Degenerate PCR Primers to Reveal the Diversity of Giant Viruses in Coastal Waters

 ,
,
Abstract
1. Introduction
2. Materials and Methods
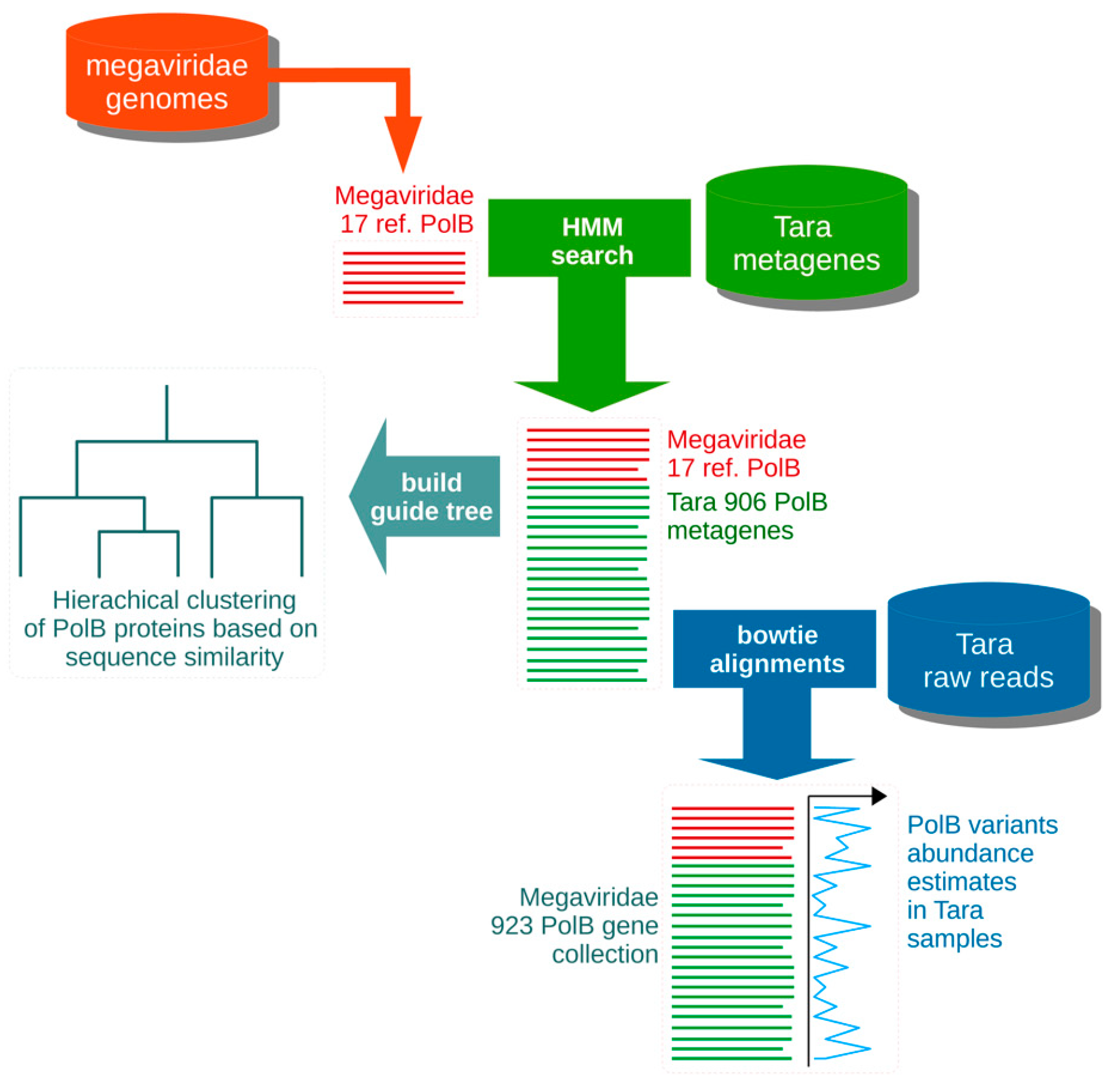
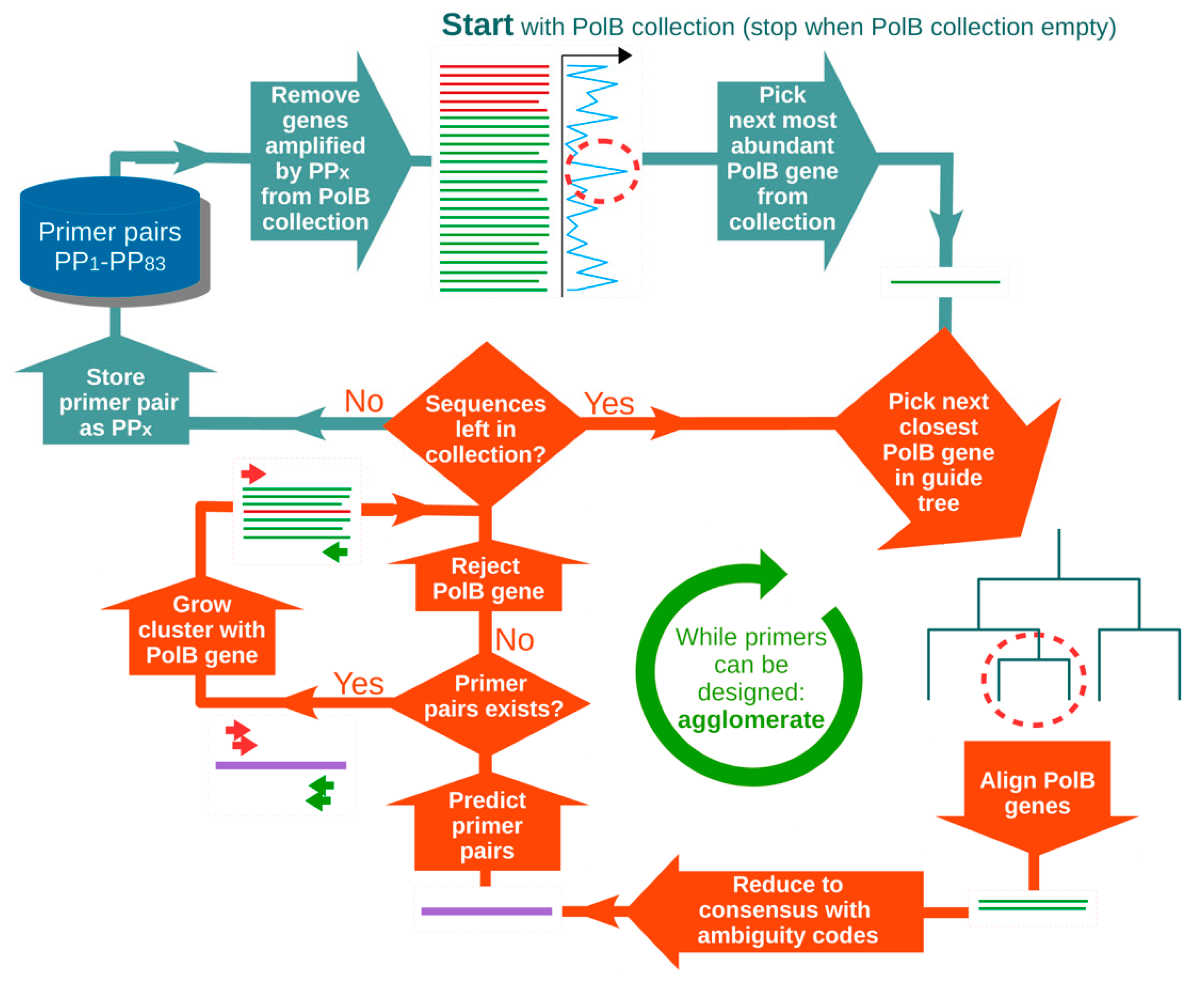
2.1. Primer Design
2.2. Sampling and DNA Extraction
2.3. PCR Amplification and Sequencing
2.4. Quality Control and Merging
2.5. Taxonomic Assignment
2.6. Clustering and Phylogeny
2.7. Data Availability
2.8. Accession Number
3. Results
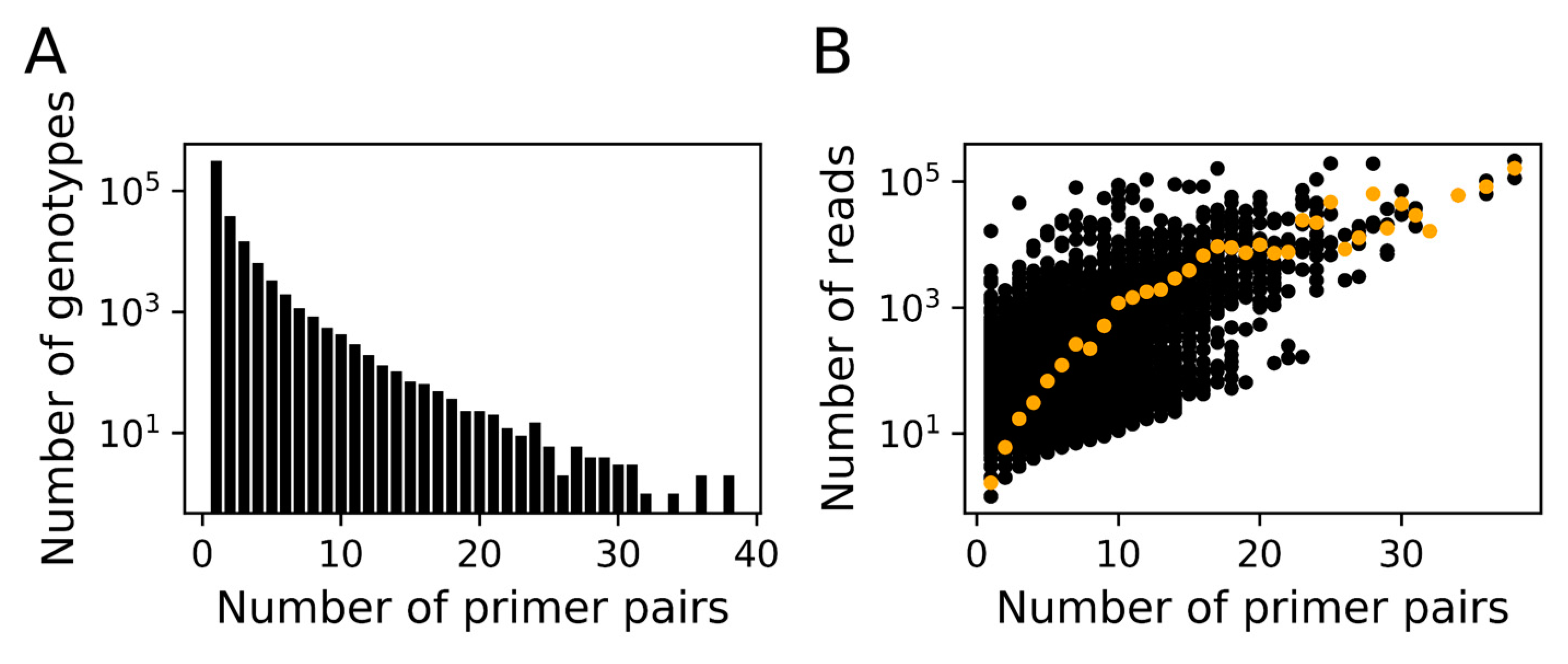
3.1. In silico Assessment of MEGAPRIMER
3.2. MEGAPRIMER Effectively Amplified Megaviridae polB
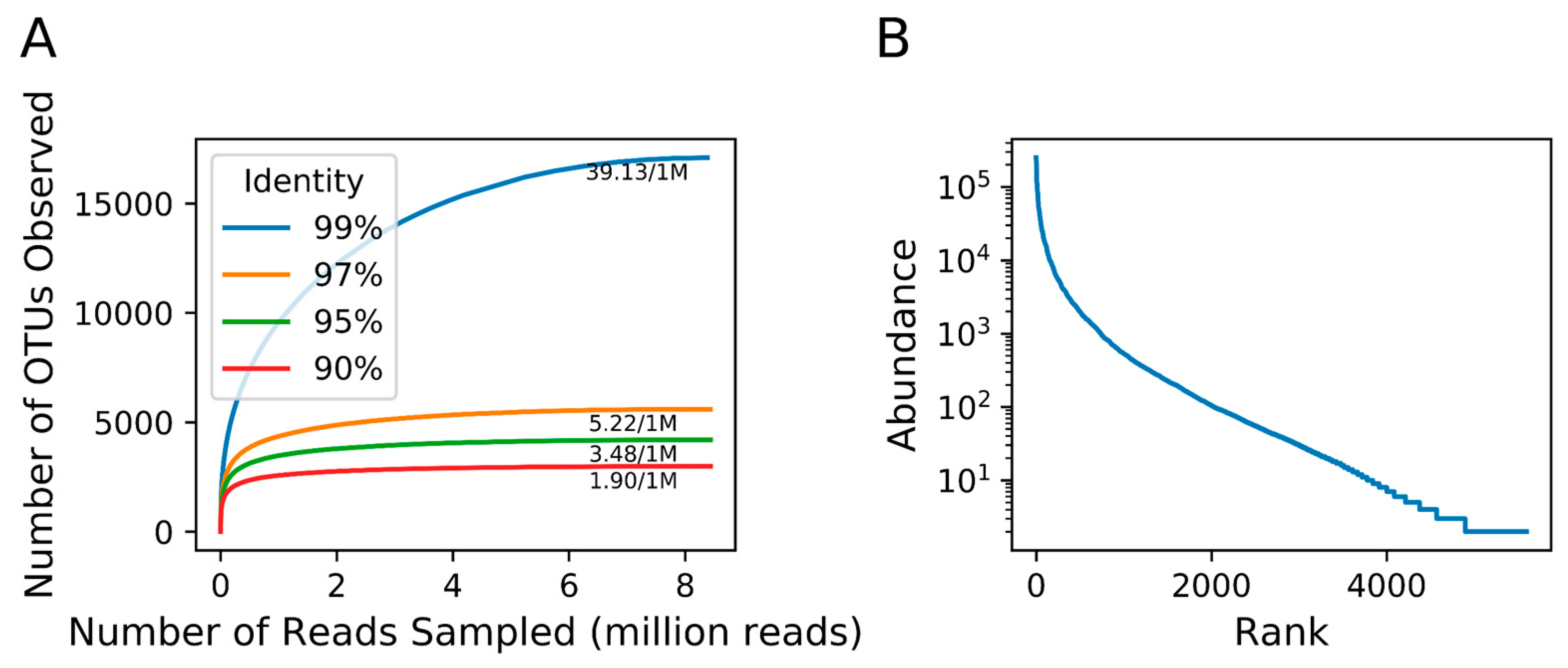
3.3. Richness of Megaviridae OTUs
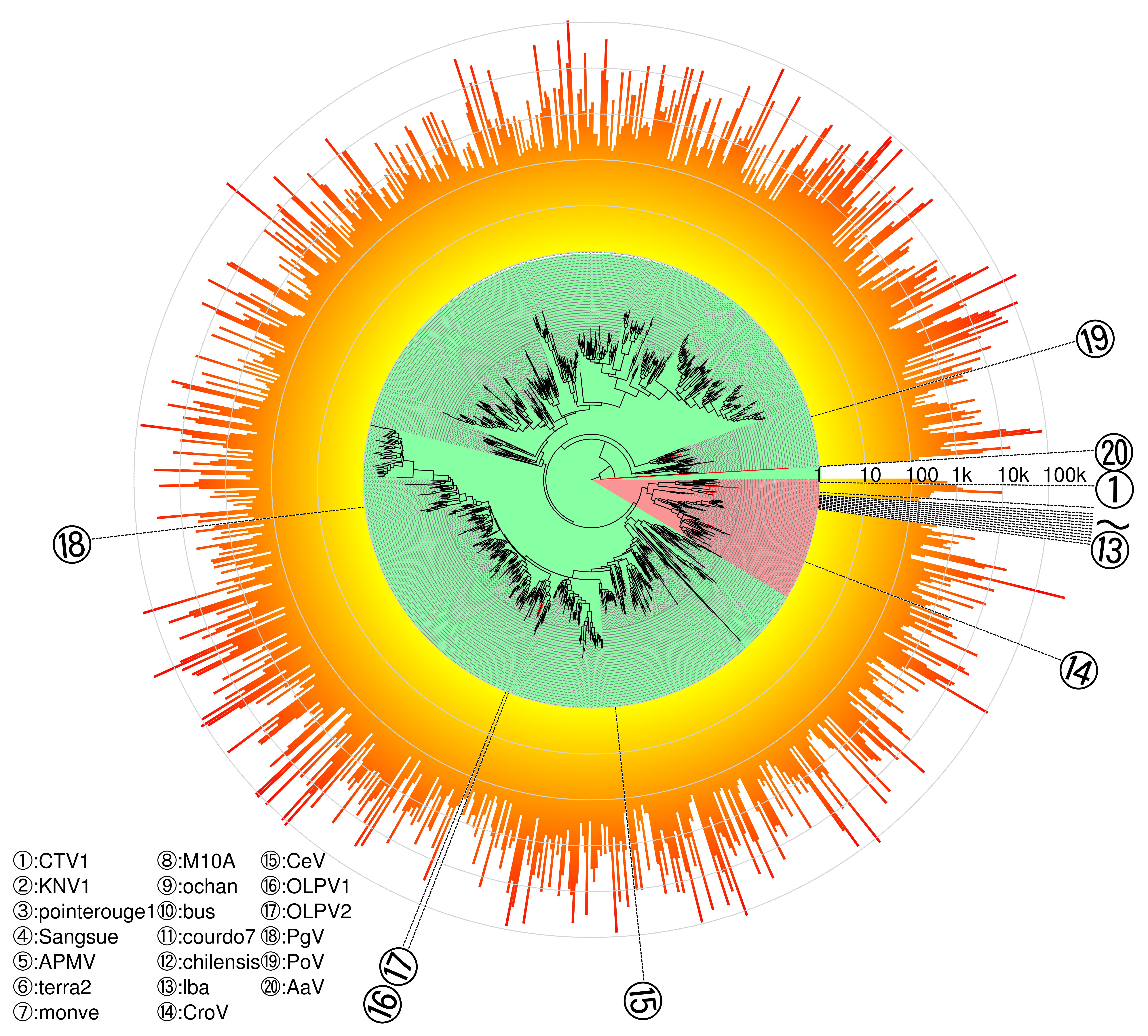
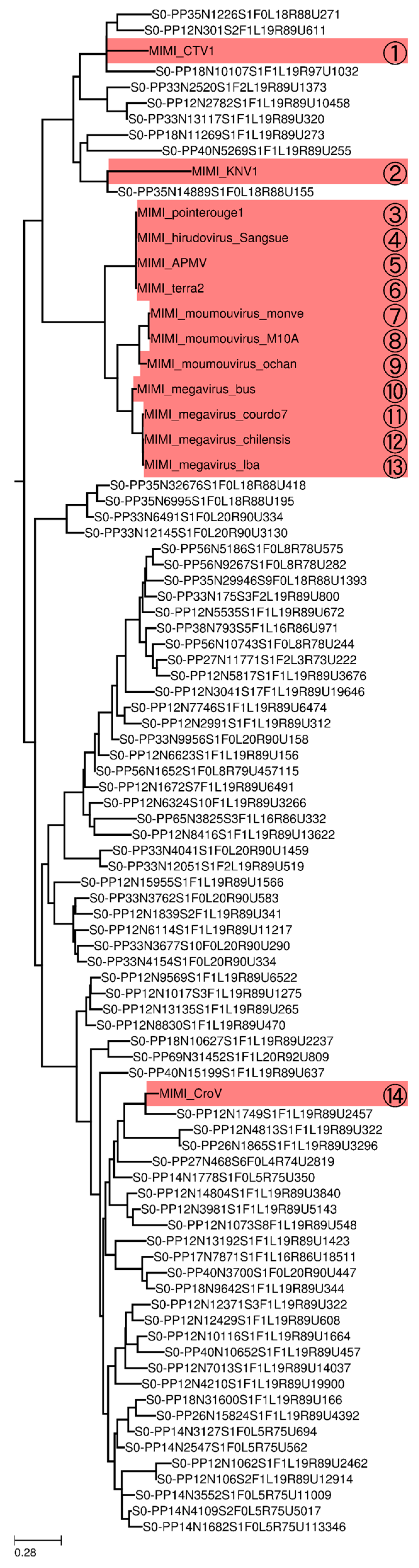
3.4. Phylogeny of Megaviridae polB Meta-Barcodes
4. Discussion
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Suttle, C.A. Marine viruses—Major players in the global ecosystem. Nat. Rev. Microbiol. 2007, 5, 801. [Google Scholar] [CrossRef] [PubMed]
- Bergh, Ø.; BØrsheim, K.Y.; Bratbak, G.; Heldal, M. High abundance of viruses found in aquatic environments. Nature 1989, 340, 467–468. [Google Scholar] [CrossRef] [PubMed]
- Wigington, C.H.; Sonderegger, D.; Brussaard, C.P.D.; Buchan, A.; Finke, J.F.; Fuhrman, J.A.; Lennon, J.T.; Middelboe, M.; Suttle, C.A.; Stock, C.; et al. Re-examination of the relationship between marine virus and microbial cell abundances. Nat. Microbiol. 2016, 1, 15024. [Google Scholar] [CrossRef] [PubMed]
- Fuhrman, J.A. Marine viruses and their biogeochemical and ecological effects. Nature 1999, 399, 541–548. [Google Scholar] [CrossRef] [PubMed]
- Rohwer, F.; Prangishvili, D.; Lindell, D. Roles of viruses in the environment. Environ. Microbiol. 2009, 11, 2771–2774. [Google Scholar] [CrossRef] [PubMed]
- Danovaro, R.; Corinaldesi, C.; Dell’Anno, A.; Fuhrman, J.A.; Middelburg, J.J.; Noble, R.T.; Suttle, C.A. Marine viruses and global climate change. FEMS Microbiol. Rev. 2011, 35, 993–1034. [Google Scholar] [CrossRef] [PubMed]
- Yoshida, T.; Nishimura, Y.; Watai, H.; Haruki, N.; Morimoto, D.; Kaneko, H.; Honda, T.; Yamamoto, K.; Hingamp, P.; Sako, Y.; et al. Locality and diel cycling of viral production revealed by a 24 h time course cross-omics analysis in a coastal region of Japan. ISME J. 2018, 12, 1287–1295. [Google Scholar] [CrossRef] [PubMed]
- Guidi, L.; Chaffron, S.; Bittner, L.; Eveillard, D.; Larhlimi, A.; Roux, S.; Darzi, Y.; Audic, S.; Berline, L.; Brum, J.; et al. Plankton networks driving carbon export in the oligotrophic ocean. Nature 2016, 532, 465–470. [Google Scholar] [CrossRef] [PubMed]
- Roux, S.; Brum, J.R.; Dutilh, B.E.; Sunagawa, S.; Duhaime, M.B.; Loy, A.; Poulos, B.T.; Solonenko, N.; Lara, E.; Poulain, J.; et al. Ecogenomics and potential biogeochemical impacts of globally abundant ocean viruses. Nature 2016, 537, 689–693. [Google Scholar] [CrossRef] [PubMed]
- Nishimura, Y.; Watai, H.; Honda, T.; Mihara, T.; Omae, K.; Roux, S.; Blanc-Mathieu, R.; Yamamoto, K.; Hingamp, P.; Sako, Y.; et al. Environmental viral genomes shed new light on virus-host interactions in the ocean. Msphere 2017, 2, e00359-16. [Google Scholar] [CrossRef] [PubMed]
- Chow, C.-E.T.; Suttle, C.A. Biogeography of viruses in the sea. Annu. Rev. Virol. 2015, 2, 41–66. [Google Scholar] [CrossRef] [PubMed]
- Hingamp, P.; Grimsley, N.; Acinas, S.G.; Clerissi, C.; Subirana, L.; Poulain, J.; Ferrera, I.; Sarmento, H.; Villar, E.; Lima-Mendez, G.; et al. Exploring nucleo-cytoplasmic large DNA viruses in Tara Oceans microbial metagenomes. ISME J. 2013, 7, 1678–1695. [Google Scholar] [CrossRef] [PubMed]
- Carradec, Q.; Pelletier, E.; Da Silva, C.; Alberti, A.; Seeleuthner, Y.; Blanc-Mathieu, R.; Lima-Mendez, G.; Rocha, F.; Tirichine, L.; Labadie, K.; et al. A global ocean atlas of eukaryotic genes. Nat. Commun. 2018, 9, 373. [Google Scholar] [CrossRef] [PubMed]
- Arslan, D.; Legendre, M.; Seltzer, V.; Abergel, C.; Claverie, J.-M. Distant Mimivirus relative with a larger genome highlights the fundamental features of Megaviridae. Proc. Natl. Acad. Sci. USA 2011, 108, 17486–17491. [Google Scholar] [CrossRef] [PubMed]
- Santini, S.; Jeudy, S.; Bartoli, J.; Poirot, O.; Lescot, M.; Abergel, C.; Barbe, V.; Wommack, K.E.; Noordeloos, A.M.A.; Brussaard, P.D.C.; et al. Genome of Phaeocystis globosa virus PgV-16T highlights the common ancestry of the largest known DNA viruses infecting eukaryotes. Proc. Natl. Acad. Sci. USA 2013, 110, 10800–10805. [Google Scholar] [CrossRef] [PubMed]
- Mihara, T.; Koyano, H.; Hingamp, P.; Grimsley, N.; Goto, S.; Ogata, H. Taxon richness of “Megaviridae” exceeds those of Bacteria and Archaea in the ocean. Microbes Env. 2018, 33, 162–171. [Google Scholar] [CrossRef] [PubMed]
- Claverie, J.-M.; Ogata, H.; Audic, S.; Abergel, C.; Suhre, K.; Fournier, P.-E. Mimivirus and the emerging concept of “giant” virus. Virus Res. 2006, 117, 133–144. [Google Scholar] [CrossRef] [PubMed]
- Van Etten, J.L. Another really, really big virus. Viruses 2011, 3, 32–46. [Google Scholar] [CrossRef] [PubMed]
- Fischer, M.G.; Condit, R.C. Editorial introduction to “Giant Viruses” special issue of Virology. Virology 2014, 466–467, 1–2. [Google Scholar] [CrossRef] [PubMed]
- Gallot-Lavallée, L.; Blanc, G.; Claverie, J.-M. Comparative genomics of Chrysochromulina ericina virus and other microalga-infecting large DNA viruses highlights their intricate evolutionary relationship with the established Mimiviridae family. J. Virol. 2017, 91. [Google Scholar] [CrossRef] [PubMed]
- Raoult, D.; Audic, S.; Robert, C.; Abergel, C.; Renesto, P.; Ogata, H.; Scola, B.L.; Suzan, M.; Claverie, J.-M. The 1.2-Megabase genome sequence of Mimivirus. Science 2004, 306, 1344–1350. [Google Scholar] [CrossRef] [PubMed]
- Fischer, M.G.; Allen, M.J.; Wilson, W.H.; Suttle, C.A. Giant virus with a remarkable complement of genes infects marine zooplankton. Proc. Natl. Acad. Sci. USA 2010, 107, 19508–19513. [Google Scholar] [CrossRef] [PubMed]
- Moniruzzaman, M.; LeCleir, G.R.; Brown, C.M.; Gobler, C.J.; Bidle, K.D.; Wilson, W.H.; Wilhelm, S.W. Genome of brown tide virus (AaV), the little giant of the Megaviridae, elucidates NCLDV genome expansion and host–virus coevolution. Virology 2014, 466–467, 60–70. [Google Scholar] [CrossRef] [PubMed]
- Gallot-Lavallée, L.; Pagarete, A.; Legendre, M.; Santini, S.; Sandaa, R.-A.; Himmelbauer, H.; Ogata, H.; Bratbak, G.; Claverie, J.-M. The 474-kilobase-pair complete genome sequence of CeV-01B, a virus infecting Haptolina (Chrysochromulina) ericina (Prymnesiophyceae). Genome Announc. 2015, 3, e01413–e01415. [Google Scholar] [CrossRef] [PubMed]
- Colson, P.; De Lamballerie, X.; Yutin, N.; Asgari, S.; Bigot, Y.; Bideshi, D.K.; Cheng, X.-W.; Federici, B.A.; Van Etten, J.L.; Koonin, E.V.; et al. “Megavirales”, a proposed new order for eukaryotic nucleocytoplasmic large DNA viruses. Arch. Virol. 2013, 158, 2517–2521. [Google Scholar] [CrossRef] [PubMed]
- Chen, F.; Suttle, C.A. Amplification of DNA polymerase gene fragments from viruses infecting microalgae. Appl. Environ. Microbiol. 1995, 61, 1274–1278. [Google Scholar] [PubMed]
- Chen, F.; Suttle, C.A.; Short, S.M. Genetic diversity in marine algal virus communities as revealed by sequence analysis of DNA polymerase genes. Appl. Environ. Microbiol. 1996, 62, 2869–2874. [Google Scholar] [PubMed]
- Larsen, J.B.; Larsen, A.; Bratbak, G.; Sandaa, R.-A. Phylogenetic analysis of members of the Phycodnaviridae virus family, using amplified fragments of the major capsid protein gene. Appl. Environ. Microbiol. 2008, 74, 3048–3057. [Google Scholar] [CrossRef] [PubMed]
- Johannessen, T.V.; Larsen, A.; Bratbak, G.; Pagarete, A.; Edvardsen, B.; Egge, E.D.; Sandaa, R.A. Seasonal dynamics of haptophytes and dsDNA algal viruses suggest complex virus-host relationship. Viruses 2017, 9, 84. [Google Scholar] [CrossRef] [PubMed]
- Wilson, W.H.; Gilg, I.C.; Duarte, A.; Ogata, H. Development of DNA mismatch repair gene, MutS, as a diagnostic marker for detection and phylogenetic analysis of algal Megaviruses. Virology 2014, 466–467, 123–128. [Google Scholar] [CrossRef] [PubMed]
- Clerissi, C.; Grimsley, N.; Subirana, L.; Maria, E.; Oriol, L.; Ogata, H.; Moreau, H.; Desdevises, Y. Prasinovirus distribution in the Northwest Mediterranean Sea is affected by the environment and particularly by phosphate availability. Virology 2014, 466–467, 146–157. [Google Scholar] [CrossRef] [PubMed]
- Sunagawa, S.; Coelho, L.P.; Chaffron, S.; Kultima, J.R.; Labadie, K.; Salazar, G.; Djahanschiri, B.; Zeller, G.; Mende, D.R.; Alberti, A.; et al. Structure and function of the global ocean microbiome. Science 2015, 348, 1261359. [Google Scholar] [CrossRef] [PubMed]
- Yamada, K.D.; Tomii, K.; Katoh, K. Application of the MAFFT sequence alignment program to large data—Reexamination of the usefulness of chained guide trees. Bioinformatics 2016, 32, 3246–3251. [Google Scholar] [CrossRef] [PubMed]
- Suyama, M.; Torrents, D.; Bork, P. PAL2NAL: Robust conversion of protein sequence alignments into the corresponding codon alignments. Nucleic Acids Res. 2006, 34, W609–W612. [Google Scholar] [CrossRef] [PubMed]
- Eddy, S.R. Profile hidden Markov models. Bioinformatics 1998, 14, 755–763. [Google Scholar] [CrossRef] [PubMed]
- La Scola, B.; Campocasso, A.; N’Dong, R.; Fournous, G.; Barrassi, L.; Flaudrops, C.; Raoult, D. Tentative Characterization of new environmental giant viruses by MALDI-TOF mass spectrometry. Intervirology 2010, 53, 344–353. [Google Scholar] [CrossRef] [PubMed]
- Matsen, F.A.; Kodner, R.B.; Armbrust, E.V. Pplacer: Linear time maximum-likelihood and Bayesian phylogenetic placement of sequences onto a fixed reference tree. BMC Bioinform. 2010, 11, 538. [Google Scholar] [CrossRef] [PubMed]
- Fu, L.; Niu, B.; Zhu, Z.; Wu, S.; Li, W. CD-HIT: Accelerated for clustering the next-generation sequencing data. Bioinformatics 2012, 28, 3150–3152. [Google Scholar] [CrossRef] [PubMed]
- Langmead, B.; Salzberg, S.L. Fast gapped-read alignment with Bowtie 2. Nat. Methods 2012, 9, 357–359. [Google Scholar] [CrossRef] [PubMed]
- Untergasser, A.; Cutcutache, I.; Koressaar, T.; Ye, J.; Faircloth, B.C.; Remm, M.; Rozen, S.G. Primer3—New capabilities and interfaces. Nucleic Acids Res. 2012, 40, e115. [Google Scholar] [CrossRef] [PubMed]
- Rice, P.; Longden, I.; Bleasby, A. EMBOSS: The European Molecular Biology Open Software Suite. Trends Genet. 2000, 16, 276–277. [Google Scholar] [CrossRef]
- Kimura, S.; Yoshida, T.; Hosoda, N.; Honda, T.; Kuno, S.; Kamiji, R.; Hashimoto, R.; Sako, Y. Diurnal infection patterns and impact of Microcystis cyanophages in a Japanese pond. Appl. Environ. Microbiol 2012, 78, 5805–5811. [Google Scholar] [CrossRef] [PubMed]
- 16S Metagenomic Sequencing Library Preparation. Available online: https://support.illumina.com/documents/documentation/chemistry_documentation/16s/16s-metagenomic-library-prep-guide-15044223-b.pdf (accessed on 13 September 2018).
- Martin, M. Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnet J. 2011, 17, 10–12. [Google Scholar] [CrossRef]
- Magoč, T.; Salzberg, S.L. FLASH: Fast length adjustment of short reads to improve genome assemblies. Bioinformatics 2011, 27, 2957–2963. [Google Scholar] [CrossRef] [PubMed]
- Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: A flexible trimmer for Illumina sequence data. Bioinformatics 2014, 30, 2114–2120. [Google Scholar] [CrossRef] [PubMed]
- Edgar, R.C.; Haas, B.J.; Clemente, J.C.; Quince, C.; Knight, R. UCHIME improves sensitivity and speed of chimera detection. Bioinformatics 2011, 27, 2194–2200. [Google Scholar] [CrossRef] [PubMed]
- Caporaso, J.G.; Kuczynski, J.; Stombaugh, J.; Bittinger, K.; Bushman, F.D.; Costello, E.K.; Fierer, N.; Pena, A.G.; Goodrich, J.K.; Gordon, J.I.; et al. QIIME allows analysis of high-throughput community sequencing data. Nat. Methods 2010, 7, 335–336. [Google Scholar] [CrossRef] [PubMed]
- Cock, P.J.A.; Antao, T.; Chang, J.T.; Chapman, B.A.; Cox, C.J.; Dalke, A.; Friedberg, I.; Hamelryck, T.; Kauff, F.; Wilczynski, B.; et al. Biopython: Freely available Python tools for computational molecular biology and bioinformatics. Bioinformatics 2009, 25, 1422–1423. [Google Scholar] [CrossRef] [PubMed]
- Altschul, S.F.; Gish, W.; Miller, W.; Myers, E.W.; Lipman, D.J. Basic local alignment search tool. J. Mol. Biol. 1990, 215, 403–410. [Google Scholar] [CrossRef]
- Hunter, J.D. Matplotlib: A 2D Graphics Environment. Comput. Sci. Eng. 2007, 9, 90–95. [Google Scholar] [CrossRef]
- Huerta-Cepas, J.; Serra, F.; Bork, P. ETE 3: Reconstruction, analysis, and visualization of phylogenomic data. Mol.Biol. Evol. 2016, 33, 1635–1638. [Google Scholar] [CrossRef] [PubMed]
- Deeg, C.M.; Chow, C.-E.T.; Suttle, C.A. The kinetoplastid-infecting Bodo saltans virus (BsV), a window into the most abundant giant viruses in the sea. eLife 2018, 7, e33014. [Google Scholar] [CrossRef] [PubMed]
- Schulz, F.; Yutin, N.; Ivanova, N.N.; Ortega, D.R.; Lee, T.K.; Vierheilig, J.; Daims, H.; Horn, M.; Wagner, M.; Jensen, G.J.; et al. Giant viruses with an expanded complement of translation system components. Science 2017, 356, 82–85. [Google Scholar] [CrossRef] [PubMed]
- Monier, A.; Larsen, J.B.; Sandaa, R.A.; Bratbak, G.; Claverie, J.M.; Ogata, H. Marine mimivirus relatives are probably large algal viruses. Virol. J. 2008, 5, 12. [Google Scholar] [CrossRef] [PubMed]
- De Vargas, C.; Audic, S.; Henry, N.; Decelle, J.; Mahé, F.; Logares, R.; Lara, E.; Berney, C.; Bescot, N.L.; Probert, I.; et al. Eukaryotic plankton diversity in the sunlit ocean. Science 2015, 348, 1261605. [Google Scholar] [CrossRef] [PubMed]
- Clerissi, C.; Grimsley, N.; Ogata, H.; Hingamp, P.; Poulain, J.; Desdevises, Y. Unveiling of the Diversity of Prasinoviruses (Phycodnaviridae) in Marine samples by using high-throughput sequencing analyses of PCR-amplified DNA polymerase and major capsid protein genes. Appl. Environ. Microbiol. 2014, 80, 3150–3160. [Google Scholar] [CrossRef] [PubMed]
- Moniruzzaman, M.; Gann, E.R.; LeCleir, G.R.; Kang, Y.; Gobler, C.J.; Wilhelm, S.W. Diversity and dynamics of algal Megaviridae members during a harmful brown tide caused by the pelagophyte, Aureococcus anophagefferens. FEMS Microbiol. Ecol. 2016, 92, fiw058. [Google Scholar] [CrossRef] [PubMed]
- Sandaa, R.A.; Heldal, M.; Castberg, T.; Thyrhaug, R.; Bratbak, G. Isolation and characterization of two viruses with large genome size infecting Chrysochromulina ericina (Prymnesiophyceae) and Pyramimonas orientalis (Prasinophyceae). Virology 2001, 290, 272–280. [Google Scholar] [CrossRef] [PubMed]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Raw | High Quality | With Primers | Merged | BLASTP Validated | Pplacer Validated |
|---|---|---|---|---|---|
| 16,677,495 | 11,217,759 | 10,211,698 | 9,377,470 | 8,475,525 | 8,440,486 |
| OTUs | Clustering Threshold | |||
|---|---|---|---|---|
| 99% | 97% | 95% | 90% | |
| Total number of OTUs | 75,923 | 13,243 | 6123 | 3426 |
| Number of singleton OTUs | 58,837 | 7648 | 1928 | 442 |
| Number of non-singleton OTUs | 17,086 | 5595 | 4195 | 2984 |
| Slope at the tail of rarefaction curve (OTU/1,000,000 reads) | 39.13 | 5.22 | 3.48 | 1.90 |
| Number of sequences included in non-singleton OTUs | 8,381,648 | 8,432,837 | 8,438,557 | 8,440,043 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Li, Y.; Hingamp, P.; Watai, H.; Endo, H.; Yoshida, T.; Ogata, H. Degenerate PCR Primers to Reveal the Diversity of Giant Viruses in Coastal Waters. Viruses 2018, 10, 496. https://doi.org/10.3390/v10090496
Li Y, Hingamp P, Watai H, Endo H, Yoshida T, Ogata H. Degenerate PCR Primers to Reveal the Diversity of Giant Viruses in Coastal Waters. Viruses. 2018; 10(9):496. https://doi.org/10.3390/v10090496
Chicago/Turabian StyleLi, Yanze, Pascal Hingamp, Hiroyasu Watai, Hisashi Endo, Takashi Yoshida, and Hiroyuki Ogata. 2018. "Degenerate PCR Primers to Reveal the Diversity of Giant Viruses in Coastal Waters" Viruses 10, no. 9: 496. https://doi.org/10.3390/v10090496
APA StyleLi, Y., Hingamp, P., Watai, H., Endo, H., Yoshida, T., & Ogata, H. (2018). Degenerate PCR Primers to Reveal the Diversity of Giant Viruses in Coastal Waters. Viruses, 10(9), 496. https://doi.org/10.3390/v10090496