Plant-Derived Purification, Chemical Synthesis, and In Vitro/In Vivo Evaluation of a Resveratrol Dimer, Viniferin, as an HCV Replication Inhibitor

 , and
, and
Abstract
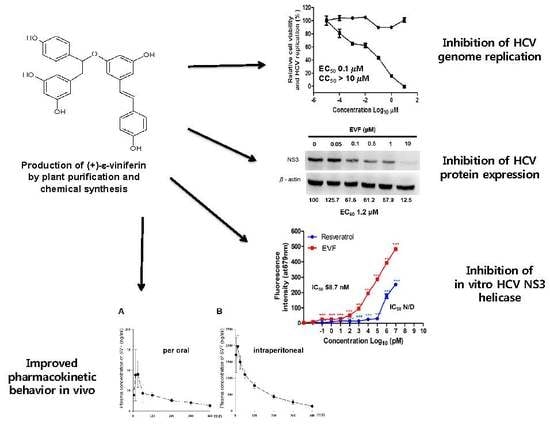
1. Introduction
2. Materials and Methods
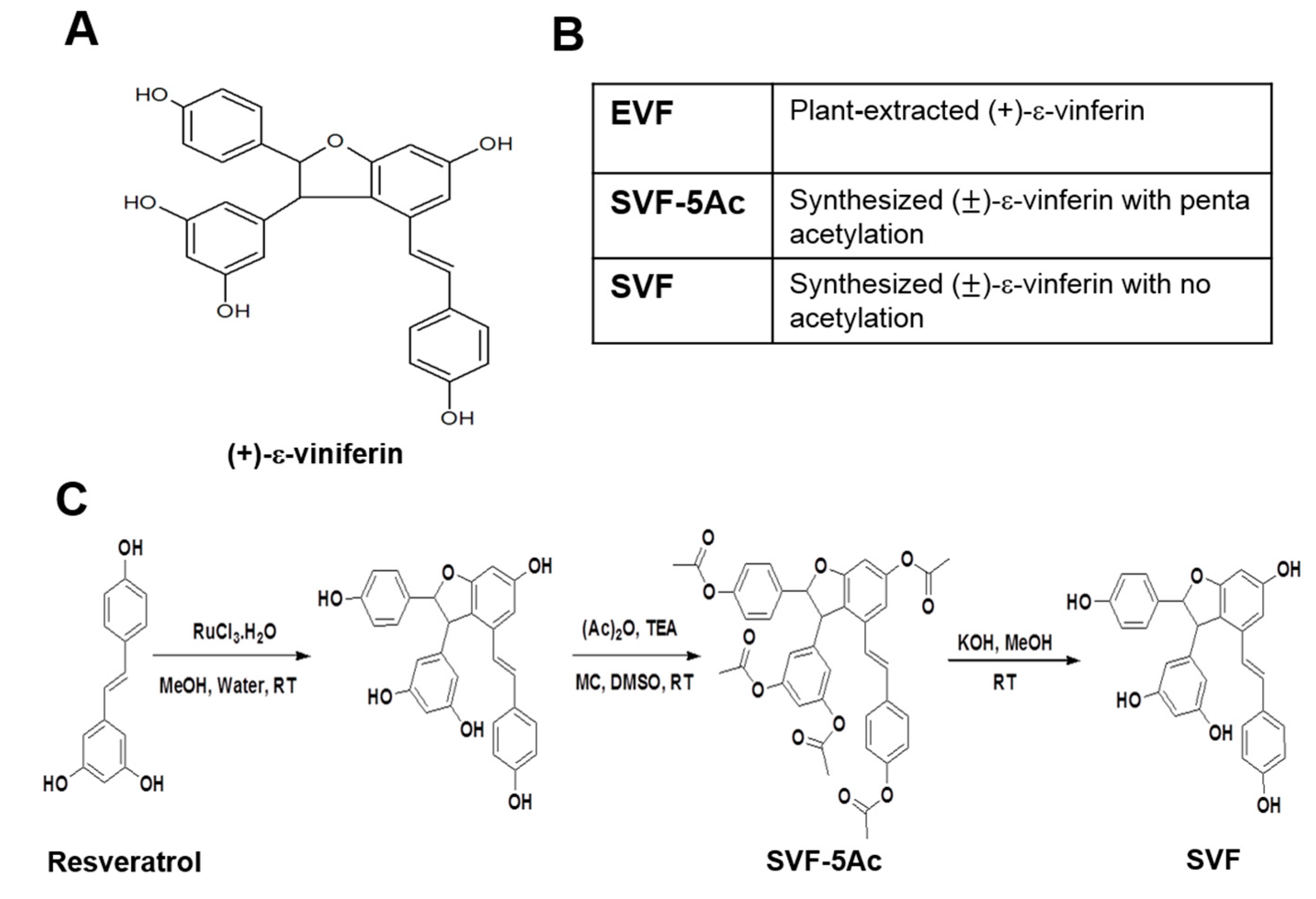
2.1. Preparation of EVF from Vitis Vinifera
2.2. Organic Synthesis of SVF-5Ac and SVF
2.3. Cell Culture
2.4. Plasmids
2.5. In Vitro Transcription for the Production of HCV RNA Genomes
2.6. Western Blot Analysis
2.7. Quantitative Real-Time RT-PCR (qRT-PCR) Analysis
2.8. Graphene Oxide (GO)-Based NS3 Helicase Inhibition Assay
2.9. In Vivo Pharmacokinetic Study of SVF
2.10. Statistical Analysis
3. Results
3.1. Isolation of Plant-Derived EVF and Organic Synthesis of SVF-5Ac and SVF
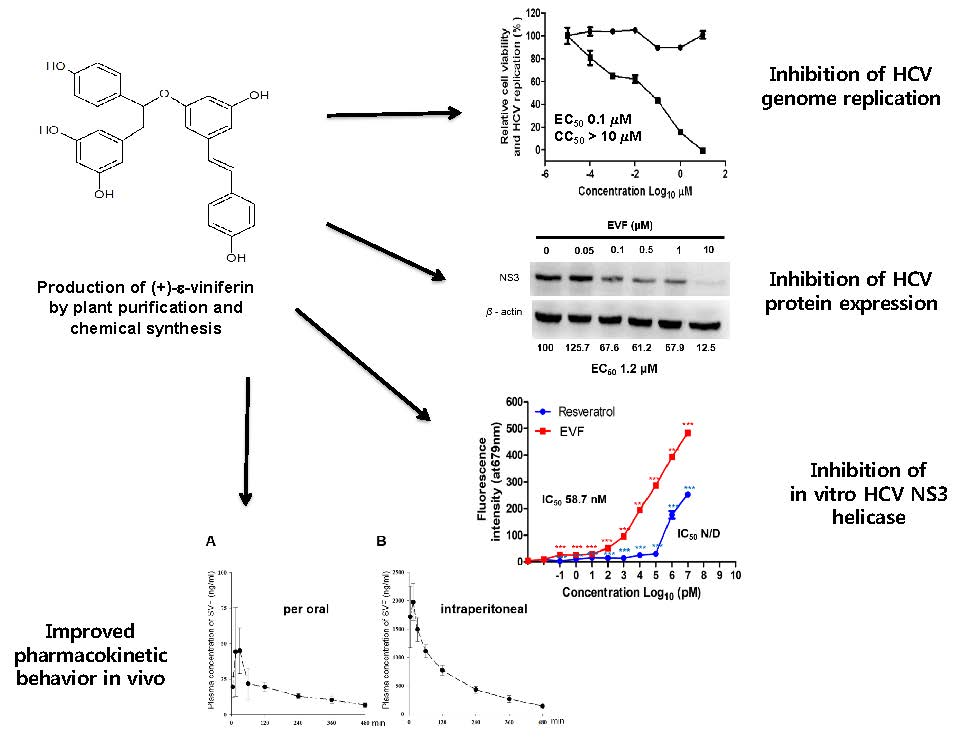
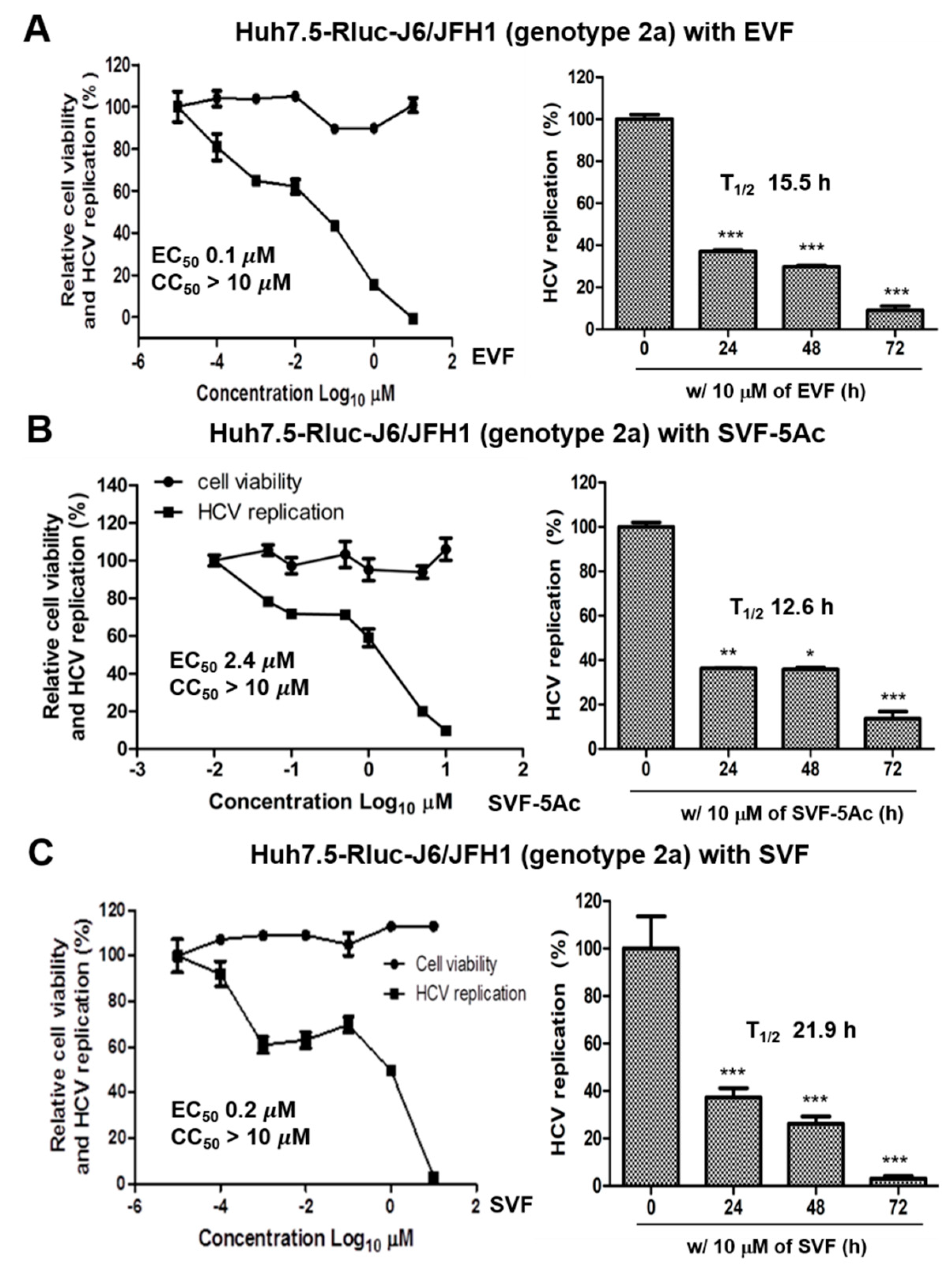
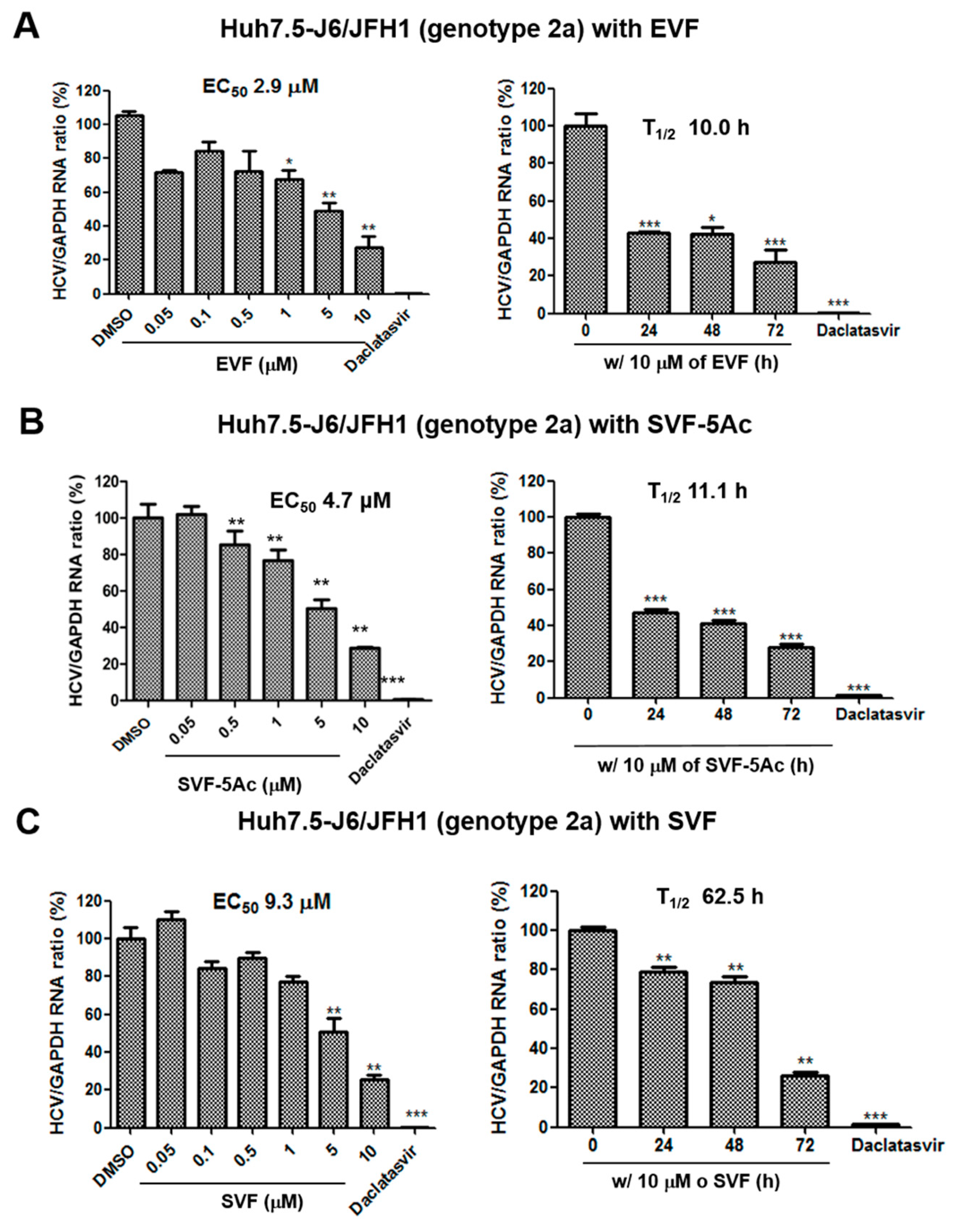
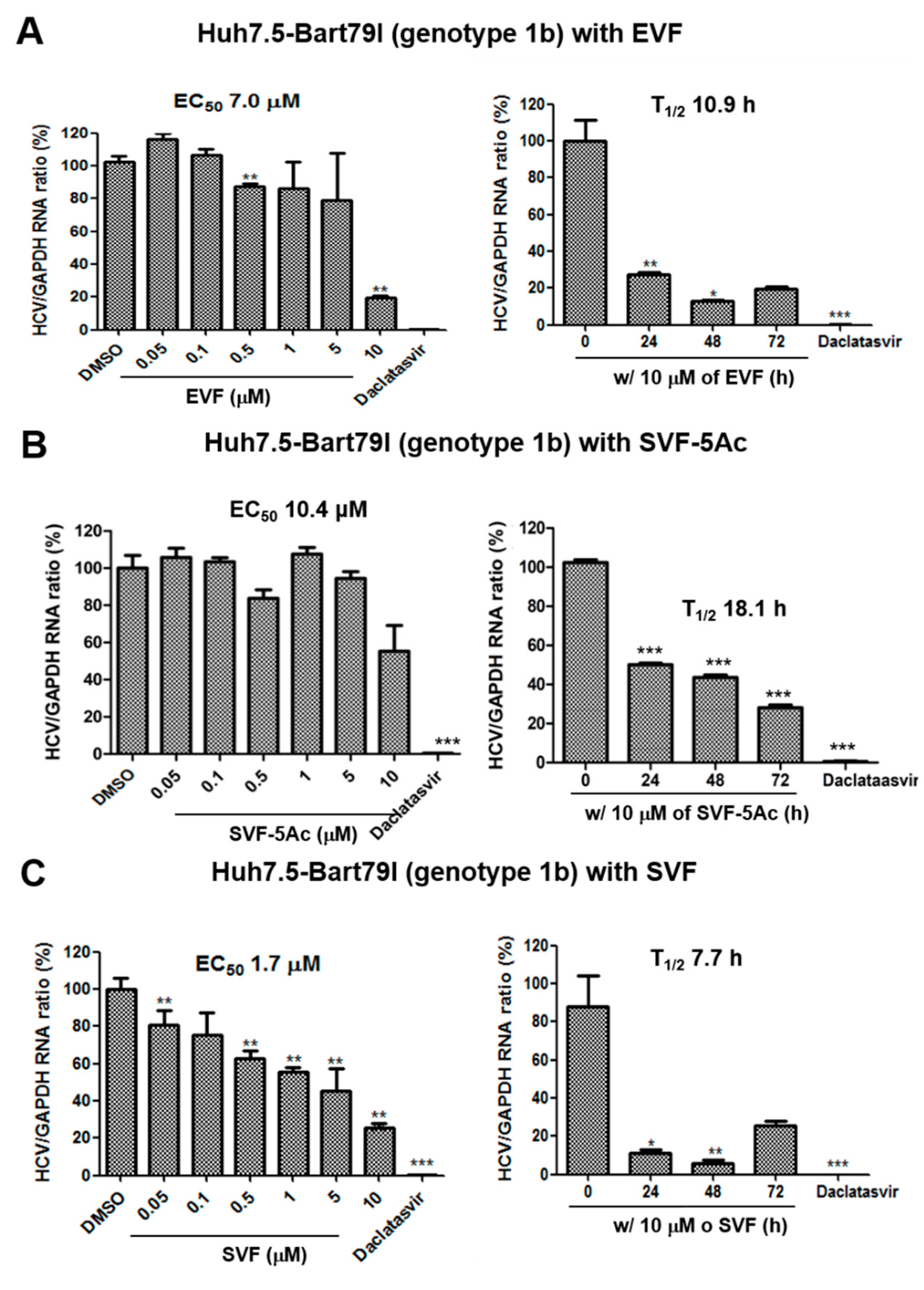
3.2. Inhibition of HCV Replication by EVF, SVF-5Ac, and SVF
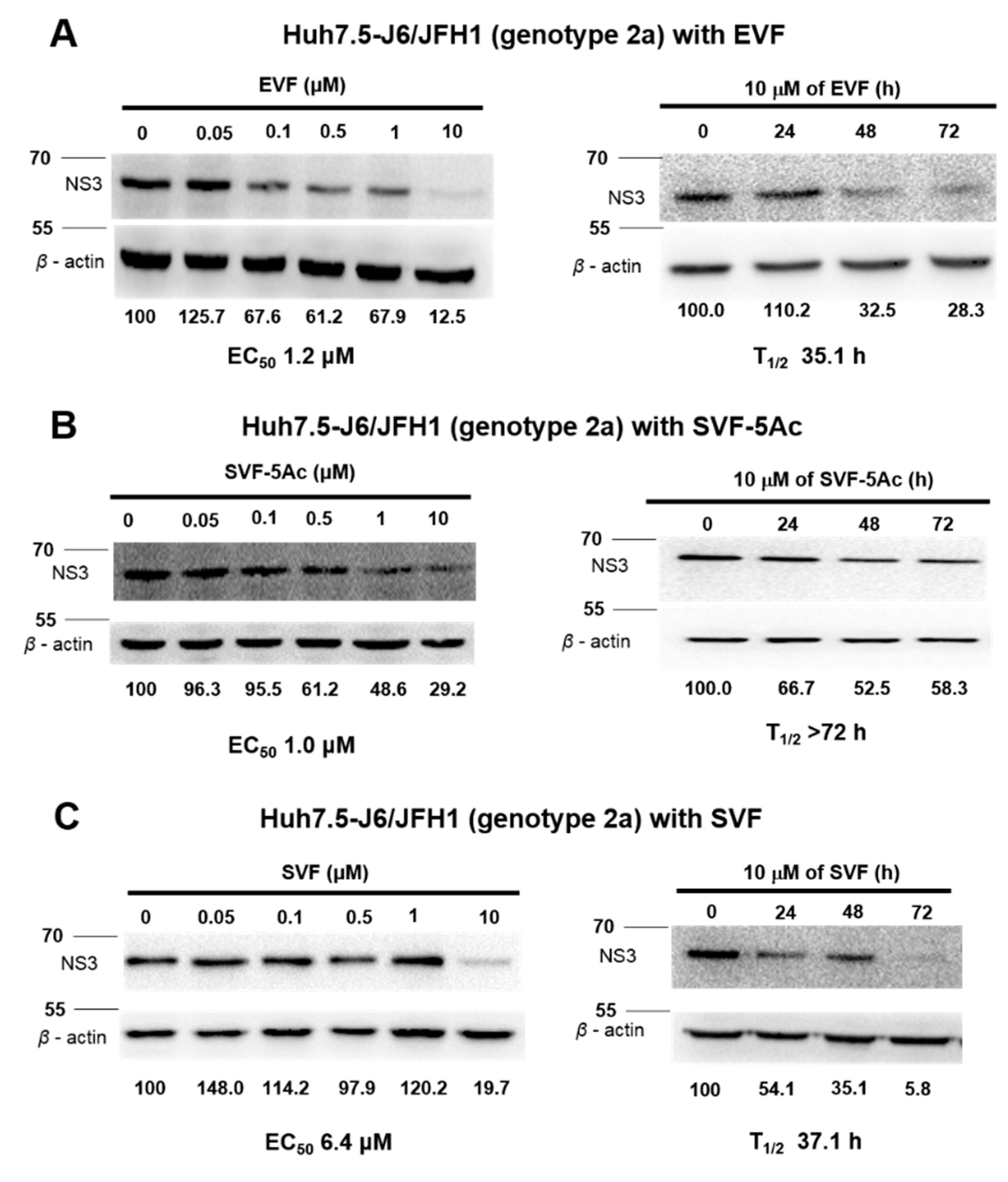
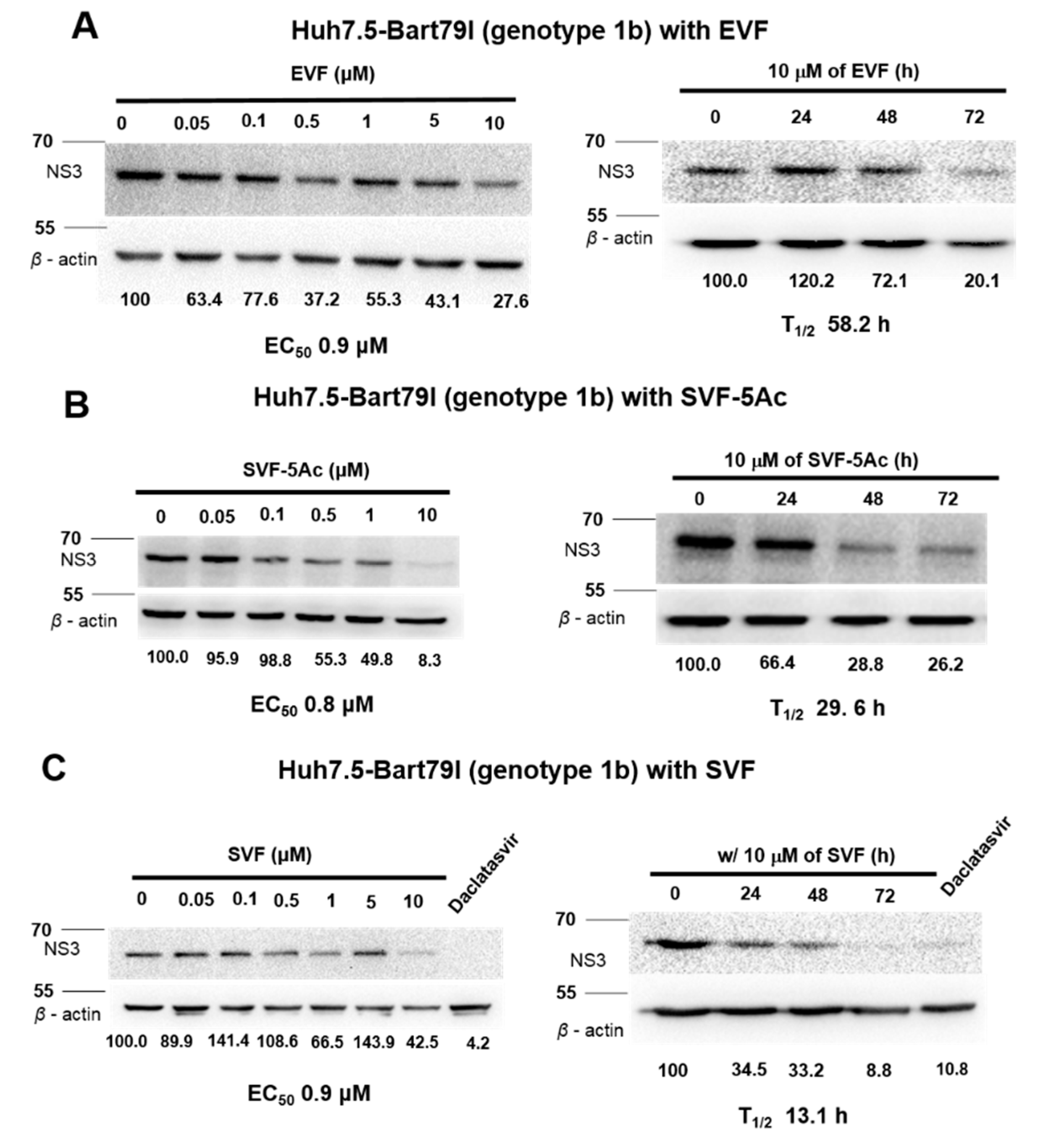
3.3. Inhibition of HCV Protein Expression by EVF, SVF-5Ac, and SVF
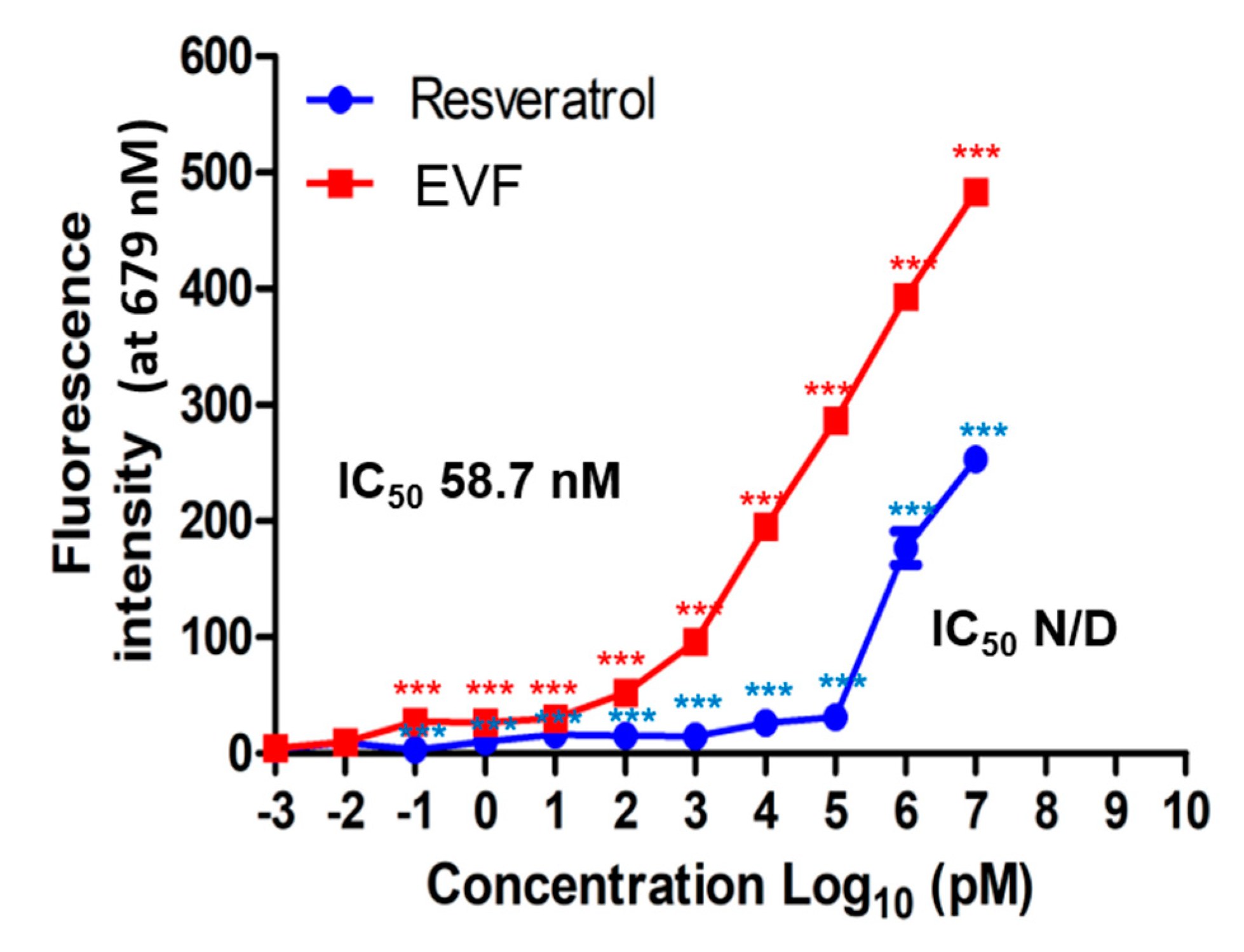
3.4. Inhibition of Genotype 1b HCV NS3 Helicase by EVF
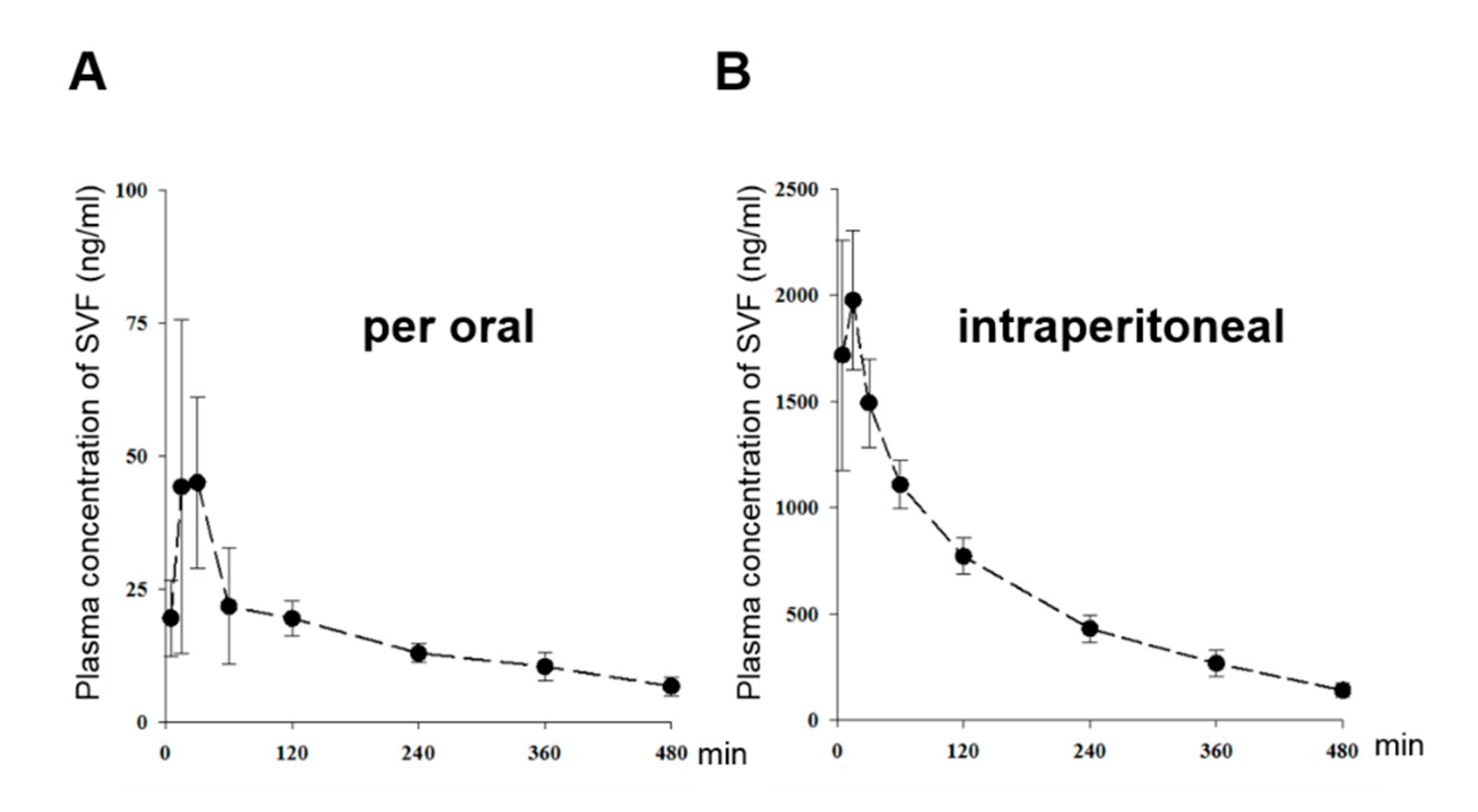
3.5. The Pharmacokinetic Characterization of SVF in Mice
4. Discussion
Author Contributions
Funding
Conflicts of Interest
References
- Grakoui, A.; McCourt, D.W.; Wychowski, C.; Feinstone, S.M.; Rice, C.M. A second hepatitis c virus-encoded proteinase. Proc. Natl. Acad. Sci. USA 1993, 90, 10583–10587. [Google Scholar] [CrossRef] [PubMed]
- Grakoui, A.; Wychowski, C.; Lin, C.; Feinstone, S.M.; Rice, C.M. Expression and identification of hepatitis c virus polyprotein cleavage products. J. Virol. 1993, 67, 1385–1395. [Google Scholar] [PubMed]
- Moradpour, D.; Penin, F.; Rice, C.M. Replication of hepatitis C virus. Nat. Rev. Microbiol. 2007, 5, 453–463. [Google Scholar] [CrossRef] [PubMed]
- Blight, K.J.; Kolykhalov, A.A.; Rice, C.M. Efficient initiation of hcv rna replication in cell culture. Science 2000, 290, 1972–1974. [Google Scholar] [CrossRef] [PubMed]
- Lohmann, V.; Korner, F.; Koch, J.; Herian, U.; Theilmann, L.; Bartenschlager, R. Replication of subgenomic hepatitis c virus rnas in a hepatoma cell line. Science 1999, 285, 110–113. [Google Scholar] [CrossRef] [PubMed]
- Belon, C.A.; Frick, D.N. Helicase inhibitors as specifically targeted antiviral therapy for hepatitis C. Future Virol. 2009, 4, 277–293. [Google Scholar] [CrossRef] [PubMed]
- Shepard, C.W.; Finelli, L.; Alter, M.J. Global epidemiology of hepatitis c virus infection. Lancet Infect. Dis. 2005, 5, 558–567. [Google Scholar] [CrossRef]
- Alter, M.J.; Kruszon-Moran, D.; Nainan, O.V.; McQuillan, G.M.; Gao, F.; Moyer, L.A.; Kaslow, R.A.; Margolis, H.S. The prevalence of hepatitis c virus infection in the united states, 1988 through 1994. N. Engl. J. Med. 1999, 341, 556–562. [Google Scholar] [CrossRef]
- Di Bisceglie, A.M. Natural history of hepatitis C: Its impact on clinical management. Hepatology 2000, 31, 1014–1018. [Google Scholar] [CrossRef]
- Mukherjee, S.; Sorrell, M.F. Controversies in liver transplantation for hepatitis C. Gastroenterology 2008, 134, 1777–1788. [Google Scholar] [CrossRef]
- Sandmann, L.; Schulte, B.; Manns, M.P.; Maasoumy, B. Treatment of chronic hepatitis C: Efficacy, side effects and complications. Visc. Med. 2019, 35, 161–170. [Google Scholar] [CrossRef] [PubMed]
- Cholankeril, G.; Joseph-Talreja, M.; Perumpail, B.J.; Liu, A.; Yoo, E.R.; Ahmed, A.; Goel, A. Timing of hepatitis c virus treatment in liver transplant candidates in the era of direct-acting antiviral agents. J. Clin. Transl. Hepatol. 2017, 5, 363–367. [Google Scholar] [CrossRef] [PubMed]
- Jakobsen, J.C.; Nielsen, E.E.; Feinberg, J.; Katakam, K.K.; Fobian, K.; Hauser, G.; Poropat, G.; Djurisic, S.; Weiss, K.H.; Bjelakovic, M.; et al. Direct-acting antivirals for chronic hepatitis c. Cochrane Database Syst. Rev. 2017, 6, CD012143. [Google Scholar] [PubMed]
- Lee, S.; Yoon, K.D.; Lee, M.; Cho, Y.; Choi, G.; Jang, H.; Kim, B.; Jung, D.H.; Oh, J.G.; Kim, G.W.; et al. Identification of a resveratrol tetramer as a potent inhibitor of hepatitis c virus helicase. Br. J. Pharmacol. 2016, 173, 191–211. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.G.; Wang, C.C. Preparative separation of oligostilbenes from vitis thunbergii var. Taiwaniana by centrifugal partition chromatography followed by sephadex lh-20 column chromatography. Sep. Purif. Technol. 2009, 66, 65–70. [Google Scholar] [CrossRef]
- Ha do, T.; Chen, Q.C.; Hung, T.M.; Youn, U.J.; Ngoc, T.M.; Thuong, P.T.; Kim, H.J.; Seong, Y.H.; Min, B.S.; Bae, K. Stilbenes and oligostilbenes from leaf and stem of vitis amurensis and their cytotoxic activity. Arch. Pharmacal Res. 2009, 32, 177–183. [Google Scholar]
- Jiang, L.; He, S.; Sun, C.; Pan, Y. Selective (1)o(2) quenchers, oligostilbenes, from vitis wilsonae: Structural identification and biogenetic relationship. Phytochemistry 2012, 77, 294–303. [Google Scholar] [CrossRef]
- Wang, K.T.; Chen, L.G.; Tseng, S.H.; Huang, J.S.; Hsieh, M.S.; Wang, C.C. Anti-inflammatory effects of resveratrol and oligostilbenes from vitis thunbergii var. Taiwaniana against lipopolysaccharide-induced arthritis. J. Agric. Food Chem. 2011, 59, 3649–3656. [Google Scholar] [CrossRef]
- Tscherne, D.M.; Jones, C.T.; Evans, M.J.; Lindenbach, B.D.; McKeating, J.A.; Rice, C.M. Time- and temperature-dependent activation of hepatitis c virus for low-ph-triggered entry. J. Virol. 2006, 80, 1734–1741. [Google Scholar] [CrossRef]
- Lindenbach, B.D.; Evans, M.J.; Syder, A.J.; Wolk, B.; Tellinghuisen, T.L.; Liu, C.C.; Maruyama, T.; Hynes, R.O.; Burton, D.R.; McKeating, J.A.; et al. Complete replication of hepatitis c virus in cell culture. Science 2005, 309, 623–626. [Google Scholar] [CrossRef]
- Blight, K.J.; McKeating, J.A.; Rice, C.M. Highly permissive cell lines for subgenomic and genomic hepatitis c virus rna replication. J. Virol. 2002, 76, 13001–13014. [Google Scholar] [CrossRef]
- Hwang, J.Y.; Windisch, M.P.; Jo, S.; Kim, K.; Kong, S.; Kim, H.C.; Kim, S.; Kim, H.; Lee, M.E.; Kim, Y.; et al. Discovery and characterization of a novel 7-aminopyrazolo[1,5-a]pyrimidine analog as a potent hepatitis C virus inhibitor. Bioorg. Med. Chem. Lett. 2012, 22, 7297–7301. [Google Scholar] [CrossRef]
- Lee, C. Interaction of hepatitis c virus core protein with janus kinase is required for efficient production of infectious viruses. Biomol. Ther. 2013, 21, 97–106. [Google Scholar] [CrossRef]
- Jang, H.; Ryoo, S.R.; Kim, Y.K.; Yoon, S.; Kim, H.; Han, S.W.; Choi, B.S.; Kim, D.E.; Min, D.H. Discovery of hepatitis c virus ns3 helicase inhibitors by a multiplexed, high-throughput helicase activity assay based on graphene oxide. Angew. Chem. Int. Ed. Engl. 2013, 52, 2340–2344. [Google Scholar] [CrossRef]
- Kim, J.; Min, J.S.; Kim, D.; Zheng, Y.F.; Mailar, K.; Choi, W.J.; Lee, C.; Bae, S.K. A simple and sensitive liquid chromatography-tandem mass spectrometry method for trans-epsilon-viniferin quantification in mouse plasma and its application to a pharmacokinetic study in mice. J. Pharm. Biomed. Anal. 2017, 134, 116–121. [Google Scholar] [CrossRef]
- Mishra, S.; Anand, D.; Vijayarangan, N.; Ajitkumar, P. An accurate method for the qualitative detection and quantification of mycobacterial promoter activity. Open Microbiol. J. 2013, 7, 1–5. [Google Scholar] [CrossRef]
- Takaya, Y.; Terashima, K.; Ito, J.; He, Y.H.; Tateoka, M.; Yamaguchi, N.; Niwa, M. Biomimic transformation of resveratrol. Tetrahedron Lett. 2005, 61, 10285–10290. [Google Scholar] [CrossRef]
- Yao, C.S.; Lin, M.; Wang, Y.H. Synthesis of the active stilbenoids by photooxidation reaction of trans-ϵ-viniferin. Chin. J. Chem. 2004, 22, 1350–1355. [Google Scholar] [CrossRef]
- McGivern, D.R.; Masaki, T.; Williford, S.; Ingravallo, P.; Feng, Z.; Lahser, F.; Asante-Appiah, E.; Neddermann, P.; De Francesco, R.; Howe, A.Y.; et al. Kinetic analyses reveal potent and early blockade of hepatitis c virus assembly by ns5a inhibitors. Gastroenterology 2014, 147, 453–462. [Google Scholar] [CrossRef]
- Frick, D.N. Hepatitis C Viruses: Genomes and Molecular Biology; Horizon Bioscience: Norfolk, UK, 2006. [Google Scholar]
- Lam, A.M.I.; Keeney, D.; Eckert, P.Q.; Frick, D.N. Hepatitis c virus ns3 atpases/helicases from different genotypes exhibit variations in enzymatic properties. J. Virol. 2003, 77, 3950–3961. [Google Scholar] [CrossRef]
- McGivern, D.R.; Masaki, T.; Lovell, W.; Hamlett, C.; Saalau-Bethell, S.; Graham, B. Protease inhibitors block multiple functions of the ns3/4a protease-helicase during the hepatitis c virus life cycle. J. Virol. 2015, 89, 5362–5370. [Google Scholar] [CrossRef]
- Ma, Y.; Yates, J.; Liang, Y.; Lemon, S.M.; Yi, M. Ns3 helicase domains involved in infectious intracellular hepatitis c virus particle assembly. J. Virol. 2008, 82, 7624–7639. [Google Scholar] [CrossRef]
- Kohlway, A.; Pirakitikulr, N.; Ding, S.C.; Yang, F.; Luo, D.; Lindenbach, B.D.; Pyle, A.M. The linker region of ns3 plays a critical role in the replication and infectivity of hepatitis c virus. J. Virol. 2014, 88, 10970–10974. [Google Scholar] [CrossRef]
- Zghonda, N.; Yoshida, S.; Araki, M.; Kusunoki, M.; Mliki, A.; Ghorbel, A.; Miyazaki, H. Greater effectiveness of epsilon-viniferin in red wine than its monomer resveratrol for inhibiting vascular smooth muscle cell proliferation and migration. Biosci. Biotechnol. Biochem. 2011, 75, 1259–1267. [Google Scholar] [CrossRef]
- Courtois, A.; Jourdes, M.; Dupin, A.; Lapeze, C.; Renouf, E.; Biais, B.; Teissedre, P.L.; Merillon, J.M.; Richard, T.; Krisa, S. In vitro glucuronidation and sulfation of epsilon-viniferin, a resveratrol dimer, in humans and rats. Molecules 2017, 22, 733. [Google Scholar] [CrossRef]
- Vion, E.; Page, G.; Bourdeaud, E.; Paccalin, M.; Guillard, J.; Rioux Bilan, A. Trans epsilon-viniferin is an amyloid-beta disaggregating and anti-inflammatory drug in a mouse primary cellular model of alzheimer’s disease. Mol. Cell. Neurosci. 2018, 88, 1–6. [Google Scholar] [CrossRef]
- Riviere, C.; Pawlus, A.D.; Merillon, J.M. Natural stilbenoids: Distribution in the plant kingdom and chemotaxonomic interest in vitaceae. Nat. Prod. Rep. 2012, 29, 1317–1333. [Google Scholar] [CrossRef]
- Wu, B.; Basu, S.; Meng, S.; Wang, X.; Hu, M. Regioselective sulfation and glucuronidation of phenolics: Insights into the structural basis. Curr. Drug Metab. 2011, 12, 900–916. [Google Scholar] [CrossRef]
- Aumont, V.; Krisa, S.; Battaglia, E.; Netter, P.; Richard, T.; Merillon, J.M.; Magdalou, J.; Sabolovic, N. Regioselective and stereospecific glucuronidation of trans- and cis-resveratrol in human. Arch. Biochem. Biophys. 2001, 393, 281–289. [Google Scholar] [CrossRef]
- Laxman, T.S.; Puttrevu, S.K.; Pradhan, R.; Mishra, A.; Verma, S.; Chhonker, Y.S.; Srivastava, S.; Singh, S.P.; Sashidhara, K.V.; Bhatta, R.S. Pharmacokinetics, metabolism, bioavailability, tissue distribution and excretion studies of 16alpha-hydroxycleroda-3, 13(14) z -dien-15, 16-olide-a novel hmg-coa reductase inhibitor. Naunyn Schmiedebergs Arch. Pharmacol. 2018, 391, 965–973. [Google Scholar] [CrossRef]
- Wenzel, E.; Soldo, T.; Erbersdobler, H.; Somoza, V. Bioactivity and metabolism of trans-resveratrol orally administered to wistar rats. Mol. Nutr. Food Res. 2005, 49, 482–494. [Google Scholar] [CrossRef]
- Gastaminza, P.; Whitten-Bauer, C.; Chisari, F.V. Unbiased probing of the entire hepatitis c virus life cycle identifies clinical compounds that target multiple aspects of the infection. Proc. Natl. Acad. Sci. USA 2010, 107, 291–296. [Google Scholar] [CrossRef]
- Bagaglio, S.; Uberti-Foppa, C.; Morsica, G. Resistance mechanisms in hepatitis C virus: Implications for direct-acting antiviral use. Drugs 2017, 77, 1043–1055. [Google Scholar] [CrossRef]
- Fernandez-Sanles, A.; Rios-Marco, P.; Romero-Lopez, C.; Berzal-Herranz, A. Functional information stored in the conserved structural rna domains of flavivirus genomes. Front. Microbiol. 2017, 8, 546. [Google Scholar] [CrossRef]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Antiviral Assays Used | EVF | SVF-5Ac | SVF | ||||||
|---|---|---|---|---|---|---|---|---|---|
| EC50 (M) | CC50 (M) | T1/2 (h) | EC50 (M) | CC50 (M) | T1/2 (h) | EC50 (M) | CC50 (M) | T1/2 (h) | |
| GT 2a luciferase reporter assay | 0.1 | >10 | 15.5 | 2.4 | >10 | 12.6 | 0.2 | >10 | 21.9 |
| GT 2a RT-PCR | 2.9 | N/A | 10 | 4.7 | N/A | 11.1 | 9.3 | N/A | 62.5 |
| GT 1b RT-PCR | 7 | N/A | 10.9 | 10.4 | N/A | 18.1 | 1.7 | N/A | 7.7 |
| GT 2a Western blot | 1.2 | N/A | 35.1 | 1 | N/A | >72 | 6.4 | N/A | 37.1 |
| GT 1b Western blot | 0.9 | N/A | 58.2 | 0.8 | N/A | 29.6 | 0.9 | N/A | 13.1 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lee, S.; Mailar, K.; Kim, M.I.; Park, M.; Kim, J.; Min, D.-H.; Heo, T.-H.; Bae, S.K.; Choi, W.; Lee, C. Plant-Derived Purification, Chemical Synthesis, and In Vitro/In Vivo Evaluation of a Resveratrol Dimer, Viniferin, as an HCV Replication Inhibitor. Viruses 2019, 11, 890. https://doi.org/10.3390/v11100890
Lee S, Mailar K, Kim MI, Park M, Kim J, Min D-H, Heo T-H, Bae SK, Choi W, Lee C. Plant-Derived Purification, Chemical Synthesis, and In Vitro/In Vivo Evaluation of a Resveratrol Dimer, Viniferin, as an HCV Replication Inhibitor. Viruses. 2019; 11(10):890. https://doi.org/10.3390/v11100890
Chicago/Turabian StyleLee, Sungjin, Karabasappa Mailar, Mi Il Kim, Minkyung Park, Jiseon Kim, Dal-Hee Min, Tae-Hwe Heo, Soo Kyung Bae, Wonjun Choi, and Choongho Lee. 2019. "Plant-Derived Purification, Chemical Synthesis, and In Vitro/In Vivo Evaluation of a Resveratrol Dimer, Viniferin, as an HCV Replication Inhibitor" Viruses 11, no. 10: 890. https://doi.org/10.3390/v11100890
APA StyleLee, S., Mailar, K., Kim, M. I., Park, M., Kim, J., Min, D.-H., Heo, T.-H., Bae, S. K., Choi, W., & Lee, C. (2019). Plant-Derived Purification, Chemical Synthesis, and In Vitro/In Vivo Evaluation of a Resveratrol Dimer, Viniferin, as an HCV Replication Inhibitor. Viruses, 11(10), 890. https://doi.org/10.3390/v11100890