Low-Grade Primary Splenic CD10-Positive Small B-Cell Lymphoma/Follicular Lymphoma


 and
and 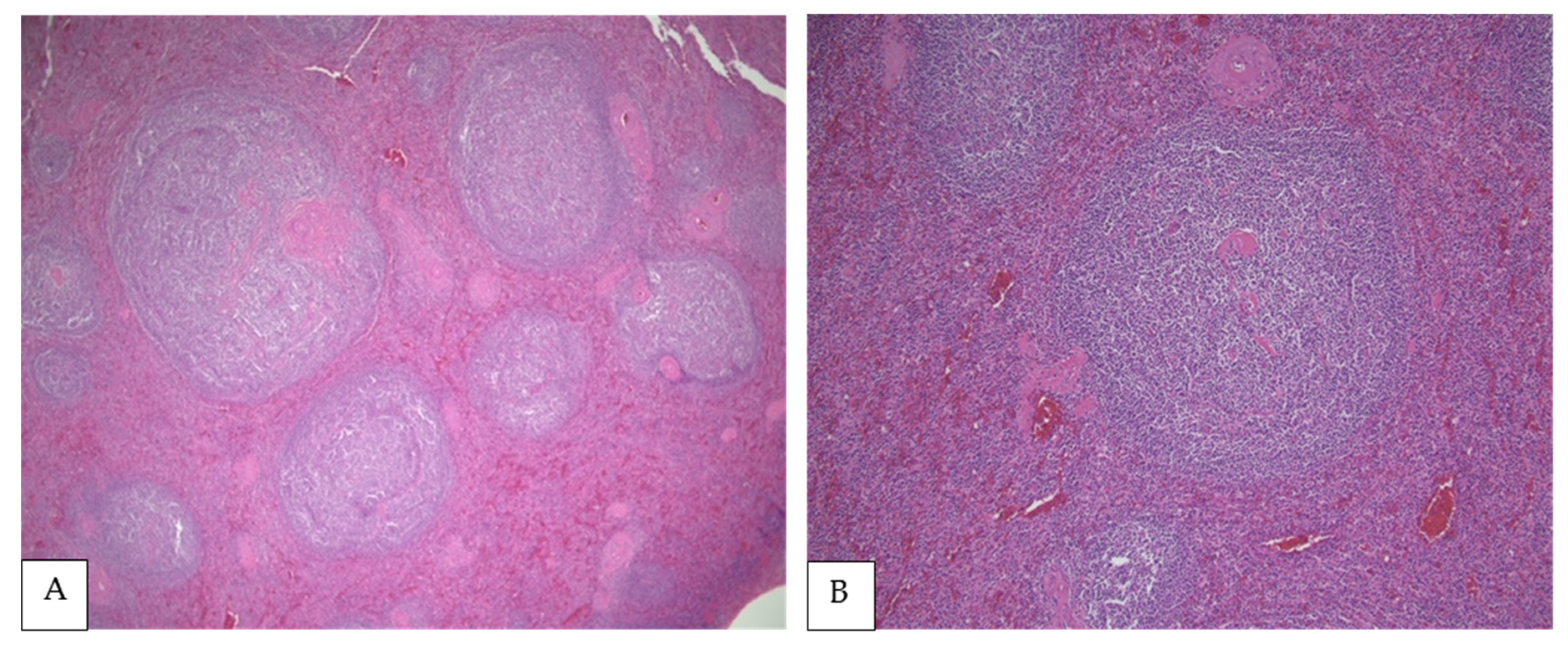{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Epidemiology
3. Clinical Presentation
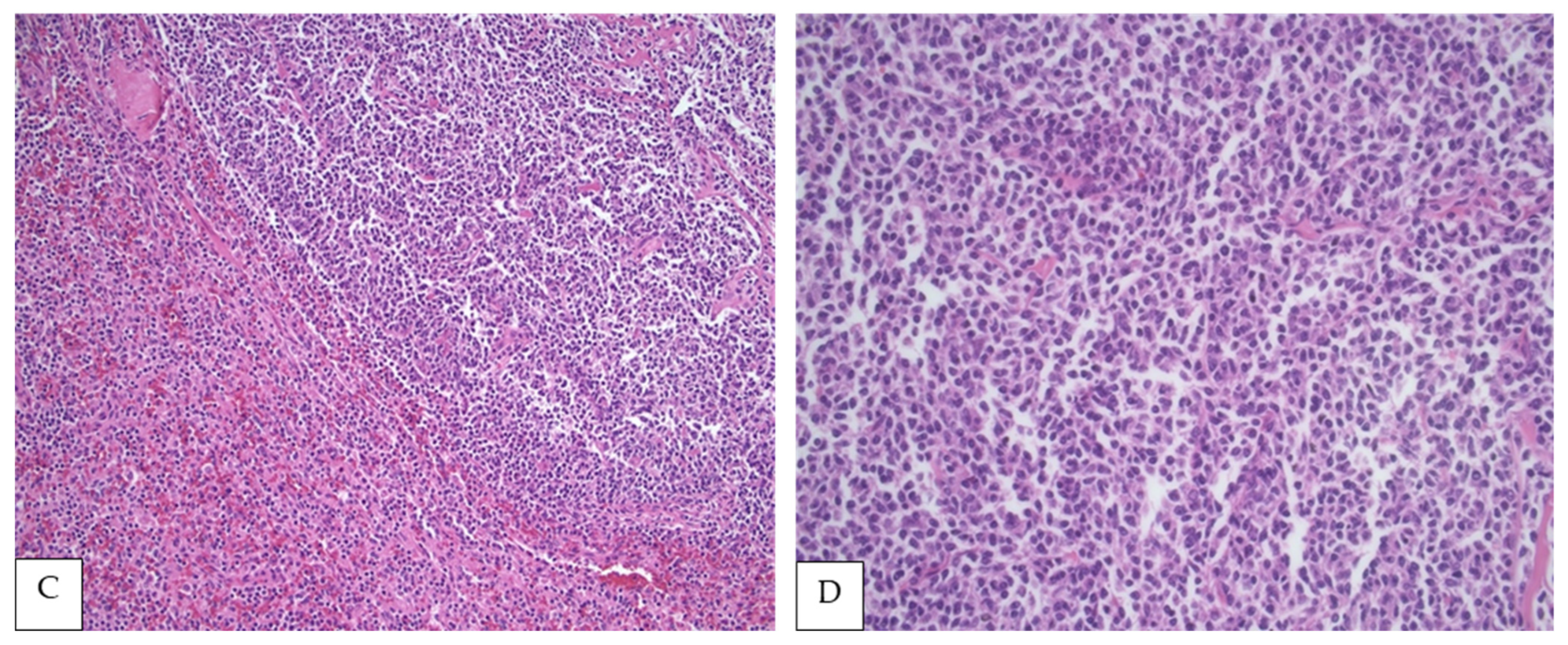
4. Morphologic Findings
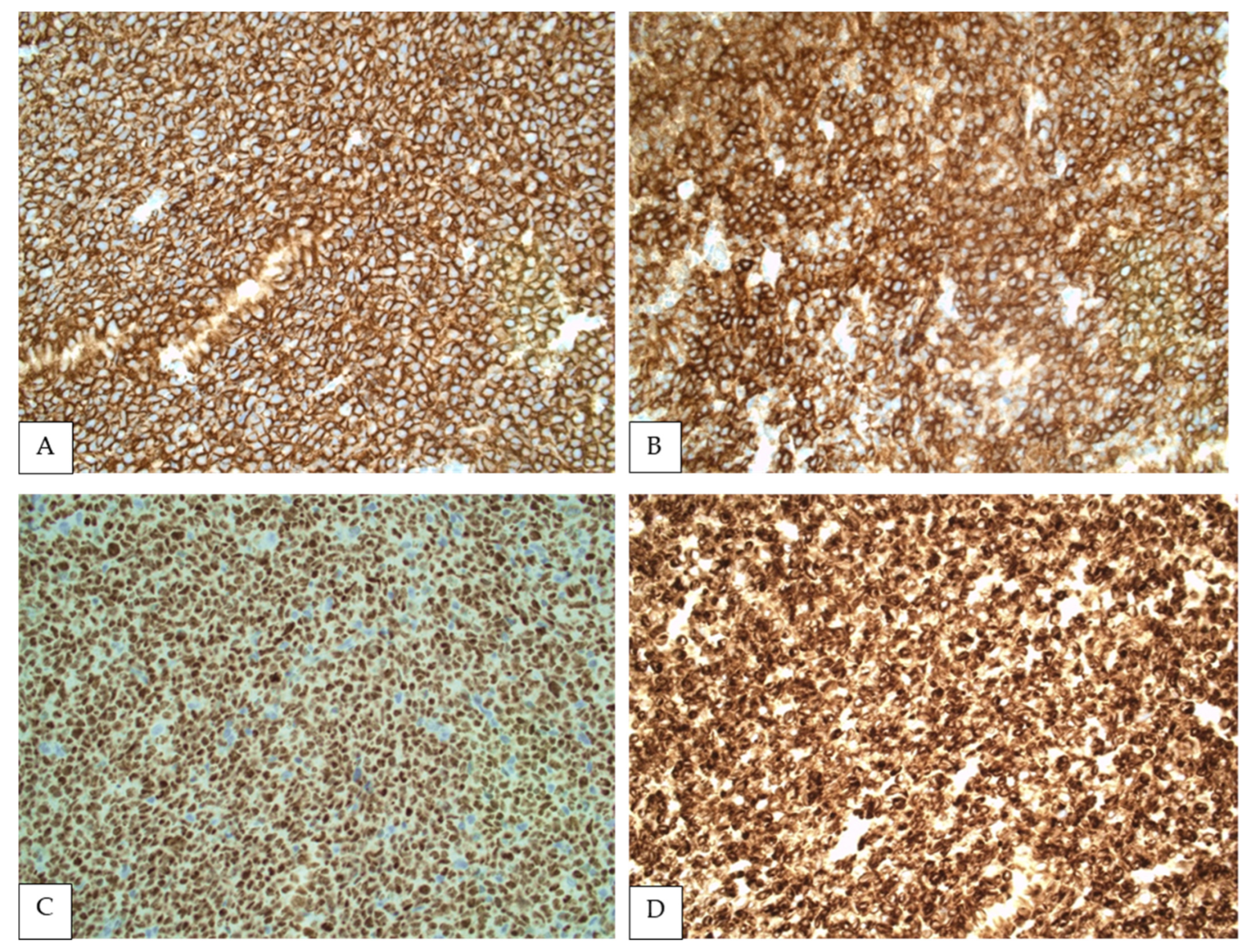
5. Immunophenotypic Findings
6. Genetic Findings
7. Diagnostic Work-Up
8. Differential Diagnosis
9. Treatment and Prognosis
10. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Maguer-Satta, V.; Besançon, R.; Bachelard-Cascales, E. Concise review: Neutral endopeptidase (CD10): A multifaceted environment actor in stem cells, physiological mechanisms, and cancer. Stem Cells 2011, 29, 389–396. [Google Scholar] [CrossRef]
- Erdös, E.G.; Skidgel, R.A. Neutral endopeptidase 24.11 (enkephalinase) and related regulators of peptide hormones. FASEB J. 1989, 3, 145–151. [Google Scholar] [CrossRef]
- McIntosh, G.G.; Lodge, A.J.; Watson, P.; Hall, A.G.; Wood, K.; Anderson, J.J.; Angus, B.; Horne, C.H.W.; Milton, I.D. NCL-CD10-270: A new monoclonal antibody recognizing CD10 in paraffin-embedded tissue. Am. J. Pathol. 1999, 154, 77–82. [Google Scholar] [CrossRef]
- Williamson, J.M.S.; Grigor, I.; Smith, M.E.F.; Holgate, C.S.; Quirke, P.; Bird, C.C.; Alison, D.L.; Child, J.A. Cluster differentiation antigen expression, proliferative activity and clinical stage in centroblastic centrocytic lymphomas. J. Pathol. 1986, 150, 51–59. [Google Scholar] [CrossRef]
- Choi, W.W.L.; Weisenburger, D.D.; Greiner, T.C.; Piris, M.A.; Banham, A.H.; Delabie, J.; Braziel, R.M.; Geng, H.; Iqbal, J.; Lenz, G.; et al. A new immunostain algorithm classifies diffuse large B-cell lymphoma into molecular subtypes with high accuracy. Clin. Cancer Res. 2009, 15, 5494–5502. [Google Scholar] [CrossRef] [PubMed]
- Younes, S.F.; Beck, A.H.; Lossos, I.S.; Levy, R.; Warnke, R.A.; Natkunam, Y. Immunoarchitectural patterns in follicular lymphoma: Efficacy of HGAL and LMO2 in the detection of the interfollicular and diffuse components. Am. J. Surg. Pathol. 2010, 34, 1266–1276. [Google Scholar] [CrossRef]
- Jasionowski, T.M.; Hartung, L.; Greenwood, J.H.; Perkins, S.L.; Bahler, D.W. Analysis of CD10+ hairy cell leukemia. Am. J. Clin. Pathol. 2003, 120, 228–235. [Google Scholar] [CrossRef] [PubMed]
- Dong, H.Y.; Gorczyca, W.; Liu, Z.; Tsang, P.; Wu, C.D.; Cohen, P.; Weisberger, J. B-cell lymphomas with coexpression of CD5 and CD10. Am. J. Clin. Pathol. 2003, 119, 218–230. [Google Scholar] [CrossRef] [PubMed]
- Swerdlow, S.H.; Campo, E.; Harris, N.L.; Jaffe, E.S.; Pileri, S.A.; Stein, H.; Thiele, J. (Eds.) WHO Classification of Tumours of Haematopoietic and Lymphoid Tissues, Revised, 4th ed.; IARC: Lyon, France, 2017. [Google Scholar]
- Ferry, J.A.; Fung, C.Y.; Zukerberg, L.; Lucarelli, M.J.; Hasserjian, R.P.; Preffer, F.I.; Harris, N.L. Lymphoma of the ocular adnexa: A study of 353 cases. Am. J. Surg. Pathol. 2007, 31, 170–184. [Google Scholar] [CrossRef] [PubMed]
- Bende, R.; Smit, L.A.; Van Noesel, C.J.M. Molecular pathways in follicular lymphoma. Leukemia 2006, 21, 18–29. [Google Scholar] [CrossRef]
- Gobbi, P.G.; Grignani, G.E.; Pozzetti, U.; Bertoloni, D.; Pieresca, C.; Montagna, G.; Ascari, E. Primary splenic lymphoma: Does it exist? Haematologica 1994, 79, 286–293. [Google Scholar]
- Healy, N.A.; Conneely, J.B.; Mahon, S.; O’Riardon, C.; McAnena, O.J. Primary splenic lymphoma presenting with ascites. Rare Tumors 2011, 3, 81–82. [Google Scholar] [CrossRef] [PubMed]
- Shimizu-Kohno, K.; Kimura, Y.; Kiyasu, J.; Miyoshi, H.; Yoshida, M.; Ichikawa, R.; Niino, D.; Ohshima, K. Malignant lymphoma of the spleen in Japan: A clinicopathological analysis of 115 cases. Pathol. Int. 2012, 62, 577–582. [Google Scholar] [CrossRef] [PubMed]
- Özsan, N.; Bedke, B.J.; Law, M.E.; Inwards, D.J.; Ketterling, R.P.; Knudson, R.A.; Keeney, G.L.; Dogan, A.; Feldman, A.L. Clinicopathologic and genetic characterization of follicular lymphomas presenting in the ovary reveals 2 distinct subgroups. Am. J. Surg. Pathol. 2011, 35, 1691–1699. [Google Scholar] [CrossRef]
- Vang, R.; Medeiros, L.J.; Ha, C.S.; Deavers, M.T. Non-Hodgkin’s lymphomas involving the uterus: A clinicopathologic analysis of 26 cases. Mod. Pathol. 2000, 13, 19–28. [Google Scholar] [CrossRef] [PubMed]
- Howard, M.T.; Dufresne, S.; Swerdlow, S.H.; Cook, J.R. Follicular lymphoma of the spleen: Multiparameter analysis of 16 cases. Am. J. Clin. Pathol. 2009, 131, 656–662. [Google Scholar] [CrossRef] [PubMed]
- Shimono, J.; Miyoshi, H.; Kamimura, T.; Eto, T.; Miyagishima, T.; Sasaki, Y.; Kurita, D.; Kawamoto, K.; Nagafuji, K.; Seto, M.; et al. Clinicopathological features of primary splenic follicular lymphoma. Ann. Hematol. 2017, 96, 2063–2070. [Google Scholar] [CrossRef]
- Jhuang, J.Y.; Hsieh, Y.C.; Kuo, C.C.; Su, Y.Z.; Chuang, S.S. Primary splenic low-grade follicular lymphoma presenting with leukaemia and large cell transformation in the marrow. Pathology 2017, 49, 649–652, Erratum in 2017, 49, 819. [Google Scholar] [CrossRef]
- Le, M.; Ghazawi, F.; Alakel, A.; Netchiporouk, E.; Rahme, E.; Zubarev, A.; Powell, M.; Moreau, L.; Roshdy, O.; Glassman, S.J.; et al. Incidence and Mortality Trends and Geographic Patterns of Follicular Lymphoma in Canada. Curr. Oncol. 2019, 26, 473–481. [Google Scholar] [CrossRef]
- Junlén, H.R.; Peterson, S.E.; Kimby, E.; Lockmer, S.; Linden, O.; Nilssonehle, H.; Erlanson, M.; Hagberg, H.; Rådlund, A.; Hagberg, O.; et al. Follicular lymphoma in Sweden: Nationwide improved survival in the rituximab era, particularly in elderly women: A Swedish Lymphoma Registry study. Leukemia 2015, 29, 668–676. [Google Scholar] [CrossRef]
- Mollejo, M.; Rodríguez-Pinilla, M.S.; Montes-Moreno, S.; Algara, P.; Dogan, A.; Cigudosa, J.C.; Juarez, R.; Flores, T.; Forteza, J.; Arribas, A.; et al. Splenic follicular lymphoma: Clinicopathologic characteristics of a series of 32 cases. Am. J. Surg. Pathol. 2009, 33, 730–738. [Google Scholar] [CrossRef] [PubMed]
- Fallah, J.; Olszewski, A.J. Diagnostic and therapeutic splenectomy for splenic lymphomas: Analysis of the National Cancer Data Base. Hematology 2019, 24, 378–386. [Google Scholar] [CrossRef] [PubMed]
- Matsuda, I.; Okada, M.; Inoue, T.; Tokugawa, T.; Ogawa, H.; Hirota, S. Primary follicular lymphoma of the spleen incidentally found in a patient with alcohol- and hepatitis C-related liver cirrhosis. Int. J. Clin. Exp. Pathol. 2014, 7, 4484–4488. [Google Scholar]
- Dayama, A.P.; Kapoor, R.; Dass, J.; Singh, G.; Mahapatra, M.; Pati, H. Pathologic splenic rupture in a patient with follicular lymphoma. Mediterr. J. Hematol. Infect. Dis. 2011, 3, e2011051. [Google Scholar] [CrossRef] [PubMed]
- Bilalovic, N.; Blystad, A.K.; Golouh, R.; Nesland, J.M.; Selak, I.; Trinh, D.; Torlakovic, E. Expression of bcl-6 and CD10 protein is associated with longer overall survival and time to treatment failure in follicular lymphoma. Am. J. Clin. Pathol. 2004, 121, 34–42. [Google Scholar] [CrossRef]
- Dogan, A.; Bagdi, E.; Munson, P.; Isaacson, P.G. CD10 and BCL-6 expression in paraffin sections of normal lymphoid tissue and b-cell lymphomas. Am. J. Surg. Pathol. 2000, 24, 846–852. [Google Scholar] [CrossRef]
- Gaulard, P.; D’Agay, M.F.; Peuchmaur, M.; Brousse, N.; Gisselbrecht, C.; Solal-Celigny, P.; Diebold, J.; Mason, D.Y. Expression of the bcl-2 gene product in follicular lymphoma. Am. J. Pathol. 1992, 140, 1089–1095. [Google Scholar]
- Younes, S.F.; Beck, A.H.; Ohgami, R.S.; Lossos, I.S.; Levy, R.; Warnke, R.A.; Natkunam, Y. The efficacy of HGAL and LMO2 in the separation of lymphomas derived from small B cells in nodal and extranodal sites, including the bone marrow. Am. J. Clin. Pathol. 2011, 135, 697–708. [Google Scholar] [CrossRef]
- Lai, R.; Weiss, L.M.; Chang, K.L.; Arber, D.A. Frequency of CD43 expression in non-Hodgkin lymphoma. A survey of 742 cases and further characterization of rare CD43+ follicular lymphomas. Am. J. Clin. Pathol. 1999, 111, 488–494. [Google Scholar] [CrossRef][Green Version]
- De Sanjose, S.; Benavente, Y.; Vajdic, C.; Engels, E.A.; Morton, L.M.; Bracci, P.M.; Spinelli, J.J.; Zheng, T.; Zhang, Y.; Franceschi, S.; et al. Hepatitis C and non-Hodgkin lymphoma among 4784 cases and 6269 controls from the International Lymphoma Epidemiology Consortium. Clin. Gastroenterol. Hepatol. 2008, 6, 451–458. [Google Scholar] [CrossRef]
- Yamamoto, E.; Tomita, N.; Sakata, S.; Tsuyama, N.; Takeuchi, K.; Nakajima, Y.; Miyashita, K.; Tachibana, T.; Takasaki, H.; Tanaka, M.; et al. MIB-1 labeling index as a prognostic factor for patients with follicular lymphoma treated with rituximab plus CHOP therapy. Cancer Sci. 2013, 104, 1670–1674. [Google Scholar] [CrossRef] [PubMed]
- Koster, A.; Tromp, H.A.; Raemaekers, J.M.; Borm, G.F.; Hebeda, K.; MacKenzie, M.A.; Van Krieken, J.H. The prognostic significance of the intra-follicular tumor cell proliferative rate in follicular lymphoma. Haematologica 2007, 92, 184–190. [Google Scholar] [CrossRef]
- Wang, S.A.; Wang, L.; Hochberg, E.P.; Muzikansky, A.; Harris, N.L.; Hasserjian, R.P. Low histologic grade follicular lymphoma with high proliferation index: Morphologic and clinical features. Am. J. Surg. Pathol. 2005, 29, 1490–1496. [Google Scholar] [CrossRef] [PubMed]
- Vaandrager, J.W.; Schuuring, E.; Raap, T.; Philippo, K.; Kleiverda, K.; Kluin, P. Interphase FISH detection of BCL2 rearrangement in follicular lymphoma using breakpoint-flanking probes. Genes Chromosomes Cancer 2000, 27, 85–94. [Google Scholar] [CrossRef]
- Karube, K.; Martinez, D.; Royo, C.; Navarro, A.; Pinyol, M.; Cazorla, M.; Castillo, P.; Valera, A.; Carrió, A.; Costa, D.; et al. Recurrent mutations ofNOTCHgenes in follicular lymphoma identify a distinctive subset of tumours. J. Pathol. 2014, 234, 423–430. [Google Scholar] [CrossRef] [PubMed]
- Dreyling, M.; Ghielmini, M.; Rule, S.; Salles, G.; Ladetto, M.; Tonino, S.; Herfarth, K.; Seymour, J.; Jerkeman, M. Newly diagnosed and relapsed follicular lymphoma: ESMO Clinical Practice Guidelines for diagnosis, treatment and follow-up. Ann. Oncol. 2021, 32, 298–308. [Google Scholar] [CrossRef]
- Iannitto, E.; Tripodo, C. How I diagnose and treat splenic lymphomas. Blood 2011, 117, 2585–2595. [Google Scholar] [CrossRef] [PubMed]
- Choi, S.M.; Betz, B.L.; Perry, A.M. Follicular Lymphoma Diagnostic Caveats and Updates. Arch. Pathol. Lab. Med. 2018, 142, 1330–1340. [Google Scholar] [CrossRef]
- Kashimura, M.; Noro, M.; Akikusa, B.; Okuhara, A.; Momose, S.; Miura, I.; Kojima, M.; Tamaru, J.-I. Primary splenic diffuse large B-cell lymphoma manifesting in red pulp. Virchows Arch. 2008, 453, 501–509. [Google Scholar] [CrossRef]
- Davidson, T.; Priel, E.; Schiby, G.; Raskin, S.; Chikman, B.; Nissan, E.; Benjamini, O.; Nissan, J.; Goshen, E.; Ben-Haim, S.; et al. Low rate of spleen involvement in sporadic Burkitt lymphoma at staging on PET-CT. Abdom. Radiol. 2018, 43, 2369–2374. [Google Scholar] [CrossRef]
- Zhang, X.M.; Aguilera, N. New immunohistochemistry for B-cell lymphoma and Hodgkin lymphoma. Arch. Pathol. Lab. Med. 2014, 138, 1666–1672. [Google Scholar] [CrossRef] [PubMed]
- Boyd, S.D.; Natkunam, Y.; Allen, J.R.; Warnke, R.A. Selective immunophenotyping for diagnosis of B-cell neoplasms: Immunohistochemistry and flow cytometry strategies and results. Appl. Immunohistochem. Mol. Morphol. 2013, 21, 116–131. [Google Scholar] [CrossRef]
- Gaidano, V.; Tenace, V.; Santoro, N.; Varvello, S.; Cignetti, A.; Prato, G.; Saglio, G.; De Rosa, G.; Geuna, M. A clinically applicable approach to the classification of B-cell non-hodgkin lymphomas with flow cytometry and machine learning. Cancers 2020, 12, 1684. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Cook, J.R. IRTA1 and MNDA Expression in Marginal Zone Lymphoma: Utility in Differential Diagnosis and Implications for Classification. Am. J. Clin. Pathol. 2018, 151, 337–343. [Google Scholar] [CrossRef] [PubMed]
- Donzel, M.; Baseggio, L.; Fontaine, J.; Pesce, F.; Ghesquières, H.; Bachy, E.; Verney, A.; Traverse-Glehen, A. New Insights into the Biology and Diagnosis of Splenic Marginal Zone Lymphomas. Curr. Oncol. 2021, 28, 3430–3447. [Google Scholar] [CrossRef]
- Yu, X.; Li, W.; Deng, Q.; Li, L.; Hsi, E.D.; Young, K.H.; Zhang, M.; Li, Y. MYD88 L265P Mutation in Lymphoid Malignancies. Cancer Res. 2018, 78, 2457–2462. [Google Scholar] [CrossRef]
- Maciocia, N.; O’Brien, A.; Ardeshna, K. Remission of Follicular Lymphoma after Treatment for Hepatitis C Virus Infection. N. Engl. J. Med. 2016, 375, 1699–1701. [Google Scholar] [CrossRef]
- Arcaini, L.; Besson, C.; Frigeni, M.; Fontaine, H.; Goldaniga, M.; Casato, M.; Visentini, M.; Torres, H.A.; Loustaud-Ratti, V.; Peveling-Oberhag, J.; et al. Interferon-free antiviral treatment in B-cell lymphoproliferative disorders associated with hepatitis C virus infection. Blood 2016, 128, 2527–2532. [Google Scholar] [CrossRef]
- Alric, L.; Besson, C.; Lapidus, N.; Jeannel, J.; Michot, J.-M.; Cacoub, P.; Canioni, D.; Pol, S.; Davi, F.; Rabiega, P.; et al. Antiviral Treatment of HCV-Infected Patients with B-Cell Non-Hodgkin Lymphoma: ANRS HC-13 Lympho-C Study. PLoS ONE 2016, 11, e0162965. [Google Scholar] [CrossRef]
- Friedberg, J.W.; Taylor, M.D.; Cerhan, J.; Flowers, C.R.; Dillon, H.; Farber, C.M.; Rogers, E.S.; Hainsworth, J.D.; Wong, E.K.; Vose, J.M.; et al. Follicular lymphoma in the United States: First report of the national lymphocare study. J. Clin. Oncol. 2009, 27, 1202–1208. [Google Scholar] [CrossRef]
- MacManus, M.; Fisher, R.; Roos, D.; O’Brien, P.; Macann, A.; Davis, S.; Tsang, R.; Christie, D.; McClure, B.; Joseph, D.; et al. Randomized Trial of Systemic Therapy after Involved-Field Radiotherapy in Patients with early-Stage Follicular Lymphoma: TROG 99.03. J. Clin. Oncol. 2018, 36, 2918–2925. [Google Scholar] [CrossRef] [PubMed]
- Advani, R.; Rosenberg, S.A.; Horning, S.J. Stage I and II follicular non-Hodgkin’s lymphoma: Long-term follow-up of no initial therapy. J. Clin. Oncol. 2004, 22, 1454–1459. [Google Scholar] [CrossRef] [PubMed]
- Marcus, R.; Imrie, K.; Solal-Celigny, P.; Catalano, J.V.; Dmoszynska, A.; Raposo, J.C.; Offner, F.C.; Gomez-Codina, J.; Belch, A.; Cunningham, D.; et al. Phase III study of R-CVP compared with cyclophosphamide, vincristine, and prednisone alone in patients with previously untreated advanced follicular lymphoma. J. Clin. Oncol. 2008, 26, 4579–4586. [Google Scholar] [CrossRef] [PubMed]
- Hiddemann, W.; Kneba, M.; Dreyling, M.; Schmitz, N.; Lengfelder, E.; Schmits, R.; Reiser, M.; Metzner, B.; Harder, H.; Hegewisch-Becker, S.; et al. Frontline therapy with rituximab added to the combination of cyclophosphamide, doxorubicin, vincristine, and prednisone (CHOP) significantly improves the outcome for patients with advanced-stage follicular lymphoma compared with therapy with CHOP alone: Results of a prospective randomized study of the German Low-Grade Lymphoma Study Group. Blood 2005, 106, 3725–3732. [Google Scholar] [CrossRef] [PubMed]
- Marcus, R.; Davies, A.; Ando, K.; Klapper, W.; Opat, S.; Owen, C.; Phillips, E.; Sangha, R.; Schlag, R.; Seymour, J.F.; et al. Obinutuzumab for the First-Line Treatment of Follicular Lymphoma. N. Engl. J. Med. 2017, 377, 1331–1344. [Google Scholar] [CrossRef]
- Salles, G.A.; Seymour, J.F.; Feugier, P.; Offner, F.; Lopez-Guillermo, A.; Belada, D.; Xerri, L.; Bouabdallah, R.; Catalano, J.; Brice, P.; et al. Long term follow-up of the PRIMA study: Half of patients receiving rituximab maintenance remain progression free at 10 years. Blood 2017, 130 (Suppl. S1), 486. [Google Scholar]
- Sarkozy, C.; Baseggio, L.; Feugier, P.; Callet-Bauchu, E.; Karlin, L.; Seymour, J.F.; Lebras, L.; Michallet, A.-S.; Offner, F.; Dumas, O.; et al. Peripheral blood involvement in patients with follicular lymphoma: A rare disease manifestation associated with poor prognosis. Br. J. Haematol. 2013, 164, 659–667. [Google Scholar] [CrossRef] [PubMed]
- Sarkozy, C.; Seymour, J.F.; Ferme, C.; Caballero, L.; Ghesquieres, H.; Leppä, S.; Delarue, R.; Pedersen, L.M.; Mounier, C.; da Silva, M.G.; et al. Rituximab maintenance obviates the poor prognosis associated with circulating lymphoma cells in patients with follicular lymphoma. Blood 2014, 123, 2740–2742. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Apostolidis, J.; Mokhtar, N.; Al Omari, R.; Darweesh, M.; Al Hashmi, H. Follicular lymphoma: Update on management and emerging therapies at the dawn of the new decade. Hematol. Oncol. 2020, 38, 213–222. [Google Scholar] [CrossRef]
- Matasar, M.J.; Luminari, S.; Barr, P.M.; Barta, S.K.; Danilov, A.V.; Hill, B.T.; Phillips, T.J.; Jerkeman, M.; Magagnoli, M.; Nastoupil, L.J.; et al. Follicular lymphoma: Recent and emerging therapies, treatment strategies, and remaining unmet needs. Oncologist 2019, 24, e1236–e1250. [Google Scholar] [CrossRef]
- Solal-Céligny, P.; Roy, P.; Colombat, P.; White, J.; Armitage, J.O.; Arranz-Saez, R.; Au, W.Y.; Bellei, M.; Brice, P.; Caballero, D.; et al. Follicular lymphoma international prognostic index. Blood 2004, 104, 1258–1265. [Google Scholar] [CrossRef] [PubMed]
- Federico, M.; Bellei, M.; Marcheselli, L.; Luminari, S.; Lopez-Guillermo, A.; Vitolo, U.; Pro, B.; Pileri, S.; Pulsoni, A.; Soubeyran, P.; et al. Follicular lymphoma international prognostic index 2: A new prognostic index for follicular lymphoma developed by the international follicular lymphoma prognostic factor project. J. Clin. Oncol. 2009, 27, 4555–4562. [Google Scholar] [CrossRef] [PubMed]



Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Abdulbaki, R.; Tizro, P.; Nava, V.E.; Gomes da Silva, M.; Ascensão, J.L. Low-Grade Primary Splenic CD10-Positive Small B-Cell Lymphoma/Follicular Lymphoma. Curr. Oncol. 2021, 28, 4821-4831. https://doi.org/10.3390/curroncol28060407
Abdulbaki R, Tizro P, Nava VE, Gomes da Silva M, Ascensão JL. Low-Grade Primary Splenic CD10-Positive Small B-Cell Lymphoma/Follicular Lymphoma. Current Oncology. 2021; 28(6):4821-4831. https://doi.org/10.3390/curroncol28060407
Chicago/Turabian StyleAbdulbaki, Rami, Parastou Tizro, Victor E. Nava, Maria Gomes da Silva, and João L. Ascensão. 2021. "Low-Grade Primary Splenic CD10-Positive Small B-Cell Lymphoma/Follicular Lymphoma" Current Oncology 28, no. 6: 4821-4831. https://doi.org/10.3390/curroncol28060407
APA StyleAbdulbaki, R., Tizro, P., Nava, V. E., Gomes da Silva, M., & Ascensão, J. L. (2021). Low-Grade Primary Splenic CD10-Positive Small B-Cell Lymphoma/Follicular Lymphoma. Current Oncology, 28(6), 4821-4831. https://doi.org/10.3390/curroncol28060407