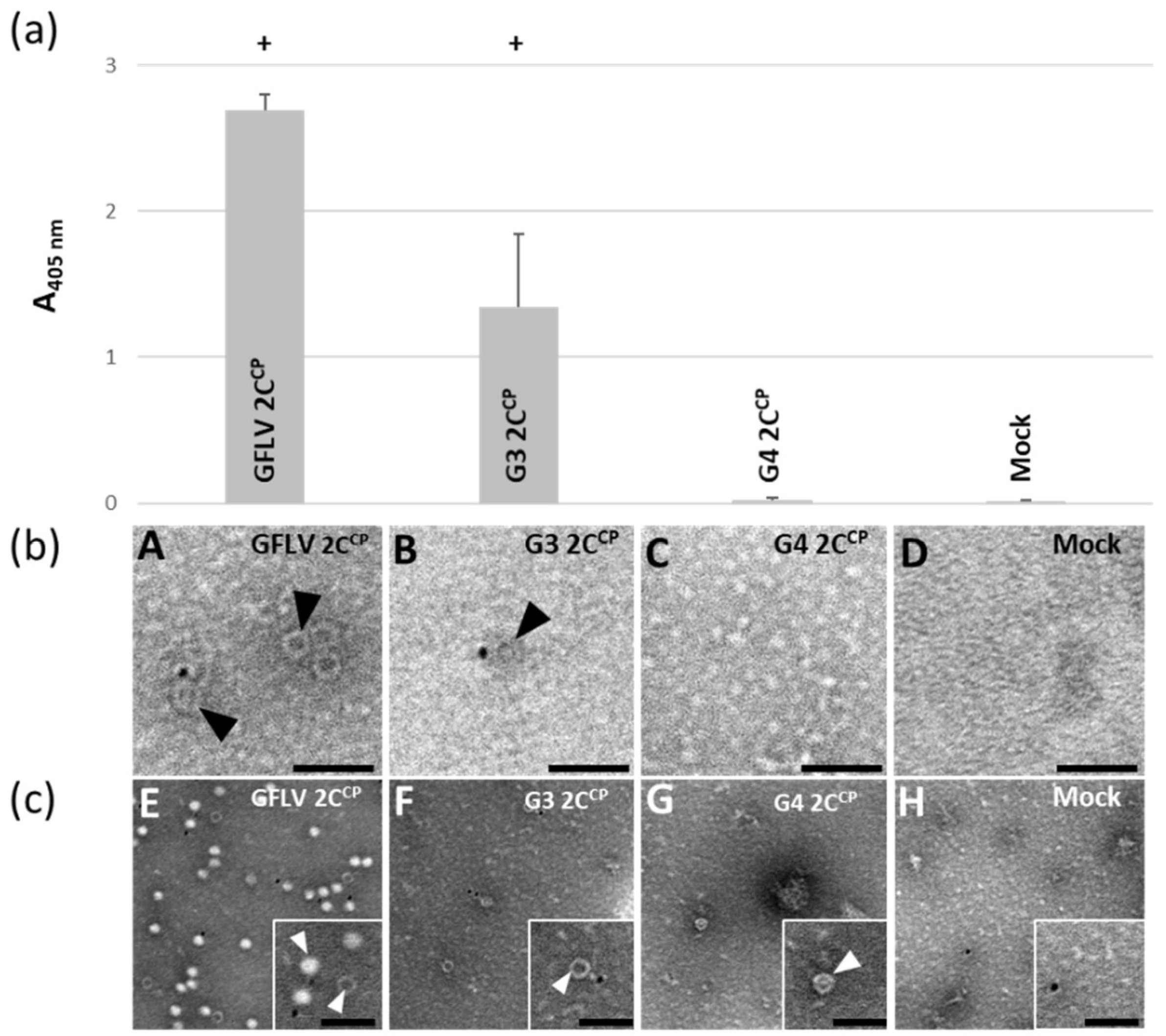
3.4. Chimeric Virus G4-EGFP Like G3-EGFP Fails to Produce Stable Capsids
3.5. E264 Is Critical for Capsid Formation
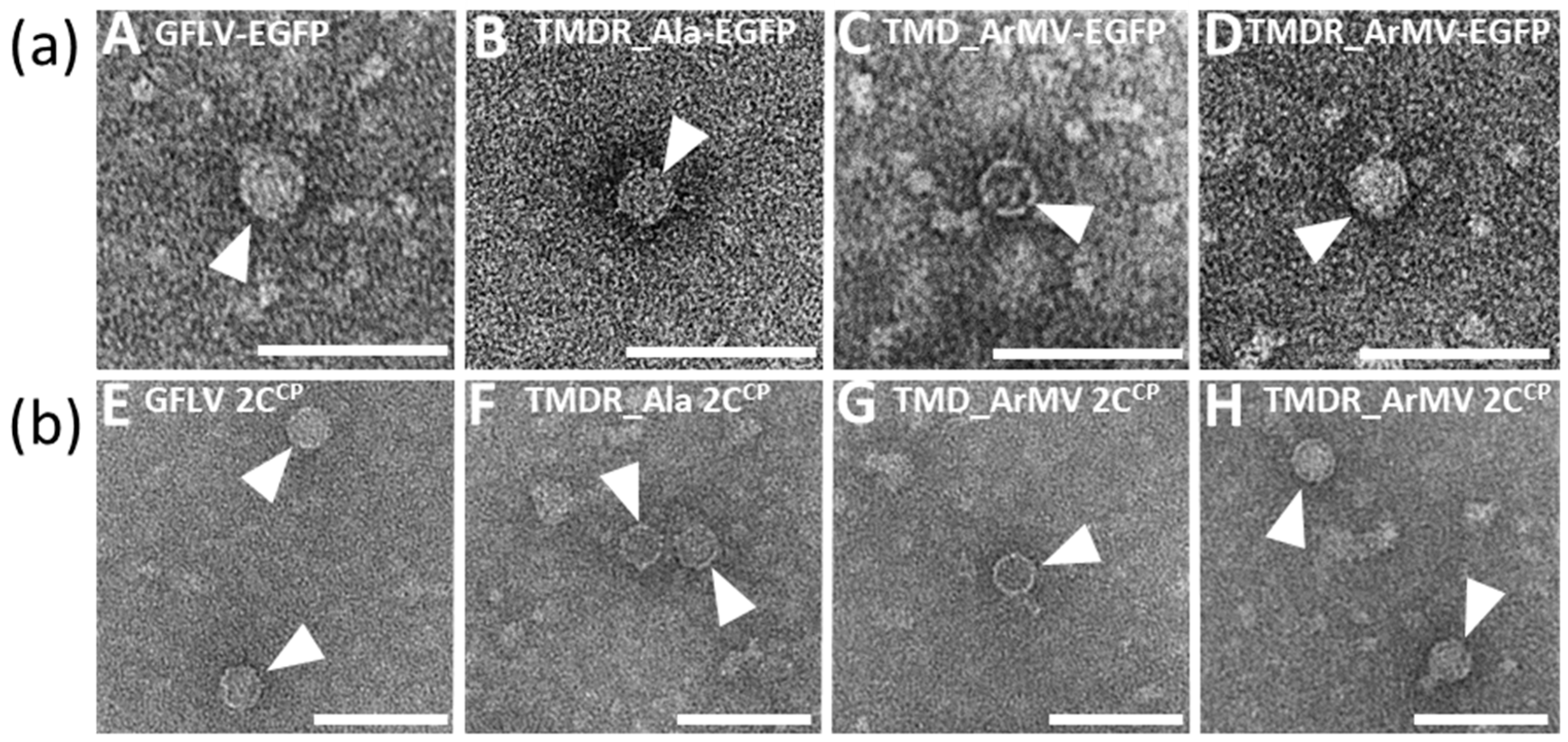
3.6. Generation of an Infectious Chimera in Region R4
3.7. The Four Residues T258, M260, D261, and R301 are Critical for GFLV Transmission by X. index
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Weber, P.H.; Bujarski, J.J. Multiple functions of capsid proteins in (+) stranded RNA viruses during plant–virus interactions. Virus Res. 2015, 196, 140–149. [Google Scholar] [CrossRef] [PubMed]
- Melcher, U. The ‘30K’ superfamily of viral movement proteins. J. Gen. Virol. 2000, 81, 257–266. [Google Scholar] [CrossRef] [PubMed]
- Atreya, P.L.; Atreya, C.D.; Pirone, T.P. Amino acid substitutions in the coat protein result in loss of insect transmissibility of a plant virus. Proc. Natl. Acad. Sci. USA 1991, 88, 7887–7891. [Google Scholar] [CrossRef] [PubMed]
- Blanc, S.; López-Moya, J.J.; Wang, R.; García-Lampasona, S.; Thornbury, D.W.; Pirone, T.P. A specific interaction between coat protein and helper component correlates with aphid transmission of a potyvirus. Virology 1997, 231, 141–147. [Google Scholar] [CrossRef]
- Bricault, C.A.; Perry, K.L. Alteration of intersubunit acid–base pair interactions at the quasi-threefold axis of symmetry of cucumber mosaic virus disrupts aphid vector transmission. Virology 2013, 440, 160–170. [Google Scholar] [CrossRef][Green Version]
- Liu, S.; He, X.; Park, G.; Josefsson, C.; Perry, K.L. A conserved capsid protein surface domain of cucumber mosaic virus is essential for efficient aphid vector transmission. J. Virol. 2002, 76, 9756–9762. [Google Scholar] [CrossRef]
- Seo, J.K.; Kang, S.H.; Seo, B.Y.; Jung, J.K.; Kim, K.H. Mutational analysis of interaction between coat protein and helper component-proteinase of soybean mosaic virus involved in aphid transmission. Mol. Plant Pathol. 2010, 11, 265–276. [Google Scholar] [CrossRef]
- Sanfaçon, H.; Wellink, J.; Le Gall, O.; Karasev, A.; van der Vlugt, R.; Wetzel, T. Secoviridae: A proposed family of plant viruses within the order Picornavirales that combines the families Sequiviridae and Comoviridae, the unassigned genera Cheravirus and Sadwavirus, and the proposed genus Torradovirus. Arch. Virol. 2009, 154, 899–907. [Google Scholar] [CrossRef]
- Andret-Link, P.; Laporte, C.; Valat, L.; Ritzenthaler, C.; Demangeat, G.; Vigne, E.; Laval, V.; Pfeiffer, P.; Stussi-Garaud, C.; Fuchs, M. Grapevine fanleaf virus: Still a major threat to the grapevine industry. J. Plant Pathol. 2004, 86, 183–195. [Google Scholar]
- Martelli, G.P. An overview on grapevine viruses, viroids, and the diseases they cause. In Grapevine Viruses: Molecular Biology, Diagnostics and Management; Meng, B., Martelli, G.P., Golino, D.A., Fuchs, M., Eds.; Springer: Cham, Switzerland, 2017; pp. 31–46. [Google Scholar]
- Harrison, B.D.; Cadman, C.H. Role of a dagger nematode (Xiphinema sp.) in outbreaks of plant diseases caused by arabis mosaic virus. Nature 1959, 184, 1624–1626. [Google Scholar]
- Wyss, U. Xiphinema index, Maintenance and feeding in monoxenic cultures. In Maintenance of Human, Animal and Plant Pathogen Vectors; Maramorosch, K., Mahmood, F., Eds.; Science Publishers Inc.: Enfield, NH, USA, 2000; pp. 251–281. [Google Scholar]
- Quacquarelli, A.; Gallitelli, D.; Savino, V.; Martelli, G.P. Properties of grapevine fanleaf virus. J. Gen. Virol. 1976, 32, 349–360. [Google Scholar] [CrossRef]
- Margis, R.; Viry, M.; Pinck, M.; Pinck, L. Cloning and in vitro characterization of the grapevine fanleaf virus proteinase cistron. Virology 1991, 185, 779–787. [Google Scholar] [CrossRef]
- Ritzenthaler, C.; Viry, M.; Pinck, M.; Margis, R.; Fuchs, M.; Pinck, L. Complete nucleotide sequence and genetic organization of grapevine fanleaf nepovirus RNA1. J. Gen. Virol. 1991, 72, 2357–2365. [Google Scholar] [CrossRef] [PubMed]
- Vigne, E.; Gottula, J.; Schmitt-Keichinger, C.; Komar, V.; Ackerer, L.; Belval, L.; Rakotomalala, L.; Lemaire, O.; Ritzenthaler, C.; Fuchs, M. A strain-specific segment of the RNA-dependent RNA polymerase of grapevine fanleaf virus determines symptoms in Nicotiana species. J. Gen. Virol. 2013, 94, 2803–2813. [Google Scholar] [CrossRef] [PubMed]
- Gaire, F.; Schmitt, C.; Stussi-Garaud, C.; Pinck, L.; Ritzenthaler, C. Protein 2A of grapevine fanleaf nepovirus is implicated in RNA2 replication and colocalizes to the replication site. Virology 1999, 264, 25–36. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Martin, I.R.; Vigne, E.; Berthold, F.; Komar, V.; Lemaire, O.; Fuchs, M.; Schmitt-Keichinger, C. The 50 distal amino acids of the 2AHP homing protein of grapevine fanleaf virus elicit a hypersensitive reaction on Nicotiana occidentalis. Mol. Plant Pathol. 2018, 19, 731–743. [Google Scholar] [CrossRef] [PubMed]
- Ritzenthaler, C.; Schmit, A.C.; Michler, P.; Stussi-Garaud, C.; Pinck, L. Grapevine fanleaf nepovirus P38 putative movement protein is located on tubules in vivo. Mol. Plant Microbe Interact. 1995, 8, 379–387. [Google Scholar] [CrossRef]
- Serghini, M.; Fuchs, M.; Pinck, M.; Reinbolt, J.; Walter, B.; Pinck, L. RNA2 of grapevine fanleaf virus: Sequence analysis and coat protein cistron location. J. Gen. Virol. 1990, 71, 1433–1441. [Google Scholar] [CrossRef]
- Schellenberger, P.; Sauter, C.; Lorber, B.; Bron, P.; Trapani, S.; Bergdoll, M.; Marmonier, A.; Schmitt-Keichinger, C.; Lemaire, O.; Demangeat, G.; et al. Structural insights into viral determinants of nematode mediated grapevine fanleaf virus transmission. PLoS Pathog. 2011, 7, e1002034. [Google Scholar] [CrossRef]
- Belin, C.; Schmitt, C.; Gaire, F.; Walter, B.; Demangeat, G.; Pinck, L. The nine C-terminal residues of the grapevine fanleaf nepovirus movement protein are critical for systemic virus spread. J. Gen. Virol. 1999, 80, 1347–1356. [Google Scholar] [CrossRef][Green Version]
- Andret-Link, P.; Marmonier, A.; Belval, L.; Hleibieh, K.; Ritzenthaler, C.; Demangeat, G. Ectoparasitic nematode vectors of grapevine viruses. In Grapevine Viruses: Molecular Biology, Diagnostics and Management; Meng, B., Martelli, G.P., Golino, D.A., Fuchs, M., Eds.; Springer: Cham, Switzerland, 2017; pp. 505–529. [Google Scholar]
- Andret-Link, P.; Schmitt-Keichinger, C.; Demangeat, G.; Komar, V.; Fuchs, M. The specific transmission of Grapevine fanleaf virus by its nematode vector Xiphinema index is solely determined by the viral coat protein. Virology 2004, 320, 12–22. [Google Scholar] [CrossRef] [PubMed]
- Marmonier, A.; Schellenberger, P.; Esmenjaud, D.; Schmitt- Keichinger, C.; Ritzenthaler, C.; Andret-Link, P.; Lemaire, O.; Fuchs, M.; Demangeat, G. The coat protein determines the specificity of virus transmission by Xiphinema diversicaudatum. J. Plant Pathol. 2010, 92, 275–279. [Google Scholar]
- Taylor, C.; Robertson, W. Sites of virus retention in the alimentary tract of the nematode vectors, Xiphinema diversicaudatum (Micol.) and X. index (Thorne and Allen). Ann. Appl. Biol. 1970, 66, 375–380. [Google Scholar] [CrossRef] [PubMed]
- Schellenberger, P.; Andret-Link, P.; Schmitt-Keichinger, C.; Bergdoll, M.; Marmonier, A.; Vigne, E.; Lemaire, O.; Fuchs, M.; Demangeat, G.; Ritzenthaler, C. A stretch of 11 amino acids in the ßB-ßC loop of the coat protein of grapevine fanleaf virus is essential for transmission by the nematode Xiphinema index. J. Virol. 2010, 84, 7924–7933. [Google Scholar] [CrossRef] [PubMed]
- Vuittenez, A.; Munck, M.C.; Kuszala, J. Souches de virus à haute agressivités isolées de vignes atteintes de dégénérescence infectieuse. Etud. Virol. Appl. 1964, 5, 68–78. [Google Scholar]
- Loudes, A.M.; Ritzenthaler, C.; Pinck, M.; Serghini, M.A.; Pinck, L. The 119 kDa and 124 kDa polyproteins of arabis mosaic nepovirus (isolate S) are encoded by two distinct RNA2 species. J. Gen. Virol. 1995, 76, 899–906. [Google Scholar] [CrossRef] [PubMed]
- Viry, M.; Serghini, M.A.; Hans, F.; Ritzenthaler, C.; Pinck, M.; Pinck, L. Biologically active transcripts from cloned cDNA of genomic grapevine fanleaf nepovirus RNAs. J. Gen. Virol. 1993, 74, 169–174. [Google Scholar] [CrossRef]
- Schmitt-Keichinger, C.; Hemmer, C.; Berthold, F.; Ritzenthaler, C. Molecular, cellular and structural biology of grapevine fanleaf virus. In Grapevine Viruses: Molecular Biology, Diagnostics and Management; Meng, B., Martelli, G.P., Golino, D.A., Fuchs, M., Eds.; Springer: Cham, Switzerland, 2017; pp. 83–107. [Google Scholar]
- Pettersen, E.F.; Goddard, T.D.; Huang, C.C.; Couch, G.S.; Greenblatt, D.M.; Meng, E.C.; Ferrin, T.E. UCSF Chimera—A visualization system for exploratory research and analysis. J. Comput. Chem. 2004, 25, 1605–1612. [Google Scholar] [CrossRef]
- Sainsbury, F.; Thuenemann, E.C.; Lomonossoff, G.P. pEAQ: Versatile expression vectors for easy and quick transient expression of heterologous proteins in plants. Plant Biotech. J. 2009, 7, 682–693. [Google Scholar] [CrossRef]
- Merzlyak, E.M.; Goedhart, J.; Shcherbo, D.; Bulina, M.E.; Shcheglov, A.S.; Fradkov, A.F.; Gaintzeva, A.; Lukyanov, K.A.; Lukyanov, S.; Gadella, T.W.J.; et al. Bright monomeric red fluorescent protein with an extended fluorescence lifetime. Nat. Methods 2007, 4, 555–557. [Google Scholar] [CrossRef]
- Berthold, F.; Roujol, D.; Hemmer, C.; Jamet, E.; Ritzenthaler, C.; Hoffmann, L.; Schmitt-Keichinger, C. Inside or outside? A new collection of Gateway vectors allowing plant protein subcellular localization or over-expression. Plasmid 2019, 105, 102436. [Google Scholar] [CrossRef] [PubMed]
- Koncz, C.; Schell, J. The promoter of TL-DNA gene 5 controls the tissue-specific expression of chimaeric genes carried by a novel type of Agrobacterium binary vector. Mol. Gen. Genet. 1986, 204, 383–396. [Google Scholar] [CrossRef]
- Belval, L.; Hemmer, C.; Sauter, C.; Reinbold, C.; Fauny, J.D.; Berthold, F.; Ackerer, L.; Schmitt-Keichinger, C.; Lemaire, O.; Demangeat, G.; et al. Display of whole proteins on inner and outer surfaces of grapevine fanleaf virus-like particles. Plant Biotech. J. 2016, 14, 2288–2299. [Google Scholar] [CrossRef] [PubMed]
- Laemmli, U.K. Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature 1970, 227, 680–685. [Google Scholar] [CrossRef]
- Lai-Kee-Him, J.; Schellenberger, P.; Dumas, C.; Richard, E.; Trapani, S.; Komar, V.; Demangeat, G.; Ritzenthaler, C.; Bron, P. The backbone model of the arabis mosaic virus reveals new insights into functional domains of nepovirus capsid. J. Struct. Biol. 2013, 182, 1–9. [Google Scholar] [CrossRef]
- Jansen, K.A.J.; Wolfs, C.J.A.M.; Lohuis, H.; Goldbach, R.W.; Verduin, B.J.M. Characterization of the brome mosaic virus movement protein expressed in E. coli. Virology 1998, 242, 387–394. [Google Scholar] [CrossRef]
- Kasteel, D.; van der Wel, N.; Jansen, K.; Goldbach, R.; van Lent, J. Tubule-forming capacity of the movement proteins of alfalfa mosaic virus and brome mosaic virus. J. Gen. Virol. 1997, 78, 2089–2093. [Google Scholar] [CrossRef]
- Takeda, A.; Nakamura, W.; Sasaki, N.; Goto, K.; Kaido, M.; Okuno, T.; Mise, K. Natural isolates of brome mosaic virus with the ability to move from cell to cell independently of coat protein. J. Gen. Virol. 2005, 86, 1201–1211. [Google Scholar] [CrossRef]
- Dolja, V.V.; Haldeman, R.; Robertson, N.L.; Dougherty, W.G.; Carrington, J.C. Distinct functions of capsid protein in assembly and movement of tobacco etch potyvirus in plants. EMBO J. 1994, 13, 1482–1491. [Google Scholar] [CrossRef]
- Salánki, K.; Kiss, L.; Gellért, Á.; Balázs, E. Identification a coat protein region of cucumber mosaic virus (CMV) essential for long-distance movement in cucumber. Arch. Virol. 2011, 156, 2279–2283. [Google Scholar] [CrossRef]
- Pantaleo, V.; Grieco, F.; Di Franco, A.; Martelli, G.P. The role of the C-terminal region of olive latent virus 1 coat protein in host systemic infection. Arch. Virol. 2006, 151, 1973–1983. [Google Scholar] [CrossRef] [PubMed]
- Karran, R.; Sanfaçon, H. Tomato ringspot virus coat protein binds to ARGONAUTE 1 and suppresses the translation repression of a reporter gene. Mol. Plant Microbe Interact. 2014, 27, 933–943. [Google Scholar] [CrossRef] [PubMed]
- Powers, J.G.; Sit, T.L.; Qu, F.; Morris, T.J.; Kim, K.H.; Lommel, S.A. A versatile assay for the identification of RNA silencing suppressors based on complementation of viral movement. Mol. Plant Microbe Interact. 2008, 21, 879–890. [Google Scholar] [CrossRef] [PubMed]









© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Belval, L.; Marmonier, A.; Schmitt-Keichinger, C.; Gersch, S.; Andret-Link, P.; Komar, V.; Vigne, E.; Lemaire, O.; Ritzenthaler, C.; Demangeat, G. From a Movement-Deficient Grapevine Fanleaf Virus to the Identification of a New Viral Determinant of Nematode Transmission. Viruses 2019, 11, 1146. https://doi.org/10.3390/v11121146
Belval L, Marmonier A, Schmitt-Keichinger C, Gersch S, Andret-Link P, Komar V, Vigne E, Lemaire O, Ritzenthaler C, Demangeat G. From a Movement-Deficient Grapevine Fanleaf Virus to the Identification of a New Viral Determinant of Nematode Transmission. Viruses. 2019; 11(12):1146. https://doi.org/10.3390/v11121146
Chicago/Turabian StyleBelval, Lorène, Aurélie Marmonier, Corinne Schmitt-Keichinger, Sophie Gersch, Peggy Andret-Link, Véronique Komar, Emmanuelle Vigne, Olivier Lemaire, Christophe Ritzenthaler, and Gérard Demangeat. 2019. "From a Movement-Deficient Grapevine Fanleaf Virus to the Identification of a New Viral Determinant of Nematode Transmission" Viruses 11, no. 12: 1146. https://doi.org/10.3390/v11121146
APA StyleBelval, L., Marmonier, A., Schmitt-Keichinger, C., Gersch, S., Andret-Link, P., Komar, V., Vigne, E., Lemaire, O., Ritzenthaler, C., & Demangeat, G. (2019). From a Movement-Deficient Grapevine Fanleaf Virus to the Identification of a New Viral Determinant of Nematode Transmission. Viruses, 11(12), 1146. https://doi.org/10.3390/v11121146