Abstract
Genomic instability facilitates the evolution of cells, tissues, organs, and species. The progression of human malignancies can be regarded as the accumulation of genomic instability, which confers a high evolutionary potential for tumor cells to adapt to continuous changes in the tumor microenvironment. Nasopharyngeal carcinoma (NPC) is a head-and-neck squamous-cell carcinoma closely associated with Epstein–Barr virus (EBV) infection. NPC progression is driven by a combination of accumulated genomic instability and persistent EBV infection. Here, we present a review of the key characteristics of genomic instability in NPC and the profound implications of EBV infection. We further discuss the significance of profiling genomic instability for the assessment of disease progression and treatment efficacy, as well as the opportunities and challenges of targeted therapies for NPC based on its unique genomic instability.
1. Introduction
Nasopharyngeal carcinoma (NPC) is a head-and-neck squamous-cell carcinoma (HNSCC) that originates in the nasopharyngeal epithelium [1]. According to the World Health Organization (WHO) histopathological classification, NPC is classified as keratinizing squamous-cell carcinoma, non-keratinizing carcinoma, and basaloid squamous-cell carcinoma [2]. In addition, non-keratinizing nasopharyngeal carcinoma is subdivided into differentiated non-keratinizing carcinoma (WHO classification, type II) and undifferentiated non-keratinizing carcinoma (WHO classification, type III) [2,3]. NPC has a high incidence in southern China and some Southeast Asian regions, yet is extremely rare in most parts of the world [1]. In areas with a high incidence of NPC in China, more than 90% of patients are histopathologically diagnosed with undifferentiated non-keratinizing squamous-cell carcinoma, and latent Epstein–Barr virus (EBV) infection can be detected in almost all of these cases [1].
The progressive accumulation of genomic instability and persistent EBV infection have encouraged the development of NPC [4]. First, exposure to carcinogens such as nitrosamines and polycyclic aromatic hydrocarbons impairs DNA damage repair and induces a series of molecular genetic pathological alterations in the nasopharyngeal epithelium, which in turn induces genomic instability and creates favorable conditions for EBV infection [5]. The EBV genome, EBV-encoded proteins, and EBV non-coding RNAs further exacerbate genomic instability in the host cells [5]. NPC cells continuously accumulate mutations, maintain EBV infection, enhance their resilience in the tumor microenvironment (TME), and acquire the capacity for malignant biological behaviors such as maintaining dysregulated proliferation, inducing tumor-associated angiogenesis, generating regional invasion, and eventually forming remote metastasis. Exploring the mechanism through which EBV infection promotes genomic instability in host cells could not only provide a comprehensive understanding of NPC pathogenesis, but also provide clinical translational insight for exploring tumor-targeted therapeutic strategies. In this review, we summarize the linkage mechanism between host genomic instability and EBV infection as well as how this cooperation drives the multistep progression of NPC carcinogenesis.
2. Characteristics of Genomic Instability in NPC
The characteristics of genomic instability in NPC are well-recognized. Although a wide variety of somatic mutations are found, copy-number variations (CNVs) are more frequent in NPC than in other HNSCCs, as determined by whole-genome sequencing (WGS) [6,7]. During NPC development, functional genes enriched in various signaling pathways are altered through different mutational forms or structural changes (Figure 1), leading to different phenotypic switches in cellular biological functions. During NPC progression, genomic instability is aggravated by somatic mutations, CNVs, and structural changes that gradually accumulate in nasopharyngeal epithelial cells, accompanied by the enhancement of adaptive plasticity in response to the TME. Consequently, cellular malignant transformation becomes irreversible and nasopharyngeal epithelial cells evolve into invasive NPC cells.
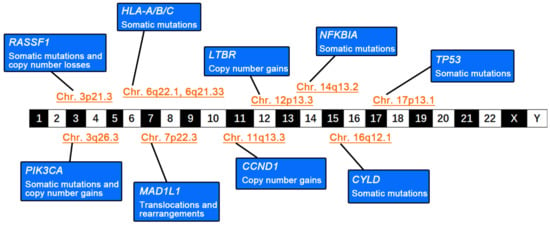
Figure 1.
Genomic instability in nasopharyngeal carcinoma (NPC). The schematic diagram summarizes the genetic abnormalities and alterations in various tumor-associated functional genes and their locations on the chromosome.
2.1. Diverse Somatic Mutations
Somatic mutations in oncogene-encoding genes, such as epidermal growth factor receptor (EGFR), phosphatidylinositol-4,5-bisphosphate 3-kinase catalytic subunit alpha (PIK3CA), Kirsten rat sarcoma viral oncogene homolog (KRAS), and Akt serine/threonine kinase 1 (AKT1), are more diverse in NPC than in other cancers [7]. Somatic mutations in NPC include missense mutations, nonsense mutations, frameshift mutations, and splicing regions [7]. Functional genes responsible for maintaining genomic stability are frequently mutated in NPC. The tumor protein p53 (TP53) gene functions to repair DNA damage and removes cells with unfixable injured genetic integrity by arresting them in the G1/S cell phase [8]. Bruce et al. reported a high rate (28.6%, 20/70) of somatic mutations in TP53 in patients with NPC [9]. The enrichment of somatic mutations in the classical NF-κB signaling axis is considered the key driver for the malignant transformation of epithelial cells. Dysfunction of the negative regulation of the NF-κB pathway caused by somatic mutations, combined with overactivated NF-κB signaling stimulated by the expression of EBV-encoded latent membrane protein 1 (LMP1), contributes to NPC pathogenesis [10]. Li et al. identified multiple somatic mutations in the genes encoding negative regulatory molecules of the NF-κB pathway in 41% (43/105) of patients with NPC by performing whole-exon sequencing (WES), including the cylindromatosis (CYLD), TNF receptor-associated factor 3 (TRAF3), and NFKB inhibitor alpha (NFKBIA) genes [11]. CYLD encodes deubiquitinating enzymes (DUBs), which regulate cell growth and invasiveness by untying K63-linked ubiquitin chains through deubiquitination [12]. TRAF3 exerts a variety of biological functions by interacting with membrane receptors, E3 ubiquitin ligases, and other molecules [13]. NFKBIA negatively regulates NF-κB activity by inhibiting its translocation from the cytoplasm to the nucleus [11]. High-frequency mutations in CYLD (10.5%, 11/105), TRAF3 (8.6%, 9/105), and NFKBIA (6.7%, 7/105) lead to constitutive activation of the nonclassical NF-κB pathway in NPC [11]. Frequent somatic mutations in genes involved in the PI3K/Akt/mTOR and MARK signaling pathways, such as phosphatase and tensin homolog (PTEN), PIK3CA, and fibroblast growth factor receptor 3 (FGFR3) have been found in NPC, and the most frequently mutated pathways are considered to be responsible for NPC progression [6,14]. PIK3CA encodes the p110α catalytic subunit, which activates Akt signaling and downstream molecules by generating phosphatidylinositol (3,4,5)-triphosphate (PIP3) [15,16]. The majority of mutations in PIK3CA (83.3%, 5/6) occur in the E542K, E545K, and H1047 hotspots [17]. It is speculated that the E545K and E542K mutations disrupt the interaction between PIK3CA and the p85 regulatory subunit, whereas the H1047 mutation increases the binding affinity of PIK3CA for the phosphatidylinositol substrate [17,18,19]. Mutations in PIK3CA promote cell proliferation and migration, and inhibit apoptosis in NPC cells [15]. In addition, high-frequency somatic mutations in chromatin-remodeling-related genes (for example, lysine (K)-specific methyltransferase 2C (KMT2C), lysine (K)-specific methyltransferase 2D (KMT2D), E1A binding protein p300 (EP300), and lysine (K)-specific demethylase 5A (KDM5A)), have also been documented in NPC [11]. Interestingly, mutations in the EBV genome have also been shown to affect the biological behavior of EBV-infected cells [20]. The expression of mutant 2117-LMP1 results in enhanced cell proliferation and migration ability and weaker cell–cell adhesion in the normal nasopharyngeal epithelial cell line NP69 through the activation of the NF-κB signaling pathway [20]. Other LMP1 variants (Alaskan, China 1, China 2, Med+, Med−, and North Carolina) have also been demonstrated to block cellular contact inhibition in Rat-1 fibroblasts and enhance the motility of human foreskin keratinocytes (HFKs) by stimulating the PI3K-Akt and NF-κB pathways [21]. The conserved mutation of the EBV gene BamH1-A fragment rightward reading frame 1 (BARF1) is common in Indonesia; however, its functional role remains poorly understood [22].
2.2. High-Frequency CNVs
With the development of high-throughput sequencing, gene chips, and comparative genomic hybridization (CGH) technologies, there is increasing evidence that CNVs constitute the most common genetic variant in NPC. Increased copy numbers of chromosomes 1q, 8p, 8q, 11q, 12p, 12q, 17q, 19p, 19q, 20p, and 20q and copy-number loss of 1p, 3p, 9p, 9q, 11q, 13q, 14q, and 16q are frequently detected in NPC [23,24]. High-frequency copy-number amplification was detected in cyclin D1 (CCND1) (73.7%, 28/38) on chromosome 11q and transforming growth factor-beta receptor 2 (TGFBR2) (16.7%, 2/12) on chromosome 3p [25,26]. The copy-number loss of cyclin-dependent kinase inhibitor 2A (CDKN2A) on chromosome 9p was also frequent (86.8%, 33/38) [25]. The copy-number loss of 3p and 9p is closely associated with latent EBV infection and has been established as a pivotal early molecular event during the evolution of the normal nasopharyngeal epithelium into NPC [27,28]. Genetic damage to tumor suppressors located in these chromosomal regions (such as CDKN2A, Ras association domain family member 1 (RASSF1A), and TGFBR2) is a key component of genomic instability in NPC [10]. The tumor suppressor gene RASSF1 regulates mitosis and maintains genomic stability by repressing cell-cycle-related protein cell division cycle 20 (CDC20) and microtubule proteins [29,30,31]. RASSF1A inhibits self-renewal and tumorigenicity and reduces the invasiveness of NPC cells in vitro [32]. The copy-number loss of RASSF1A destabilizes the host genome and is used as a metric to assess the severity of genomic instability in NPC [33]. Alterations in CCND1 and CDKN2A promote NPC progression by disturbing the cell cycle [34,35].
2.3. Gene Translocations and Rearrangements
Chromosome breakage and gene rearrangement occur when an organism is exposed to pathological or harmful external environmental factors, resulting in the formation of new gene fragments that lead to the development of malignant neoplastic diseases [36]. Fusion genes with both breakage and rearrangement have been detected in NPC, such as UBR5-ZNF423, RARS-MAD1L1, and FGFR3-TACC3 [37,38,39]. The expression of the UBR5-ZNF423 and RARS-MAD1L1 fusion genes enables NPC cells to proliferate and form colonies in vitro and induce tumorigenesis in vivo [37,38]. Despite the low incidence of gene translocations and rearrangements, their functional roles in NPC progression require further clarification.
2.4. Unique Genomic Alterations in Relapse Lesions
The genomic instability of recurrent NPC exhibits unique features compared to those of primary tumors. For instance, all mutations of TRAF3, CYLD, and NFKBIA are clonal (100.0%) in recurrent tumors, whereas they only range from 55.6% to 57.9% in primary tumors [11,40]. This suggests that clonal mutations in the NF-κB pathway are closely correlated with the local recurrence of NPC. The TP53 mutation rate is significantly higher in recurrent samples than in primary tumors (15.2% vs. 6.4%) [11]. Moreover, mutations in genes such as ABL proto-oncogene 1 (ABL1), BUB1 mitotic checkpoint serine/threonine kinase B (BUB1B), nuclear receptor corepressor 1 (NCOR1), chimeric antigen receptors (CARS), heat shock protein 90 alpha family class B member 1 (HSP90AB1), and nuclear receptor coactivator 1 (NCOA1) have been found only in recurrent diseases [11,41]. Mutations in phosphatidylinositol-4,5-bisphosphate 3-kinase catalytic subunit alpha (PIK3CA) were only detected in the recurrent lesions (37.5%, 3/8), but none in their paired primary tumors (0%, 0/7) [42]. In addition, gains in cyclin-dependent kinase 2 (CDK2) and erb-b2 receptor tyrosine kinase 3 (ERBB3) were found only in recurrent lesions (37.5% and 37.5%, respectively) but not in the corresponding primary tumor controls [42]. Somatic mutations enriched in the PI3K and MAPK pathways, such as Harvey-RAS (HRAS) and neuroblastoma-RAS (NRAS), are considered key factors driving NPC recurrence [11,14]. Tumor suppressor in lung cancer 1 (TSLC1) was downregulated or absent in 81% (21/26) of lymph-node metastases, which was significantly higher than that in the primary tumor of NPC (35%, 9/26) [43]. Thymus cell antigen 1 (THY1), a tumor suppressor mainly involved in T-cell activation, neurite outgrowth, and cell apoptosis, was downregulated or lost in 74% (20/27) of metastatic lymph nodes, which was significantly higher than that in primary NPC (40%, 17/43) [44,45]. Alterations in fibronectin type III domain containing 3B (FNDC3B), cytoplasmic linker-associated protein 1 (CLASP1), annexin A1 (ANXA1), and C-X-C motif chemokine ligand 10 (CXCL10) have been correlated with metastasis in NPC [46]. In addition, mutations in EP300, PIK3CA, and splicing factor 3b subunit 1 (SF3B1) also contribute to metastasis in NPC [7,47]. Specifically, mutations in the BRCA1-associated protein 1 (BAP1) are found only in NPC patients with metastatic disease [48].
2.5. Deleterious Germline Variants
Although most cases of NPC occur sporadically, family genetic predisposition to NPC is widely recognized. Unlike somatic mutations, germline variants in susceptibility genes contribute to familial nasopharyngeal carcinoma [49]. Deleterious germline variants lead to genomic instability and promote tumor progression by increasing somatic mutations and altering gene expression [49]. Germline variation in NPC mainly focuses on the pathways of DNA damage repair (such as BRCA2 DNA-repair-associated (BRCA2), protein kinase, DNA-activated, catalytic subunit (PRKDC), mutL homolog 1 (MLH1), and lysine methyltransferase 2C (KMT2C)), host defense (macrophage-stimulating 1 receptor (MST1R)), EBV virus infection (BCL2-like 12 (BCL2L12), NEDD4-like E3 ubiquitin protein ligase (NEDD4L)), and NOTCH signaling (notch receptor 1 (NOTCH1), DLL3 delta-like canonical notch ligand 3 (DLL3)) [50,51,52,53]. In particular, multiple familial linkage studies have shown that major histocompatibility complex (MHC) class I genes with genetic susceptibility on chromosome 6p are highly associated with NPC, suggesting a critical role for EBV antigen presentation to host immune cells in this disease [33]. Mutations in MHC class I genes, such as NLR family caspase recruitment domain containing 5 (NLRC5), human leukocyte antigen A (HLA-A), human leukocyte antigen B (HLA-B), and human leukocyte antigen C (HLA-C), cause disruption of the antigen-presentation machinery and enable NPC cells to escape immune surveillance [33]. Germline variants in MST1R have also been identified in NPC, and the MST1R-encoded protein is predominantly expressed in tissue-resident macrophages to protect organs from inflammatory injury [50]. However, most germline variations in NPC are focused on a limited number of specific genes, and the lack of large-scale studies limits our understanding of the risk of germline variations in NPC [54].
3. Synergy of Host Genomic Instability and EBV Infection
3.1. Host Genomic Alterations Enable Stable EBV Infection
EBV infection can be detected in almost all NPC cases with undifferentiated non-keratinizing carcinomas (WHO type III) [1]. However, EBV infection is rarely detected in normal nasopharyngeal epithelial cells [34]. Paradoxically, EBV infection inhibits the proliferation of immortalized nasopharyngeal epithelial cells [34]. The establishment and maintenance of latent EBV infection in nasopharyngeal epithelial cells is a critical step in the malignant transformation of the nasopharyngeal epithelium. Normal nasopharyngeal epithelial cells exposed to carcinogens such as nitrosamines and polycyclic aromatic hydrocarbons induce specific genomic alterations that enable EBV infection [41]. The copy-number loss of CDKN2A (86.8%, 33/38) and copy-number increase of CCND1 (73.7%, 28/38) are crucial genomic alterations in the transformation from nasopharyngeal epithelium to carcinoma [25]. CDKN2A inactivates retinoblastoma protein (RB) by binding to cyclin-dependent kinase 4 (CDK4) or cyclin-dependent kinase 6 (CDK6), resulting in cell-cycle arrest [35]. CCND1 also regulates the cell-cycle transition from the G1 to the S phase by binding to CDK4 or CDK6, which phosphorylates RB and releases E2F transcription factor [55]. The inactivation of CDKN2A and the hyperactivation of CCND1 eliminates the inhibitory effect on cell growth induced by EBV, converts the lytic infection state to a latent infection state, and induces the sustained expression of the latent EBV genome [34,56]. Clonal expansion can be initiated even if stable and persistent latent EBV infection is present only in a single nasopharyngeal epithelial cell [57]. In contrast, EBV-infected cells lose their potential for malignant transformation once the stable expression of the latent EBV genome is not maintained [15].
3.2. EBV Genes and Their Products Promote Host Genomic Instability
Integration of the EBV genome into host cells affects the stability of the host genome through various mechanisms. EBV genes and their products include EBV-encoded RNAs (EBERs), LMP1, latent membrane protein 2 (LMP2), EBV nuclear antigen 1 (EBNA1), BamH1 A rightward transcript miRNAs (BART-miRNAs), BamH1 A rightward transcripts (BARTs), and BARF1. EBV cleavage genes encoding proteins (e.g., BGLF5 and BALF3) promote host genomic instability by inducing the formation of micronuclei (MN) and DNA double-strand breaks and inhibiting the repair of damaged DNA. Micronuclei are derived from acentric chromatids and serve as classic markers of genomic instability [58]. BGLF5 has been proven to be a strong inducer of micronucleus formation and DNA damage [58]. BGLF5 causes DNA damage, inhibits DNA repair, and increases chromosomal aberration (CA), microsatellite instability (MSI), and mutation frequency, thereby contributing to the early stages of the malignant transformation of nasopharyngeal epithelial cells [59]. Another EBV cleavage gene encoding the protein BALF3 induces micronucleus formation and DNA strand breaks, promotes cell migration in vitro, and enhances the tumorigenicity of NPC cells in vivo [60]. However, micronucleus formation is not necessary for EBV cleavage of genes and their products to induce genomic instability. The early cleavage gene BRLF1 promotes genomic instability in NPC cells by interfering with mitosis to induce chromosomal mis-segregation without causing a significant increase in micronuclei [61]. The EBV cleavage gene encoding the protein BGLF4 induces genomic instability by prolonging the S phase of the cell cycle and causing chromosomal abnormalities [62]. In addition, the EBV product BNRF1 triggers centromeric amplification, thereby causing chromosomal instability [63].
The expression of latent EBV genes exacerbates host genome instability. EBNA1 binds to ubiquitin-specific protease 7 (USP7) to form a p53 binding domain that protects infected cells from apoptosis and inhibits DNA repair [64]. EBNA1 also attenuates the repair of the damage repair via p53 [65]. Promyelocytic leukemia (PML) protein maintains genomic stability by regulating cell survival, DNA damage and repair, and p53 gene-related apoptosis [66]. EBNA1 induces PML protein degradation by binding to USP7 or casein kinase 2 (CK2), thus contributing to genomic instability [65,67,68]. In addition to affecting the stability of the host genome, EBNA1 also maintains the stability of the EBV genome by regulating the replication and mitotic segregation of EBV episomes [67]. Indeed, EBNA1 is a key factor in sustaining EBV genome activation in all EBV-associated malignancies [67]. EBV-encoded LMP1, which is actively expressed in NPC, interferes with DNA damage repair. LMP1 inhibits the expression of DNA-damage-binding protein 1 (DDB1) through the inactivation of forkhead box O3A (FOXO3a) by activating the NF-κB and PI3K/Akt pathways [69]. Additionally, LMP1 activates telomerase through the expression of c-Myc in nasopharyngeal epithelial cells, induces micronucleus formation, and enhances the sensitivity of host cells to pathological factors that induce DNA damage, thereby promoting the accumulation of genomic instability in NPC cells [70,71]. LMP1 also inhibits p53-mediated DNA repair and transcriptional activity through its C-terminal-activating region 1 (CTAR1) [72]. Moreover, the expression of LMP1 in nasopharyngeal epithelial cells results in the impairment of the G2 checkpoint through the activation of defective checkpoint kinase 1 (Chk1) [73].
The EBV genome has been implicated in the methylation of the host genome and the inactivation of suppressor genes [33]. EBV induces the DNA methylation of the host genome by affecting DNA methyltransferases (DNMTs) [33]. LMP1 activates DNMTs through c-Jun NH2-terminal kinase (JNK) signaling, induces the promoter methylation of calmodulin 1 (CDH-1), and downregulates calmodulin E (E-cadherin) expression [74,75]. LMP1 also disrupts the microtubule structure and induces chromosomal aberrations in nasopharyngeal epithelial cells by repressing RASS1A expression [76]. LMP1 manipulates the infected cell cycle by inactivating the suppressor CDKN2A by transferring E2F transcription factor 4 (E2F4)/E2F transcription factor 5 (E2F5) and ETS proto-oncogene 2 (Ets2) to the cytoplasm [77]. In addition to LMP1, LMP2A inactivates PTEN, another well-characterized suppressor, by phosphorylating the signal transducer and activator of transcription 3 (STAT3) and inducing DNMT1 transcription [78].
In summary, on the one hand, host genomic instability, such as the inactivation of CDKN2A and the hyperactivation of CCND1 provides a prerequisite for latent EBV infection in the nasopharyngeal epithelium. In contrast, the consistent activation of the EBV genome transcript, the expression of EBV-encoded proteins, and the targeting effects of EBV non-coding RNAs on the genome of infected cells further aggravate host genomic instability. The transformation from normal nasopharyngeal epithelium to carcinoma is a continuous, multistep, and complex process. Malignant transformation can be conceptualized as a combined driving model of genomic alteration and EBV infection, in which host genomic instability is at a fundamental and central position (Figure 2). Once EBV latently infects nasopharyngeal epithelial cells, the EBV genes and their related products begin to exhibit the potential to drive malignant cellular transformation [10]. Along with stable EBV infection in the host nasopharyngeal epithelium, EBV-encoded genes and their products deteriorate genomic instability by activating functional gene expression and stimulating the related signaling pathways, which in turn triggers the hallmarks of cancer (Supplementary Table S1) [5]. The oncogenic effects of EBV genes and their products are mediated by the various molecular events of genomic instability that occur during NPC progression, culminating in dysregulated malignant proliferative neoplastic lesions.
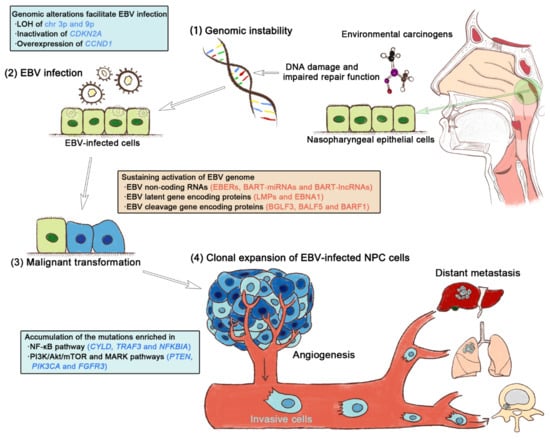
Figure 2.
The synergy of host genomic instability and Epstein–Barr virus (EBV) infection drives the multistep NPC progression. (1) Regional environmental carcinogens, such as nitrosamines and polycyclic aromatic hydrocarbons, induce DNA damage and repair function impairment, thereby compromising genomic stability. (2) Early genetic abnormalities resulting from host genomic instability allow the EBV infection of nasopharyngeal epithelial cells. (3) Persistent EBV infection sustainably activates EBV genes and their products and actively contributes to the transformation of non-malignant epithelium to carcinoma. (4) The clonal proliferation of EBV-infected cells leads to the accumulation of gene mutations enriched in key oncogenic signaling pathways, eventually triggering the hallmarks of cancer.
4. The Clinical Translational Value of Genomic Instability in NPC
Distant metastasis and recurrence are the primary causes of treatment failure and mortality in patients with NPC. Despite significant improvements in the clinical outcomes of NPC patients in recent years, approximately 30% of patients experience tumor relapse or distant metastasis [79]. Research on individualized and precise therapies based on the characteristics of genomic instability is needed to further improve the prognosis of NPC.
4.1. Targeted Therapeutic Strategies for Mutated Genes
The therapeutic strategy for TP53 mutations is to restore the normal physiological function of TP53 and inhibit the TP53–MDM2 interaction by introducing the wild-type TP53 gene into tumor cells [8,80,81,82,83]. Gendicine consists of the TP53 gene and a recombinant adenoviral vector, which is used for the treatment of HNSCCs by ectopically expressing TP53 to exhibit anti-tumor effects [80]. In addition, the small-molecule compound APR-246 and the novel condensed aminothiourea derivative COTI-2 can reactivate the TP53 gene by promoting the refolding of the mutated TP53, thus restoring the tumor suppressor effect of TP53 [8]. Mouse double minute 2 (MDM2) promotes tumor progression by inhibiting the transcriptional activity and stability of TP53 and impairing its ability of TP53 to induce cell apoptosis [81]. An MDM2 inhibitor, Nutlin-3, is a small-molecule imidazoline analog that exerts antitumor effects by competing with MDM2 for the TP53 binding site and inhibiting p53–MDM2 interactions to reactivate the p53 pathway [83,84]. Nutlin-3 also enhances the sensitivity to cisplatin and upregulates the expression of the apoptosis regulator BCL2-associated X (BAX) and p53-upregulated modulator of apoptosis (PUMA) in C666-1 cells, an NPC cell line [85]. Theoretically, because the majority of mutations in PIK3CA are located in the E542K, E545K, and H1047 hot spots, a therapy targeting these mutations can serve as an effective treatment strategy for NPC. However, so far, no selective inhibitors have been developed for the mutations in the hotspots or PI3K inhibitors specific for the p110α subtype [17]. Regarding the most commonly mutated pathway in NPC, a single drug or a combination of drugs targeting the PI3K/Akt/mTOR signaling pathway has been clinically used or studied in preclinical trials. Inhibitors are mainly divided into three categories: PI3K, Akt, and mTOR inhibitors [17]. The PI3K inhibitors alpelisib, copanlisib, duvelisib, idelalisib, and umbralisib have been approved for use in the treatment of breast cancer and hematologic malignancies [86]. However, the clinical benefits of PI3K inhibitors in NPC patients with PIK3CA mutations require further testing [11]. Akt inhibitors (ipatasertib and uprosertib) and mTOR inhibitors (everolimus) have also been investigated in clinical trials in breast cancer [87,88]. The Akt inhibitor MK-2206 was tested in a multicenter phase II study of patients with recurrent or metastatic NPC [89]. In addition, the mTOR inhibitor NVP-BEZ235 and the PI3K-MTOR inhibitor PF-04691502 are currently being tested as new targeted drugs for NPC [90,91]. The KIT proto-oncogene), a gene encoding a tyrosine kinase receptor, is frequently mutated or overexpressed in NPC [92,93]. The pharmacological blockage of KIT with imatinib inhibits the proliferation of NPC cell lines in a dose-dependent manner [94]. However, the effect of imatinib in NPC patients with mutated KIT remains to be elucidated. Furthermore, EGFR and KRAS mutations can be used to predict the clinical efficacy of tyrosine kinase inhibitors (TKIs) [95]. However, compared with that in lung cancer, the efficacy of TKIs is limited because of the low mutation rates of EGFR and KRAS in NPC patients, which is one of the main barriers hampering the development of targeted therapies for the disease [93,96,97].
4.2. Genomic Instability and Cancer Immunotherapy
Genomic stability variants such as TMB (tumor mutation burden), MSI, and CNVs are closely associated with the tumor immune microenvironment, and severe genomic instability suggests a poor clinical prognosis in NPC [98]. During the malignant transformation of nasopharyngeal epithelial cells, EBV-infected host cells accumulate somatic mutations, and a small part of the mutated genes are presented to MHC to form tumor neoantigens and are recognized by activated CD8+ T cells, stimulating the tumor immune response [99]. TMB is defined as the number of non-synonymous single-nucleotide variants (SNVs) per megabase, which indirectly reflects the ability of tumors to produce neoantigens and can be used to predict the effectiveness of treatment with immune checkpoint inhibitors (ICIs) [99]. ICIs include programmed death-1/programmed death-ligand 1 (PD-1/PD-L1) and cytotoxic T-lymphocyte-associated antigen-4 (CTLA-4) inhibitors, which can restore the immune response against tumor-derived antigens by restoring T-cell activity and promoting the T-cell recognition of tumor-derived antigens [100,101]. Growing evidence indicates that TMB is positively correlated with tumor-derived neoantigen load and high-TMB (TMB-H) cases are more likely to benefit from ICI treatment [101]. A genome-wide analysis of 190 patients with NPC showed that high TMB was strongly associated with EBV infection and that the degree of TMB was significantly correlated with each subtype of NPC [102]. Low TMB limits the production of tumor-derived antigens through somatic mutations, resulting in a lack of tumor neoantigens on the surface of NPC cells and decreased sensitivity to cancer immunotherapy [103]. However, neither tolipalimab monotherapy in the Polaris-02 study nor camlizumab monotherapy in the CAPTAIN study showed a significant correlation between TMB levels and treatment response in NPC [104,105]. Thus, the clinical significance of TMB for predicting prognosis or evaluating treatment efficacy is still controversial, as TMB in NPC is usually lower than that in other cancers [106]. In addition, the value of TMB for assessing the responsiveness of ICIs in NPC requires further validation.
DNA mismatch repair (MMR) genes are mainly responsible for correcting DNA replication errors to maintain genomic stability, whereas MMR mutations often lead to MSI [107]. As an important indicator of genomic instability, high microsatellite instability (MSI-H) or MMR functional deficiency (dMMR) is generally accompanied by high TMB [108]. Chen et al. reported that at least one of the four MMR gene-encoded mismatch repair proteins, including MLH1, MSH2, MSH6, and PMS2, was lost in 49.3% (34/69) of pre-treatment NPC biopsies [109]. Pembrolizumab, a PD-1 inhibitor used to treat patients with MSI-H/dMMR and unresectable or metastatic solid tumors, has shown proven efficacy in colorectal cancer, melanoma, and NSCLC [110,111,112]. An overall response rate (ORR) of 17.7% (34/192) was observed in a multicenter, non-randomized, phase Ib trial in patients with recurrent/metastatic HNSCC treated with pembrolizumab (KEYNOTE-012) [113]. However, the efficacy of pembrolizumab in the treatment of NPC remains to be evaluated in randomized controlled clinical trials.
Somatic CNVs, also known as segmental aneuploidies, are associated with responsiveness to cancer immunotherapy [114,115]. Patients with relatively frequent genomic amplification of the genes on chromosome 11q13, such as CCND1, FGF14, FGF3, and FGF4, as well as ETV6 genomic alterations, exhibit poor clinical response to toripalimab [105,116]. In another clinical trial of patients with metastatic melanoma treated with ipilimumab, a monoclonal antibody against CTLA-4, a negative correlation between CNV frequency and survival rate was established (hazard ratio = 2.24, p = 0.0004) [115]. The frequency of CNVs in HNSCC is strongly correlated with T-cell infiltration [117]. Two phase I clinical trials showed a significant benefit of anti-PD-1 therapy for long-term survival in patients with recurrent/metastatic NPC, and copy-number loss in granzyme genes leads to resistance to ICIs and is associated with a reduced survival rate [106].
5. Conclusions and Perspective
Genomic instability is an intrinsic driving force that promotes the progression of NPC. An in-depth exploration of the underlying mechanism through which genomic instability occurs, especially in terms of how EBV infection is implicated in host genomic instability, will enable a better understanding of the unique pathogenesis of NPC. The characterization of genomic instability could provide a theoretical basis for the development of novel approaches and strategies for the diagnosis and treatment of NPC. The detection of genomic-instability-related indicators (such as TMB, MSI, and CNVs) allows for a comprehensive evaluation of the effectiveness of immunotherapy for NPC. Targeted therapy based on genomic instability, particularly the genomic instability characteristics of recurrent and metastatic lesions, is crucial for precision NPC therapy in the future. However, the complexity of genomic instability and individual genomic variability are the greatest challenges in precision therapy for NPC.
In the past decade, much work has been carried out to clarify the interaction between the host genome and EBV infection, which consistently contributes to NPC pathogenesis. However, a model of genomic alteration integrated with EBV infection that drives TME evolution allowing NPC progression remains to be established. Solving this mystery could provide valuable biomarkers and effective targets for the development of treatment strategies, especially immunotherapy, for NPC.
Supplementary Materials
The following supporting information can be downloaded from https://www.mdpi.com/article/10.3390/curroncol29090475/s1. Table S1: Hallmarks of cancer triggered by EBV genes and their products [64,70,74,118,119,120,121,122,123,124,125,126,127,128,129,130,131,132,133,134,135,136,137,138,139,140,141,142,143,144,145,146,147,148,149,150,151,152,153,154,155,156,157,158,159,160,161,162,163].
Author Contributions
Conceptualization, J.W. and J.Z.; methodology, X.L. and J.W.; software, X.L. and J.W.; validation, X.L., J.W. and J.Z.; formal analysis, X.L. and J.W.; investigation, X.L., Y.D., Y.H., J.Y., S.X., Y.C., Y.L. (Yan Lin), R.L., Q.H., Y.L. (Yongqiang Li), J.W. and J.Z.; resources, Y.L. (Yongqiang Li), J.W. and J.Z.; data curation, X.L., J.W. and J.Z.; writing—original draft preparation, X.L. and J.W.; writing—review and editing, X.L., Y.L. (Yongqiang Li), J.W. and J.Z.; visualization, X.L., S.X., J.W. and J.Z.; supervision, Y.L. (Yongqiang Li), J.W. and J.Z.; project administration, J.W. and J.Z.; funding acquisition, J.Y., J.W. and J.Z. All authors have read and agreed to the published version of the manuscript.
Funding
This research was funded by the National Natural Science Foundation of China (82002859 and 82073004), the Guangxi Natural Science Foundation (2020GXNSFBA297024 and 2020GXNSFBA297059), and the Open Research Project of Key Laboratory of Early Prevention and Treatment for Regional High Frequency Tumor (Guangxi Medical University), Ministry of Education (GKE2d9-KF01).
Conflicts of Interest
The authors declare no conflict of interest.
References
- Chen, Y.-P.; Chan, A.T.C.; Le, Q.-T.; Blanchard, P.; Sun, Y.; Ma, J. Nasopharyngeal carcinoma. Lancet 2019, 394, 64–80. [Google Scholar] [CrossRef]
- Badoual, C. Update from the 5th Edition of the World Health Organization Classification of Head and Neck Tumors: Oropharynx and Nasopharynx. Head Neck Pathol. 2022, 16, 19–30. [Google Scholar] [CrossRef] [PubMed]
- Shanmugaratnam, K.; Sobin, L.H. The World Health Organization histological classification of tumours of the upper respiratory tract and ear. A commentary on the second edition. Cancer 1993, 71, 2689–2697. [Google Scholar] [CrossRef]
- Hau, P.M.; Lung, H.L.; Wu, M.; Tsang, C.M.; Wong, K.L.; Mak, N.K.; Lo, K.W. Targeting Epstein-Barr Virus in Nasopharyngeal Carcinoma. Front. Oncol. 2020, 10, 600. [Google Scholar] [CrossRef] [PubMed]
- Wong, K.C.W.; Hui, E.P.; Lo, K.W.; Lam, W.K.J.; Johnson, D.; Li, L.; Tao, Q.; Chan, K.C.A.; To, K.F.; King, A.D.; et al. Nasopharyngeal carcinoma: An evolving paradigm. Nat. Rev. Clin. Oncol. 2021, 18, 679–695. [Google Scholar] [CrossRef]
- Dai, W.; Zheng, H.; Cheung, A.K.; Lung, M.L. Genetic and epigenetic landscape of nasopharyngeal carcinoma. Chin. Clin. Oncol. 2016, 5, 16. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Z.; Li, P.; Zhang, X.; Xu, J.; Xu, J.; Yu, S.; Wang, D.; Dong, W.; Cao, X.; Yan, H.; et al. Mutational landscape of nasopharyngeal carcinoma based on targeted next-generation sequencing: Implications for predicting clinical outcomes. Mol. Med. 2022, 28, 55. [Google Scholar] [CrossRef]
- Zhu, G.; Pan, C.; Bei, J.X.; Li, B.; Liang, C.; Xu, Y.; Fu, X. Mutant p53 in Cancer Progression and Targeted Therapies. Front. Oncol. 2020, 10, 595187. [Google Scholar] [CrossRef] [PubMed]
- Bruce, J.P.; To, K.F.; Lui, V.W.Y.; Chung, G.T.Y.; Chan, Y.Y.; Tsang, C.M.; Yip, K.Y.; Ma, B.B.Y.; Woo, J.K.S.; Hui, E.P.; et al. Whole-genome profiling of nasopharyngeal carcinoma reveals viral-host co-operation in inflammatory NF-kappaB activation and immune escape. Nat. Commun. 2021, 12, 4193. [Google Scholar] [CrossRef]
- Tsang, C.M.; Lui, V.W.Y.; Bruce, J.P.; Pugh, T.J.; Lo, K.W. Translational genomics of nasopharyngeal cancer. Semin. Cancer Biol. 2020, 61, 84–100. [Google Scholar] [CrossRef]
- Li, Y.Y.; Chung, G.T.; Lui, V.W.; To, K.F.; Ma, B.B.; Chow, C.; Woo, J.K.; Yip, K.Y.; Seo, J.; Hui, E.P.; et al. Exome and genome sequencing of nasopharynx cancer identifies NF-kappaB pathway activating mutations. Nat. Commun. 2017, 8, 14121. [Google Scholar] [CrossRef]
- Massoumi, R. CYLD: A deubiquitination enzyme with multiple roles in cancer. Future Oncol. 2011, 7, 285–297. [Google Scholar] [CrossRef]
- Park, H.H. Structure of TRAF Family: Current Understanding of Receptor Recognition. Front. Immunol. 2018, 9, 1999. [Google Scholar] [CrossRef]
- Lin, D.C.; Meng, X.; Hazawa, M.; Nagata, Y.; Varela, A.M.; Xu, L.; Sato, Y.; Liu, L.Z.; Ding, L.W.; Sharma, A.; et al. The genomic landscape of nasopharyngeal carcinoma. Nat. Genet. 2014, 46, 866–871. [Google Scholar] [CrossRef]
- Lo, K.W.; Chung, G.T.; To, K.F. Deciphering the molecular genetic basis of NPC through molecular, cytogenetic, and epigenetic approaches. Semin. Cancer Biol. 2012, 22, 79–86. [Google Scholar] [CrossRef]
- Samuels, Y.; Wang, Z.; Bardelli, A.; Silliman, N.; Ptak, J.; Szabo, S.; Yan, H.; Gazdar, A.; Powell, S.M.; Riggins, G.J.; et al. High frequency of mutations of the PIK3CA gene in human cancers. Science 2004, 304, 554. [Google Scholar] [CrossRef]
- Gustin, J.P.; Cosgrove, D.P.; Park, B.H. The PIK3CA gene as a mutated target for cancer therapy. Curr. Cancer Drug. Targets 2008, 8, 733–740. [Google Scholar] [CrossRef]
- Miled, N.; Yan, Y.; Hon, W.C.; Perisic, O.; Zvelebil, M.; Inbar, Y.; Schneidman-Duhovny, D.; Wolfson, H.J.; Backer, J.M.; Williams, R.L. Mechanism of two classes of cancer mutations in the phosphoinositide 3-kinase catalytic subunit. Science 2007, 317, 239–242. [Google Scholar] [CrossRef]
- Kang, S.; Bader, A.G.; Zhao, L.; Vogt, P.K. Mutated PI 3-kinases—Cancer targets on a silver platter. Cell Cycle 2005, 4, 578–581. [Google Scholar] [CrossRef][Green Version]
- Lo, A.K.; Huang, D.P.; Lo, K.W.; Chui, Y.L.; Li, H.M.; Pang, J.C.; Tsao, S.W. Phenotypic alterations induced by the Hong Kong-prevalent Epstein-Barr virus-encoded LMP1 variant (2117-LMP1) in nasopharyngeal epithelial cells. Int. J. Cancer 2004, 109, 919–925. [Google Scholar] [CrossRef]
- Mainou, B.A.; Raab-Traub, N. LMP1 strain variants: Biological and molecular properties. J. Virol. 2006, 80, 6458–6468. [Google Scholar] [CrossRef] [PubMed]
- Hutajulu, S.H.; Hoebe, E.K.; Verkuijlen, S.A.; Fachiroh, J.; Hariwijanto, B.; Haryana, S.M.; Stevens, S.J.; Greijer, A.E.; Middeldorp, J.M. Conserved mutation of Epstein-Barr virus-encoded BamHI-A Rightward Frame-1 (BARF1) gene in Indonesian nasopharyngeal carcinoma. Infect. Agent. Cancer 2010, 5, 16. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.J.; Ko, J.Y.; Chen, P.J.; Shu, C.H.; Hsu, M.T.; Tsai, S.F.; Lin, C.H. Chromosomal aberrations in nasopharyngeal carcinoma analyzed by comparative genomic hybridization. Genes. Chromosomes Cancer 1999, 25, 169–175. [Google Scholar] [CrossRef]
- Hui, A.B.; Lo, K.W.; Leung, S.F.; Teo, P.; Fung, M.K.; To, K.F.; Wong, N.; Choi, P.H.; Lee, J.C.; Huang, D.P. Detection of recurrent chromosomal gains and losses in primary nasopharyngeal carcinoma by comparative genomic hybridisation. Int. J. Cancer 1999, 82, 498–503. [Google Scholar] [CrossRef]
- Hui, A.B.; Or, Y.Y.; Takano, H.; Tsang, R.K.; To, K.F.; Guan, X.Y.; Sham, J.S.; Hung, K.W.; Lam, C.N.; van Hasselt, C.A.; et al. Array-based comparative genomic hybridization analysis identified cyclin D1 as a target oncogene at 11q13.3 in nasopharyngeal carcinoma. Cancer Res. 2005, 65, 8125–8133. [Google Scholar] [CrossRef] [PubMed]
- Tu, C.; Zeng, Z.; Qi, P.; Li, X.; Guo, C.; Xiong, F.; Xiang, B.; Zhou, M.; Liao, Q.; Yu, J.; et al. Identification of genomic alterations in nasopharyngeal carcinoma and nasopharyngeal carcinoma-derived Epstein-Barr virus by whole-genome sequencing. Carcinogenesis 2018, 39, 1517–1528. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Fu, L.; Zhang, L.Y.; Kwong, D.L.; Yan, L.; Guan, X.Y. Tumor suppressor genes on frequently deleted chromosome 3p in nasopharyngeal carcinoma. Chin. J. Cancer 2012, 31, 215–222. [Google Scholar] [CrossRef]
- Chan, A.S.; To, K.F.; Lo, K.W.; Ding, M.; Li, X.; Johnson, P.; Huang, D.P. Frequent chromosome 9p losses in histologically normal nasopharyngeal epithelia from southern Chinese. Int. J. Cancer 2002, 102, 300–303. [Google Scholar] [CrossRef]
- Song, M.S.; Song, S.J.; Ayad, N.G.; Chang, J.S.; Lee, J.H.; Hong, H.K.; Lee, H.; Choi, N.; Kim, J.; Kim, H.; et al. The tumour suppressor RASSF1A regulates mitosis by inhibiting the APC-Cdc20 complex. Nat. Cell Biol. 2004, 6, 129–137. [Google Scholar] [CrossRef]
- Dallol, A.; Cooper, W.N.; Al-Mulla, F.; Agathanggelou, A.; Maher, E.R.; Latif, F. Depletion of the ras association domain family 1, isoform A-associated novel microtubule-associated protein, C190RF5/MAP1S, causes mitotic abnormalities. Cancer Res. 2007, 67, 492–500. [Google Scholar] [CrossRef]
- van der Weyden, L.; Tachibana, K.K.; Gonzalez, M.A.; Adams, D.J.; Ng, B.L.; Petty, R.; Venkitaraman, A.R.; Arends, M.J.; Bradley, A. The RASSF1A isoform of RASSF1 promotes microtubule stability and suppresses tumorigenesis. Mol. Cell. Biol. 2005, 25, 8356–8367. [Google Scholar] [CrossRef]
- Liang, Y.Y.; Deng, X.B.; Lin, X.T.; Jiang, L.L.; Huang, X.T.; Mo, Z.W.; Yuan, Y.W.; Teh, M.T. RASSF1A inhibits PDGFB-driven malignant phenotypes of nasopharyngeal carcinoma cells in a YAP1-dependent manner. Cell Death Dis. 2020, 11, 1–12. [Google Scholar] [CrossRef]
- Tsao, S.W.; Tsang, C.M.; Lo, K.W. Epstein-Barr virus infection and nasopharyngeal carcinoma. Philos. Trans. R. Soc. Lond. B Biol. Sci. 2017, 372, 20160270. [Google Scholar] [CrossRef]
- Tsang, C.M.; Yip, Y.L.; Lo, K.W.; Deng, W.; To, K.F.; Hau, P.M.; Lau, V.M.; Takada, K.; Lui, V.W.; Lung, M.L.; et al. Cyclin D1 overexpression supports stable EBV infection in nasopharyngeal epithelial cells. Proc. Natl. Acad. Sci. USA 2012, 109, E3473–E3482. [Google Scholar] [CrossRef]
- Liggett, W.H., Jr.; Sidransky, D. Role of the p16 tumor suppressor gene in cancer. J. Clin. Oncol. 1998, 16, 1197–1206. [Google Scholar] [CrossRef]
- Lu, B.; Jiang, R.; Xie, B.; Wu, W.; Zhao, Y. Fusion genes in gynecologic tumors: The occurrence, molecular mechanism and prospect for therapy. Cell Death Dis. 2021, 12, 783. [Google Scholar] [CrossRef]
- Chung, G.T.Y.; Lung, R.W.M.; Hui, A.B.Y.; Yip, K.Y.L.; Woo, J.K.S.; Chow, C.; Tong, C.Y.K.; Lee, S.D.; Yuen, J.W.F.; Lun, S.W.M.; et al. Identification of a recurrent transforming UBR5-ZNF423 fusion gene in EBV-associated nasopharyngeal carcinoma. J. Pathol. 2013, 231, 158–167. [Google Scholar] [CrossRef]
- Zhong, Q.; Liu, Z.H.; Lin, Z.R.; Hu, Z.D.; Yuan, L.; Liu, Y.M.; Zhou, A.J.; Xu, L.H.; Hu, L.J.; Wang, Z.F.; et al. The RARS-MAD1L1 Fusion Gene Induces Cancer Stem Cell-like Properties and Therapeutic Resistance in Nasopharyngeal Carcinoma. Clin. Cancer Res. 2018, 24, 659–673. [Google Scholar] [CrossRef]
- Yuan, L.; Liu, Z.H.; Lin, Z.R.; Xu, L.H.; Zhong, Q.; Zeng, M.S. Recurrent FGFR3-TACC3 fusion gene in nasopharyngeal carcinoma. Cancer Biol. Ther. 2014, 15, 1613–1621. [Google Scholar] [CrossRef]
- You, R.; Liu, Y.P.; Lin, D.C.; Li, Q.; Yu, T.; Zou, X.; Lin, M.; Zhang, X.L.; He, G.P.; Yang, Q.; et al. Clonal Mutations Activate the NF-kappaB Pathway to Promote Recurrence of Nasopharyngeal Carcinoma. Cancer Res. 2019, 79, 5930–5943. [Google Scholar] [CrossRef]
- Campion, N.J.; Ally, M.; Jank, B.J.; Ahmed, J.; Alusi, G. The molecular march of primary and recurrent nasopharyngeal carcinoma. Oncogene 2021, 40, 1757–1774. [Google Scholar] [CrossRef]
- Cho, W.C.S.; Tse, K.P.; Ngan, R.K.C.; Cheuk, W.; Ma, V.W.S.; Yang, Y.T.; Yip, T.T.C.; Tan, K.T.; Chen, S.J. Genomic characterization reveals potential biomarkers in nasopharyngeal carcinoma patients with relapse. Expert. Rev. Mol. Diagn. 2020, 20, 1149–1159. [Google Scholar] [CrossRef]
- Lung, H.L.; Cheung, A.K.; Xie, D.; Cheng, Y.; Kwong, F.M.; Murakami, Y.; Guan, X.Y.; Sham, J.S.; Chua, D.; Protopopov, A.I.; et al. TSLC1 is a tumor suppressor gene associated with metastasis in nasopharyngeal carcinoma. Cancer Res. 2006, 66, 9385–9392. [Google Scholar] [CrossRef]
- Lung, H.L.; Bangarusamy, D.K.; Xie, D.; Cheung, A.K.; Cheng, Y.; Kumaran, M.K.; Miller, L.; Liu, E.T.; Guan, X.Y.; Sham, J.S.; et al. THY1 is a candidate tumour suppressor gene with decreased expression in metastatic nasopharyngeal carcinoma. Oncogene 2005, 24, 6525–6532. [Google Scholar] [CrossRef]
- Rege, T.A.; Hagood, J.S. Thy-1, a versatile modulator of signaling affecting cellular adhesion, proliferation, survival, and cytokine/growth factor responses. Biochim. Biophys. Acta 2006, 1763, 991–999. [Google Scholar] [CrossRef]
- Tang, X.R.; Li, Y.Q.; Liang, S.B.; Jiang, W.; Liu, F.; Ge, W.X.; Tang, L.L.; Mao, Y.P.; He, Q.M.; Yang, X.J.; et al. Development and validation of a gene expression-based signature to predict distant metastasis in locoregionally advanced nasopharyngeal carcinoma: A retrospective, multicentre, cohort study. Lancet Oncol. 2018, 19, 382–393. [Google Scholar] [CrossRef]
- Mi, J.L.; Xu, M.; Liu, C.; Wang, R.S. Identification of novel biomarkers and small-molecule compounds for nasopharyngeal carcinoma with metastasis. Medicine 2020, 99, e21505. [Google Scholar] [CrossRef]
- Si, J.; Huang, B.; Lan, G.; Zhang, B.; Wei, J.; Deng, Z.; Li, Y.; Qin, Y.; Li, B.; Lu, Y.; et al. Comparison of whole exome sequencing in circulating tumor cells of primitive and metastatic nasopharyngeal carcinoma. Transl. Cancer Res. 2020, 9, 4080–4092. [Google Scholar] [CrossRef] [PubMed]
- Ramroop, J.R.; Gerber, M.M.; Toland, A.E. Germline Variants Impact Somatic Events during Tumorigenesis. Trends Genet. 2019, 35, 515–526. [Google Scholar] [CrossRef]
- Dai, W.; Zheng, H.; Cheung, A.K.; Tang, C.S.; Ko, J.M.; Wong, B.W.; Leong, M.M.; Sham, P.C.; Cheung, F.; Kwong, D.L.; et al. Whole-exome sequencing identifies MST1R as a genetic susceptibility gene in nasopharyngeal carcinoma. Proc. Natl. Acad. Sci. USA 2016, 113, 3317–3322. [Google Scholar] [CrossRef]
- Sasaki, M.M.; Skol, A.D.; Bao, R.; Rhodes, L.V.; Chambers, R.; Vokes, E.E.; Cohen, E.E.; Onel, K. Integrated genomic analysis suggests MLL3 is a novel candidate susceptibility gene for familial nasopharyngeal carcinoma. Cancer Epidemiol. Biomark. Prev. 2015, 24, 1222–1228. [Google Scholar] [CrossRef] [PubMed]
- Yu, G.; Hsu, W.L.; Coghill, A.E.; Yu, K.J.; Wang, C.P.; Lou, P.J.; Liu, Z.; Jones, K.; Vogt, A.; Wang, M.; et al. Whole-Exome Sequencing of Nasopharyngeal Carcinoma Families Reveals Novel Variants Potentially Involved in Nasopharyngeal Carcinoma. Sci. Rep. 2019, 9, 9916. [Google Scholar] [CrossRef] [PubMed]
- Dai, W.; Chung, D.L.; Chow, L.K.; Yu, V.Z.; Lei, L.C.; Leong, M.M.; Chan, C.K.; Ko, J.M.; Lung, M.L. Clinical Outcome-Related Mutational Signatures Identified by Integrative Genomic Analysis in Nasopharyngeal Carcinoma. Clin. Cancer Res. 2020, 26, 6494–6504. [Google Scholar] [CrossRef] [PubMed]
- Hildesheim, A.; Wang, C.P. Genetic predisposition factors and nasopharyngeal carcinoma risk: A review of epidemiological association studies, 2000-2011: Rosetta Stone for NPC: Genetics, viral infection, and other environmental factors. Semin. Cancer Biol. 2012, 22, 107–116. [Google Scholar] [CrossRef]
- Müller, H.; Lukas, J.; Schneider, A.; Warthoe, P.; Bartek, J.; Eilers, M.; Strauss, M. Cyclin D1 expression is regulated by the retinoblastoma protein. Proc. Natl. Acad. Sci. USA 1994, 91, 2945–2949. [Google Scholar] [CrossRef] [PubMed]
- Yip, Y.L.; Pang, P.S.; Deng, W.; Tsang, C.M.; Zeng, M.; Hau, P.M.; Man, C.; Jin, Y.; Yuen, A.P.; Tsao, S.W. Efficient immortalization of primary nasopharyngeal epithelial cells for EBV infection study. PLoS ONE 2013, 8, e78395. [Google Scholar] [CrossRef]
- Tsang, C.M.; Deng, W.; Yip, Y.L.; Zeng, M.S.; Lo, K.W.; Tsao, S.W. Epstein-Barr virus infection and persistence in nasopharyngeal epithelial cells. Chin. J. Cancer 2014, 33, 549–555. [Google Scholar] [CrossRef]
- Fang, C.Y.; Lee, C.H.; Wu, C.C.; Chang, Y.T.; Yu, S.L.; Chou, S.P.; Huang, P.T.; Chen, C.L.; Hou, J.W.; Chang, Y.; et al. Recurrent chemical reactivations of EBV promotes genome instability and enhances tumor progression of nasopharyngeal carcinoma cells. Int. J. Cancer 2009, 124, 2016–2025. [Google Scholar] [CrossRef]
- Wu, C.C.; Liu, M.T.; Chang, Y.T.; Fang, C.Y.; Chou, S.P.; Liao, H.W.; Kuo, K.L.; Hsu, S.L.; Chen, Y.R.; Wang, P.W.; et al. Epstein-Barr Virus DNase (BGLF5) induces genomic instability in human epithelial cells. Nucleic Acids Res. 2010, 38, 1932–1949. [Google Scholar] [CrossRef]
- Chiu, S.H.; Wu, C.C.; Fang, C.Y.; Yu, S.L.; Hsu, H.Y.; Chow, Y.H.; Chen, J.Y. Epstein-Barr virus BALF3 mediates genomic instability and progressive malignancy in nasopharyngeal carcinoma. Oncotarget 2014, 5, 8583–8601. [Google Scholar] [CrossRef]
- Huang, S.Y.; Wu, C.C.; Cheng, Y.J.; Chou, S.P.; Jiang, Y.J.; Chu, K.C.; Tsai, C.H.; Lin, S.F.; Chen, J.Y. Epstein-Barr virus BRLF1 induces genomic instability and progressive malignancy in nasopharyngeal carcinoma cells. Oncotarget 2017, 8, 78948–78964. [Google Scholar] [CrossRef]
- Chang, Y.H.; Lee, C.P.; Su, M.T.; Wang, J.T.; Chen, J.Y.; Lin, S.F.; Tsai, C.H.; Hsieh, M.J.; Takada, K.; Chen, M.R. Epstein-Barr virus BGLF4 kinase retards cellular S-phase progression and induces chromosomal abnormality. PLoS ONE 2012, 7, e39217. [Google Scholar] [CrossRef]
- Shumilov, A.; Tsai, M.H.; Schlosser, Y.T.; Kratz, A.S.; Bernhardt, K.; Fink, S.; Mizani, T.; Lin, X.; Jauch, A.; Mautner, J.; et al. Epstein-Barr virus particles induce centrosome amplification and chromosomal instability. Nat. Commun. 2017, 8, 14257. [Google Scholar] [CrossRef]
- Saridakis, V.; Sheng, Y.; Sarkari, F.; Holowaty, M.N.; Shire, K.; Nguyen, T.; Zhang, R.G.; Liao, J.; Lee, W.; Edwards, A.M.; et al. Structure of the p53 binding domain of HAUSP/USP7 bound to Epstein-Barr nuclear antigen 1 implications for EBV-mediated immortalization. Mol. Cell 2005, 18, 25–36. [Google Scholar] [CrossRef]
- Sivachandran, N.; Sarkari, F.; Frappier, L. Epstein-Barr nuclear antigen 1 contributes to nasopharyngeal carcinoma through disruption of PML nuclear bodies. PLoS Pathog. 2008, 4, e1000170. [Google Scholar] [CrossRef]
- Chang, H.R.; Munkhjargal, A.; Kim, M.J.; Park, S.Y.; Jung, E.; Ryu, J.H.; Yang, Y.; Lim, J.S.; Kim, Y. The functional roles of PML nuclear bodies in genome maintenance. Mutat. Res. 2018, 809, 99–107. [Google Scholar] [CrossRef]
- Frappier, L. EBNA1. Curr. Top. Microbiol. Immunol. 2015, 391, 3–34. [Google Scholar] [CrossRef]
- Sivachandran, N.; Cao, J.Y.; Frappier, L. Epstein-Barr virus nuclear antigen 1 Hijacks the host kinase CK2 to disrupt PML nuclear bodies. J. Virol. 2010, 84, 11113–11123. [Google Scholar] [CrossRef]
- Chen, Y.R.; Liu, M.T.; Chang, Y.T.; Wu, C.C.; Hu, C.Y.; Chen, J.Y. Epstein-Barr virus latent membrane protein 1 represses DNA repair through the PI3K/Akt/FOXO3a pathway in human epithelial cells. J. Virol. 2008, 82, 8124–8137. [Google Scholar] [CrossRef]
- Liu, M.T.; Chen, Y.R.; Chen, S.C.; Hu, C.Y.; Lin, C.S.; Chang, Y.T.; Wang, W.B.; Chen, J.Y. Epstein-Barr virus latent membrane protein 1 induces micronucleus formation, represses DNA repair and enhances sensitivity to DNA-damaging agents in human epithelial cells. Oncogene 2004, 23, 2531–2539. [Google Scholar] [CrossRef]
- Liu, M.T.; Chang, Y.T.; Chen, S.C.; Chuang, Y.C.; Chen, Y.R.; Lin, C.S.; Chen, J.Y. Epstein-Barr virus latent membrane protein 1 represses p53-mediated DNA repair and transcriptional activity. Oncogene 2005, 24, 2635–2646. [Google Scholar] [CrossRef] [PubMed]
- Deng, W.; Pang, P.S.; Tsang, C.M.; Hau, P.M.; Yip, Y.L.; Cheung, A.L.; Tsao, S.W. Epstein-Barr virus-encoded latent membrane protein 1 impairs G2 checkpoint in human nasopharyngeal epithelial cells through defective Chk1 activation. PLoS ONE 2012, 7, e39095. [Google Scholar] [CrossRef] [PubMed]
- Tsai, C.N.; Tsai, C.L.; Tse, K.P.; Chang, H.Y.; Chang, Y.S. The Epstein-Barr virus oncogene product, latent membrane protein 1, induces the downregulation of E-cadherin gene expression via activation of DNA methyltransferases. Proc. Natl. Acad. Sci. USA 2002, 99, 10084–10089. [Google Scholar] [CrossRef] [PubMed]
- Tsai, C.L.; Li, H.P.; Lu, Y.J.; Hsueh, C.; Liang, Y.; Chen, C.L.; Tsao, S.W.; Tse, K.P.; Yu, J.S.; Chang, Y.S. Activation of DNA methyltransferase 1 by EBV LMP1 Involves c-Jun NH(2)-terminal kinase signaling. Cancer Res. 2006, 66, 11668–11676. [Google Scholar] [CrossRef] [PubMed]
- Man, C.; Rosa, J.; Lee, L.T.; Lee, V.H.; Chow, B.K.; Lo, K.W.; Doxsey, S.; Wu, Z.G.; Kwong, Y.L.; Jin, D.Y.; et al. Latent membrane protein 1 suppresses RASSF1A expression, disrupts microtubule structures and induces chromosomal aberrations in human epithelial cells. Oncogene 2007, 26, 3069–3080. [Google Scholar] [CrossRef] [PubMed]
- Ohtani, N.; Brennan, P.; Gaubatz, S.; Sanij, E.; Hertzog, P.; Wolvetang, E.; Ghysdael, J.; Rowe, M.; Hara, E. Epstein-Barr virus LMP1 blocks p16INK4a-RB pathway by promoting nuclear export of E2F4/5. J. Cell Biol. 2003, 162, 173–183. [Google Scholar] [CrossRef] [PubMed]
- Hino, R.; Uozaki, H.; Murakami, N.; Ushiku, T.; Shinozaki, A.; Ishikawa, S.; Morikawa, T.; Nakaya, T.; Sakatani, T.; Takada, K.; et al. Activation of DNA methyltransferase 1 by EBV latent membrane protein 2A leads to promoter hypermethylation of PTEN gene in gastric carcinoma. Cancer Res. 2009, 69, 2766–2774. [Google Scholar] [CrossRef] [PubMed]
- Lai, S.Z.; Li, W.F.; Chen, L.; Luo, W.; Chen, Y.Y.; Liu, L.Z.; Sun, Y.; Lin, A.H.; Liu, M.Z.; Ma, J. How does intensity-modulated radiotherapy versus conventional two-dimensional radiotherapy influence the treatment results in nasopharyngeal carcinoma patients? Int. J. Radiat. Oncol. Biol. Phys. 2011, 80, 661–668. [Google Scholar] [CrossRef]
- Peng, Z. Current status of gendicine in China: Recombinant human Ad-p53 agent for treatment of cancers. Hum. Gene Ther. 2005, 16, 1016–1027. [Google Scholar] [CrossRef]
- Haupt, Y.; Maya, R.; Kazaz, A.; Oren, M. Mdm2 promotes the rapid degradation of p53. Nature 1997, 387, 296–299. [Google Scholar] [CrossRef]
- Yee-Lin, V.; Pooi-Fong, W.; Soo-Beng, A.K. Nutlin-3, A p53-Mdm2 Antagonist for Nasopharyngeal Carcinoma Treatment. Mini Rev. Med. Chem. 2018, 18, 173–183. [Google Scholar] [CrossRef]
- Fan, X.; Wang, Y.; Song, J.; Wu, H.; Yang, M.; Lu, L.; Weng, X.; Liu, L.; Nie, G. MDM2 inhibitor RG7388 potently inhibits tumors by activating p53 pathway in nasopharyngeal carcinoma. Cancer Biol. Ther. 2019, 20, 1328–1336. [Google Scholar] [CrossRef]
- Shinohara, T.; Uesugi, M. [In-vivo activation of the p53 pathway by small-molecule antagonists of MDM2]. Tanpakushitsu Kakusan Koso 2007, 52, 1816–1817. [Google Scholar]
- Voon, Y.L.; Ahmad, M.; Wong, P.F.; Husaini, R.; Ng, W.T.; Leong, C.O.; Lane, D.P.; Khoo, A.S. Nutlin-3 sensitizes nasopharyngeal carcinoma cells to cisplatin-induced cytotoxicity. Oncol. Rep. 2015, 34, 1692–1700. [Google Scholar] [CrossRef]
- Roskoski, R., Jr. Properties of FDA-approved small molecule phosphatidylinositol 3-kinase inhibitors prescribed for the treatment of malignancies. Pharmacol. Res. 2021, 168, 105579. [Google Scholar] [CrossRef]
- Kaboli, P.J.; Salimian, F.; Aghapour, S.; Xiang, S.X.; Zhao, Q.J.; Li, M.X.; Wu, X.; Du, F.K.; Zhao, Y.S.; Shen, J.; et al. Akt-targeted therapy as a promising strategy to overcome drug resistance in breast cancer—A comprehensive review from chemotherapy to immunotherapy. Pharmacol. Res. 2020, 156, 104806. [Google Scholar] [CrossRef]
- China Anti-cancer Association Tumor Drug Clinical Research; Breast Cancer Expert Committee; National Tumor Quality Control Center; Tumor Pathology Committee of China Anti-cancer Association; Boao Institute of Oncology Innovation. [Expert consensus on the clinical application of PI3K/AKT/mTOR inhibitors in the treatment of advanced breast cancer]. Zhonghua Zhong Liu Za Zhi 2022, 44, 673–692. [Google Scholar] [CrossRef]
- Ma, B.B.; Goh, B.C.; Lim, W.T.; Hui, E.P.; Tan, E.H.; Lopes Gde, L.; Lo, K.W.; Li, L.; Loong, H.; Foster, N.R.; et al. Multicenter phase II study of the AKT inhibitor MK-2206 in recurrent or metastatic nasopharyngeal carcinoma from patients in the mayo phase II consortium and the cancer therapeutics research group (MC1079). Investig. New Drugs 2015, 33, 985–991. [Google Scholar] [CrossRef]
- Yang, F.; Qian, X.J.; Qin, W.; Deng, R.; Wu, X.Q.; Qin, J.; Feng, G.K.; Zhu, X.F. Dual phosphoinositide 3-kinase/mammalian target of rapamycin inhibitor NVP-BEZ235 has a therapeutic potential and sensitizes cisplatin in nasopharyngeal carcinoma. PLoS ONE 2013, 8, e59879. [Google Scholar] [CrossRef]
- Wong, C.H.; Loong, H.H.; Hui, C.W.; Lau, C.P.; Hui, E.P.; Ma, B.B.; Chan, A.T. Preclinical evaluation of the PI3K-mTOR dual inhibitor PF-04691502 as a novel therapeutic drug in nasopharyngeal carcinoma. Investig. New Drugs 2013, 31, 1399–1408. [Google Scholar] [CrossRef]
- Bar-Sela, G.; Kuten, A.; Ben-Eliezer, S.; Gov-Ari, E.; Ben-Izhak, O. Expression of HER2 and C-KIT in nasopharyngeal carcinoma: Implications for a new therapeutic approach. Mod. Pathol. 2003, 16, 1035–1040. [Google Scholar] [CrossRef]
- Zhang, J.W.; Qin, T.; Hong, S.D.; Zhang, J.; Fang, W.F.; Zhao, Y.Y.; Yang, Y.P.; Xue, C.; Huang, Y.; Zhao, H.Y.; et al. Multiple oncogenic mutations related to targeted therapy in nasopharyngeal carcinoma. Chin. J. Cancer 2015, 34, 177–183. [Google Scholar] [CrossRef]
- Huang, P.Y.; Hong, M.H.; Zhang, X.; Mai, H.Q.; Luo, D.H.; Zhang, L. C-KIT overexpression and mutation in nasopharyngeal carcinoma cell lines and reactivity of Imatinib on these cell lines. Chin. J. Cancer 2010, 29, 131–135. [Google Scholar] [CrossRef]
- Roberts, P.J.; Stinchcombe, T.E. KRAS mutation: Should we test for it, and does it matter? J. Clin. Oncol. 2013, 31, 1112–1121. [Google Scholar] [CrossRef]
- Chen, X.; Liang, R.; Zhu, X. Anti-EGFR therapies in nasopharyngeal carcinoma. Biomed. Pharmacother. 2020, 131, 110649. [Google Scholar] [CrossRef]
- Pinheiro, G.; Pereira, T.; Dias, C.; Freitas, C.; Hespanhol, V.; Costa, J.L.; Cunha, A.; Oliveira, H.P. Identifying relationships between imaging phenotypes and lung cancer-related mutation status: EGFR and KRAS. Sci. Rep. 2020, 10, 3625. [Google Scholar] [CrossRef]
- Jin, X.; Yan, J.; Chen, C.; Chen, Y.; Huang, W.K. Integrated Analysis of Copy Number Variation, Microsatellite Instability, and Tumor Mutation Burden Identifies an 11-Gene Signature Predicting Survival in Breast Cancer. Front. Cell Dev. Biol. 2021, 9, 721505. [Google Scholar] [CrossRef]
- Chan, T.A.; Yarchoan, M.; Jaffee, E.; Swanton, C.; Quezada, S.A.; Stenzinger, A.; Peters, S. Development of tumor mutation burden as an immunotherapy biomarker: Utility for the oncology clinic. Ann. Oncol. 2019, 30, 44–56. [Google Scholar] [CrossRef]
- Buttner, R.; Longshore, J.W.; Lopez-Rios, F.; Merkelbach-Bruse, S.; Normanno, N.; Rouleau, E.; Penault-Llorca, F. Implementing TMB measurement in clinical practice: Considerations on assay requirements. ESMO Open. 2019, 4, e000442. [Google Scholar] [CrossRef]
- Chabanon, R.M.; Pedrero, M.; Lefebvre, C.; Marabelle, A.; Soria, J.C.; Postel-Vinay, S. Mutational Landscape and Sensitivity to Immune Checkpoint Blockers. Clin. Cancer Res. 2016, 22, 4309–4321. [Google Scholar] [CrossRef]
- Ali, S.M.; Yao, M.; Yao, J.; Wang, J.; Cheng, Y.; Schrock, A.B.; Chirn, G.W.; Chen, H.; Mu, S.; Gay, L.; et al. Comprehensive genomic profiling of different subtypes of nasopharyngeal carcinoma reveals similarities and differences to guide targeted therapy. Cancer 2017, 123, 3628–3637. [Google Scholar] [CrossRef] [PubMed]
- Chung, Y.L.; Wu, M.L. Spatiotemporal homogeneity and distinctness of the T-cell receptor beta-chain repertoires in Epstein-Barr virus-associated primary and metastatic nasopharyngeal carcinomas. Int. J. Cancer 2018, 143, 610–620. [Google Scholar] [CrossRef] [PubMed]
- Fang, W.; Yang, Y.; Ma, Y.; Hong, S.; Lin, L.; He, X.; Xiong, J.; Li, P.; Zhao, H.; Huang, Y.; et al. Camrelizumab (SHR-1210) alone or in combination with gemcitabine plus cisplatin for nasopharyngeal carcinoma: Results from two single-arm, phase 1 trials. Lancet Oncol. 2018, 19, 1338–1350. [Google Scholar] [CrossRef]
- Wang, F.H.; Wei, X.L.; Feng, J.; Li, Q.; Xu, N.; Hu, X.C.; Liao, W.; Jiang, Y.; Lin, X.Y.; Zhang, Q.Y.; et al. Efficacy, Safety, and Correlative Biomarkers of Toripalimab in Previously Treated Recurrent or Metastatic Nasopharyngeal Carcinoma: A Phase II Clinical Trial (POLARIS-02). J. Clin. Oncol. 2021, 39, 704–712. [Google Scholar] [CrossRef] [PubMed]
- Ma, Y.; Chen, X.; Wang, A.; Zhao, H.; Lin, Q.; Bao, H.; Zhang, Y.; Hong, S.; Tang, W.; Huang, Y.; et al. Copy number loss in granzyme genes confers resistance to immune checkpoint inhibitor in nasopharyngeal carcinoma. J. Immunother. Cancer 2021, 9, e002014. [Google Scholar] [CrossRef] [PubMed]
- Pecina-Slaus, N.; Kafka, A.; Salamon, I.; Bukovac, A. Mismatch Repair Pathway, Genome Stability and Cancer. Front. Mol. Biosci. 2020, 7, 122. [Google Scholar] [CrossRef]
- Chalmers, Z.R.; Connelly, C.F.; Fabrizio, D.; Gay, L.; Ali, S.M.; Ennis, R.; Schrock, A.; Campbell, B.; Shlien, A.; Chmielecki, J.; et al. Analysis of 100,000 human cancer genomes reveals the landscape of tumor mutational burden. Genome Med. 2017, 9, 34. [Google Scholar] [CrossRef]
- Chen, F.M.; Zhang, Y.X.; Li, X.F.; Gao, J.F.; Ma, H.; Wang, X.L.; Li, Y.; Li, C.; Zhang, Y.N.; Zhang, Y.T.; et al. The Prognostic Value of Deficient Mismatch Repair in Stage II-IVa Nasopharyngeal Carcinoma in the Era of IMRT. Sci. Rep. 2020, 10, 9690. [Google Scholar] [CrossRef]
- Wu, X.; Gu, Z.; Chen, Y.; Chen, B.; Chen, W.; Weng, L.; Liu, X. Application of PD-1 Blockade in Cancer Immunotherapy. Comput. Struct. Biotechnol. J. 2019, 17, 661–674. [Google Scholar] [CrossRef]
- Tumeh, P.C.; Hellmann, M.D.; Hamid, O.; Tsai, K.K.; Loo, K.L.; Gubens, M.A.; Rosenblum, M.; Harview, C.L.; Taube, J.M.; Handley, N.; et al. Liver Metastasis and Treatment Outcome with Anti-PD-1 Monoclonal Antibody in Patients with Melanoma and NSCLC. Cancer Immunol. Res. 2017, 5, 417–424. [Google Scholar] [CrossRef]
- Le, D.T.; Durham, J.N.; Smith, K.N.; Wang, H.; Bartlett, B.R.; Aulakh, L.K.; Lu, S.; Kemberling, H.; Wilt, C.; Luber, B.S.; et al. Mismatch repair deficiency predicts response of solid tumors to PD-1 blockade. Science 2017, 357, 409–413. [Google Scholar] [CrossRef]
- Mehra, R.; Seiwert, T.Y.; Gupta, S.; Weiss, J.; Gluck, I.; Eder, J.P.; Burtness, B.; Tahara, M.; Keam, B.; Kang, H.; et al. Efficacy and safety of pembrolizumab in recurrent/metastatic head and neck squamous cell carcinoma: Pooled analyses after long-term follow-up in KEYNOTE-012. Br. J. Cancer 2018, 119, 153–159. [Google Scholar] [CrossRef]
- Feng, B.; Hess, J. Immune-Related Mutational Landscape and Gene Signatures: Prognostic Value and Therapeutic Impact for Head and Neck Cancer. Cancers 2021, 13, 1162. [Google Scholar] [CrossRef]
- Davoli, T.; Uno, H.; Wooten, E.C.; Elledge, S.J. Tumor aneuploidy correlates with markers of immune evasion and with reduced response to immunotherapy. Science 2017, 355, eaaf8399. [Google Scholar] [CrossRef]
- Xu, J.Y.; Wei, X.L.; Wang, Y.Q.; Wang, F.H. Current status and advances of immunotherapy in nasopharyngeal carcinoma. Ther. Adv. Med. Oncol. 2022, 14, 17588359221096214. [Google Scholar] [CrossRef]
- Mandal, R.; Senbabaoglu, Y.; Desrichard, A.; Havel, J.J.; Dalin, M.G.; Riaz, N.; Lee, K.W.; Ganly, I.; Hakimi, A.A.; Chan, T.A.; et al. The head and neck cancer immune landscape and its immunotherapeutic implications. JCI Insight 2016, 1, e89829. [Google Scholar] [CrossRef]
- Everly, D.N., Jr.; Mainou, B.A.; Raab-Traub, N. The ID proteins contribute to the growth of rodent fibroblasts during LMP1-mediated transformation. Virology 2008, 376, 258–269. [Google Scholar] [CrossRef]
- Shi, Y.; Tao, Y.; Jiang, Y.; Xu, Y.; Yan, B.; Chen, X.; Xiao, L.; Cao, Y. Nuclear epidermal growth factor receptor interacts with transcriptional intermediary factor 2 to activate cyclin D1 gene expression triggered by the oncoprotein latent membrane protein 1. Carcinogenesis 2012, 33, 1468–1478. [Google Scholar] [CrossRef]
- Xu, Y.; Shi, Y.; Yuan, Q.; Liu, X.; Yan, B.; Chen, L.; Tao, Y.; Cao, Y. Epstein-Barr Virus encoded LMP1 regulates cyclin D1 promoter activity by nuclear EGFR and STAT3 in CNE1 cells. J. Exp. Clin. Cancer Res. 2013, 32, 90. [Google Scholar] [CrossRef]
- Yang, X.; He, Z.; Xin, B.; Cao, L. LMP1 of Epstein-Barr virus suppresses cellular senescence associated with the inhibition of p16INK4a expression. Oncogene 2000, 19, 2002–2013. [Google Scholar] [CrossRef]
- Lo, A.K.; Lo, K.W.; Ko, C.W.; Young, L.S.; Dawson, C.W. Inhibition of the LKB1-AMPK pathway by the Epstein-Barr virus-encoded LMP1 promotes proliferation and transformation of human nasopharyngeal epithelial cells. J. Pathol. 2013, 230, 336–346. [Google Scholar] [CrossRef] [PubMed]
- Lo, A.K.; Dawson, C.W.; Lo, K.W.; Yu, Y.; Young, L.S. Upregulation of Id1 by Epstein-Barr virus-encoded LMP1 confers resistance to TGFbeta-mediated growth inhibition. Mol. Cancer 2010, 9, 155. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Guo, L.; Tao, Y.; Zhou, S.; Wang, Z.; Luo, W.; Hu, D.; Li, Z.; Xiao, L.; Tang, M.; et al. Latent membrane protein 1 of Epstein-Barr virus regulates p53 phosphorylation through MAP kinases. Cancer Lett. 2007, 255, 219–231. [Google Scholar] [CrossRef] [PubMed]
- Fries, K.L.; Miller, W.E.; Raab-Traub, N. Epstein-Barr virus latent membrane protein 1 blocks p53-mediated apoptosis through the induction of the A20 gene. J. Virol. 1996, 70, 8653–8659. [Google Scholar] [CrossRef]
- Yang, J.; Deng, X.; Deng, L.; Gu, H.; Fan, W.; Cao, Y. Telomerase activation by Epstein-Barr virus latent membrane protein 1 is associated with c-Myc expression in human nasopharyngeal epithelial cells. J. Exp. Clin. Cancer Res. 2004, 23, 495–506. [Google Scholar]
- Kondo, S.; Seo, S.Y.; Yoshizaki, T.; Wakisaka, N.; Furukawa, M.; Joab, I.; Jang, K.L.; Pagano, J.S. EBV latent membrane protein 1 up-regulates hypoxia-inducible factor 1alpha through Siah1-mediated down-regulation of prolyl hydroxylases 1 and 3 in nasopharyngeal epithelial cells. Cancer Res. 2006, 66, 9870–9877. [Google Scholar] [CrossRef]
- Yang, L.; Liu, L.; Xu, Z.; Liao, W.; Feng, D.; Dong, X.; Xu, S.; Xiao, L.; Lu, J.; Luo, X.; et al. EBV-LMP1 targeted DNAzyme enhances radiosensitivity by inhibiting tumor angiogenesis via the JNKs/HIF-1 pathway in nasopharyngeal carcinoma. Oncotarget 2015, 6, 5804–5817. [Google Scholar] [CrossRef]
- Wakisaka, N.; Kondo, S.; Yoshizaki, T.; Murono, S.; Furukawa, M.; Pagano, J.S. Epstein-Barr virus latent membrane protein 1 induces synthesis of hypoxia-inducible factor 1 alpha. Mol. Cell Biol. 2004, 24, 5223–5234. [Google Scholar] [CrossRef]
- Wakisaka, N.; Murono, S.; Yoshizaki, T.; Furukawa, M.; Pagano, J.S. Epstein-barr virus latent membrane protein 1 induces and causes release of fibroblast growth factor-2. Cancer Res. 2002, 62, 6337–6344. [Google Scholar]
- Murono, S.; Inoue, H.; Tanabe, T.; Joab, I.; Yoshizaki, T.; Furukawa, M.; Pagano, J.S. Induction of cyclooxygenase-2 by Epstein-Barr virus latent membrane protein 1 is involved in vascular endothelial growth factor production in nasopharyngeal carcinoma cells. Proc. Natl. Acad. Sci. USA 2001, 98, 6905–6910. [Google Scholar] [CrossRef]
- Yoshizaki, T. Promotion of metastasis in nasopharyngeal carcinoma by Epstein-Barr virus latent membrane protein-1. Histol. Histopathol. 2002, 17, 845–850. [Google Scholar] [CrossRef]
- Tsuji, A.; Wakisaka, N.; Kondo, S.; Murono, S.; Furukawa, M.; Yoshizaki, T. Induction of receptor for advanced glycation end products by EBV latent membrane protein 1 and its correlation with angiogenesis and cervical lymph node metastasis in nasopharyngeal carcinoma. Clin. Cancer Res. 2008, 14, 5368–5375. [Google Scholar] [CrossRef]
- Wei, J.; Zhang, J.; Si, Y.; Kanada, M.; Zhang, Z.; Terakawa, S.; Watanabe, H. Blockage of LMP1-modulated store-operated Ca(2+) entry reduces metastatic potential in nasopharyngeal carcinoma cell. Cancer Lett. 2015, 360, 234–244. [Google Scholar] [CrossRef]
- Wang, Z.; Luo, F.; Li, L.; Yang, L.; Hu, D.; Ma, X.; Lu, Z.; Sun, L.; Cao, Y. STAT3 activation induced by Epstein-Barr virus latent membrane protein1 causes vascular endothelial growth factor expression and cellular invasiveness via JAK3 And ERK signaling. Eur. J. Cancer 2010, 46, 2996–3006. [Google Scholar] [CrossRef]
- Horikawa, T.; Yang, J.; Kondo, S.; Yoshizaki, T.; Joab, I.; Furukawa, M.; Pagano, J.S. Twist and epithelial-mesenchymal transition are induced by the EBV oncoprotein latent membrane protein 1 and are associated with metastatic nasopharyngeal carcinoma. Cancer Res. 2007, 67, 1970–1978. [Google Scholar] [CrossRef]
- Zuo, L.L.; Zhang, J.; Liu, L.Z.; Zhou, Q.; Du, S.J.; Xin, S.Y.; Ning, Z.P.; Yang, J.; Yu, H.B.; Yue, W.X.; et al. Cadherin 6 is activated by Epstein-Barr virus LMP1 to mediate EMT and metastasis as an interplay node of multiple pathways in nasopharyngeal carcinoma. Oncogenesis 2017, 6, 402. [Google Scholar] [CrossRef]
- Aga, M.; Bentz, G.L.; Raffa, S.; Torrisi, M.R.; Kondo, S.; Wakisaka, N.; Yoshizaki, T.; Pagano, J.S.; Shackelford, J. Exosomal HIF1alpha supports invasive potential of nasopharyngeal carcinoma-associated LMP1-positive exosomes. Oncogene 2014, 33, 4613–4622. [Google Scholar] [CrossRef]
- Ye, D.; Zhu, J.; Zhao, Q.; Ma, W.; Xiao, Y.; Xu, G.; Zhang, Z. LMP1 Up-regulates Calreticulin to Induce Epithelial-mesenchymal Transition via TGF-beta/Smad3/NRP1 Pathway in Nasopharyngeal Carcinoma Cells. J. Cancer 2020, 11, 1257–1269. [Google Scholar] [CrossRef]
- Zhang, J.; Jia, L.; Liu, T.; Yip, Y.L.; Tang, W.C.; Lin, W.; Deng, W.; Lo, K.W.; You, C.; Lung, M.L.; et al. mTORC2-mediated PDHE1alpha nuclear translocation links EBV-LMP1 reprogrammed glucose metabolism to cancer metastasis in nasopharyngeal carcinoma. Oncogene 2019, 38, 4669–4684. [Google Scholar] [CrossRef]
- Horikawa, T.; Yoshizaki, T.; Kondo, S.; Furukawa, M.; Kaizaki, Y.; Pagano, J.S. Epstein-Barr Virus latent membrane protein 1 induces Snail and epithelial-mesenchymal transition in metastatic nasopharyngeal carcinoma. Br. J. Cancer 2011, 104, 1160–1167. [Google Scholar] [CrossRef]
- Kim, K.R.; Yoshizaki, T.; Miyamori, H.; Hasegawa, K.; Horikawa, T.; Furukawa, M.; Harada, S.; Seiki, M.; Sato, H. Transformation of Madin-Darby canine kidney (MDCK) epithelial cells by Epstein-Barr virus latent membrane protein 1 (LMP1) induces expression of Ets1 and invasive growth. Oncogene 2000, 19, 1764–1771. [Google Scholar] [CrossRef]
- Kondo, S.; Wakisaka, N.; Schell, M.J.; Horikawa, T.; Sheen, T.S.; Sato, H.; Furukawa, M.; Pagano, J.S.; Yoshizaki, T. Epstein-Barr virus latent membrane protein 1 induces the matrix metalloproteinase-1 promoter via an Ets binding site formed by a single nucleotide polymorphism: Enhanced susceptibility to nasopharyngeal carcinoma. Int. J. Cancer 2005, 115, 368–376. [Google Scholar] [CrossRef]
- Zeng, L.; Liu, Y.P.; Tao, Y.G.; Ai, M.D.; Zhao, X.R.; Cao, Y. [Cross-talk between c-Jun/Ets1 involved in EB virus-encoded latent membrane protein 1 regulates expression of matrix metalloproteinase-9 in nasopharyngeal carcinoma]. Zhonghua Zhong Liu Za Zhi 2005, 27, 204–208. [Google Scholar]
- Horikawa, T.; Yoshizaki, T.; Sheen, T.S.; Lee, S.Y.; Furukawa, M. Association of latent membrane protein 1 and matrix metalloproteinase 9 with metastasis in nasopharyngeal carcinoma. Cancer 2000, 89, 715–723. [Google Scholar] [CrossRef]
- Himelstein, B.P.; Lee, E.J.; Sato, H.; Seiki, M.; Muschel, R.J. Transcriptional activation of the matrix metalloproteinase-9 gene in an H-ras and v-myc transformed rat embryo cell line. Oncogene 1997, 14, 1995–1998. [Google Scholar] [CrossRef]
- Luo, X.; Hong, L.; Cheng, C.; Li, N.; Zhao, X.; Shi, F.; Liu, J.; Fan, J.; Zhou, J.; Bode, A.M.; et al. DNMT1 mediates metabolic reprogramming induced by Epstein-Barr virus latent membrane protein 1 and reversed by grifolin in nasopharyngeal carcinoma. Cell Death Dis. 2018, 9, 619. [Google Scholar] [CrossRef]
- Zhang, J.; Jia, L.; Lin, W.; Yip, Y.L.; Lo, K.W.; Lau, V.M.Y.; Zhu, D.; Tsang, C.M.; Zhou, Y.; Deng, W.; et al. Epstein-Barr Virus-Encoded Latent Membrane Protein 1 Upregulates Glucose Transporter 1 Transcription via the mTORC1/NF-kappaB Signaling Pathways. J. Virol. 2017, 91. [Google Scholar] [CrossRef]
- Xiao, L.; Hu, Z.Y.; Dong, X.; Tan, Z.; Li, W.; Tang, M.; Chen, L.; Yang, L.; Tao, Y.; Jiang, Y.; et al. Targeting Epstein-Barr virus oncoprotein LMP1-mediated glycolysis sensitizes nasopharyngeal carcinoma to radiation therapy. Oncogene 2014, 33, 4568–4578. [Google Scholar] [CrossRef]
- Klibi, J.; Niki, T.; Riedel, A.; Pioche-Durieu, C.; Souquere, S.; Rubinstein, E.; Le Moulec, S.; Guigay, J.; Hirashima, M.; Guemira, F.; et al. Blood diffusion and Th1-suppressive effects of galectin-9-containing exosomes released by Epstein-Barr virus-infected nasopharyngeal carcinoma cells. Blood 2009, 113, 1957–1966. [Google Scholar] [CrossRef]
- Xiang, T.; Lin, Y.X.; Ma, W.; Zhang, H.J.; Chen, K.M.; He, G.P.; Zhang, X.; Xu, M.; Feng, Q.S.; Chen, M.Y.; et al. Vasculogenic mimicry formation in EBV-associated epithelial malignancies. Nat. Commun. 2018, 9, 5009. [Google Scholar] [CrossRef]
- Pegtel, D.M.; Subramanian, A.; Sheen, T.S.; Tsai, C.H.; Golub, T.R.; Thorley-Lawson, D.A. Epstein-Barr-virus-encoded LMP2A induces primary epithelial cell migration and invasion: Possible role in nasopharyngeal carcinoma metastasis. J. Virol. 2005, 79, 15430–15442. [Google Scholar] [CrossRef] [PubMed]
- Fotheringham, J.A.; Coalson, N.E.; Raab-Traub, N. Epstein-Barr virus latent membrane protein-2A induces ITAM/Syk- and Akt-dependent epithelial migration through alphav-integrin membrane translocation. J. Virol. 2012, 86, 10308–10320. [Google Scholar] [CrossRef] [PubMed]
- Fotheringham, J.A.; Mazzucca, S.; Raab-Traub, N. Epstein-Barr virus latent membrane protein-2A-induced DeltaNp63alpha expression is associated with impaired epithelial-cell differentiation. Oncogene 2010, 29, 4287–4296. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Lee, H.M.; Lo, K.W.; Wei, W.; Tsao, S.W.; Chung, G.T.Y.; Ibrahim, M.H.; Dawson, C.W.; Murray, P.G.; Paterson, I.C.; Yap, L.F. Oncogenic S1P signalling in EBV-associated nasopharyngeal carcinoma activates AKT and promotes cell migration through S1P receptor 3. J. Pathol. 2017, 242, 62–72. [Google Scholar] [CrossRef]
- Fukuda, M.; Longnecker, R. Latent membrane protein 2A inhibits transforming growth factor-beta 1-induced apoptosis through the phosphatidylinositol 3-kinase/Akt pathway. J. Virol. 2004, 78, 1697–1705. [Google Scholar] [CrossRef]
- Wang, L.; Tian, W.D.; Xu, X.; Nie, B.; Lu, J.; Liu, X.; Zhang, B.; Dong, Q.; Sunwoo, J.B.; Li, G.; et al. Epstein-Barr virus nuclear antigen 1 (EBNA1) protein induction of epithelial-mesenchymal transition in nasopharyngeal carcinoma cells. Cancer 2014, 120, 363–372. [Google Scholar] [CrossRef]
- Lu, J.; Murakami, M.; Verma, S.C.; Cai, Q.; Haldar, S.; Kaul, R.; Wasik, M.A.; Middeldorp, J.; Robertson, E.S. Epstein-Barr Virus nuclear antigen 1 (EBNA1) confers resistance to apoptosis in EBV-positive B-lymphoma cells through up-regulation of survivin. Virology 2011, 410, 64–75. [Google Scholar] [CrossRef]
- Fan, C.; Tang, Y.; Wang, J.; Xiong, F.; Guo, C.; Wang, Y.; Xiang, B.; Zhou, M.; Li, X.; Wu, X.; et al. The emerging role of Epstein-Barr virus encoded microRNAs in nasopharyngeal carcinoma. J. Cancer 2018, 9, 2852–2864. [Google Scholar] [CrossRef]
- Cai, L.; Ye, Y.; Jiang, Q.; Chen, Y.; Lyu, X.; Li, J.; Wang, S.; Liu, T.; Cai, H.; Yao, K.; et al. Epstein-Barr virus-encoded microRNA BART1 induces tumour metastasis by regulating PTEN-dependent pathways in nasopharyngeal carcinoma. Nat. Commun. 2015, 6, 7353, Correction in Nat. Commun. 2020, 11, 3437. [Google Scholar]
- Choy, E.Y.; Siu, K.L.; Kok, K.H.; Lung, R.W.; Tsang, C.M.; To, K.F.; Kwong, D.L.; Tsao, S.W.; Jin, D.Y. An Epstein-Barr virus-encoded microRNA targets PUMA to promote host cell survival. J. Exp. Med. 2008, 205, 2551–2560. [Google Scholar] [CrossRef]
- Wong, T.S.; Chen, S.; Zhang, M.J.; Chan, J.Y.; Gao, W. Epstein-Barr virus-encoded microRNA BART7 downregulates major histocompatibility complex class I chain-related peptide A and reduces the cytotoxicity of natural killer cells to nasopharyngeal carcinoma. Oncol. Lett. 2018, 16, 2887–2892. [Google Scholar] [CrossRef]
- Song, Y.; Li, X.; Zeng, Z.; Li, Q.; Gong, Z.; Liao, Q.; Li, X.; Chen, P.; Xiang, B.; Zhang, W.; et al. Epstein-Barr virus encoded miR-BART11 promotes inflammation-induced carcinogenesis by targeting FOXP1. Oncotarget 2016, 7, 36783–36799. [Google Scholar] [CrossRef]
- Huang, J.; Qin, Y.; Yang, C.; Wan, C.; Dai, X.; Sun, Y.; Meng, J.; Lu, Y.; Li, Y.; Zhang, Z.; et al. Downregulation of ABI2 expression by EBV-miR-BART13-3p induces epithelial-mesenchymal transition of nasopharyngeal carcinoma cells through upregulation of c-JUN/SLUG signaling. Aging 2020, 12, 340–358. [Google Scholar] [CrossRef]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).