CD5-Negative, CD10-Negative Low-Grade B-Cell Lymphoproliferative Disorders of the Spleen

Abstract
1. Introduction
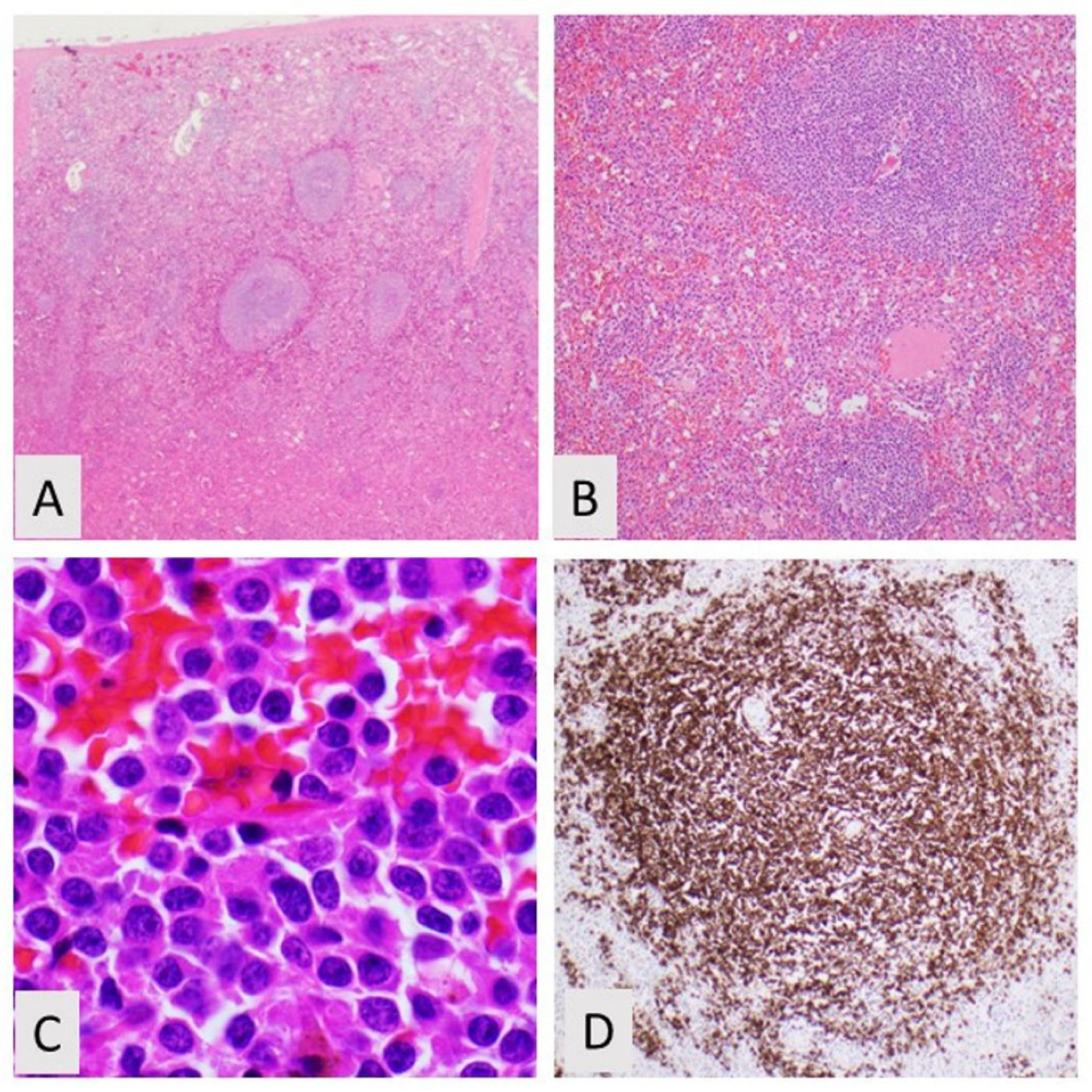
2. Splenic Marginal Zone Lymphoma
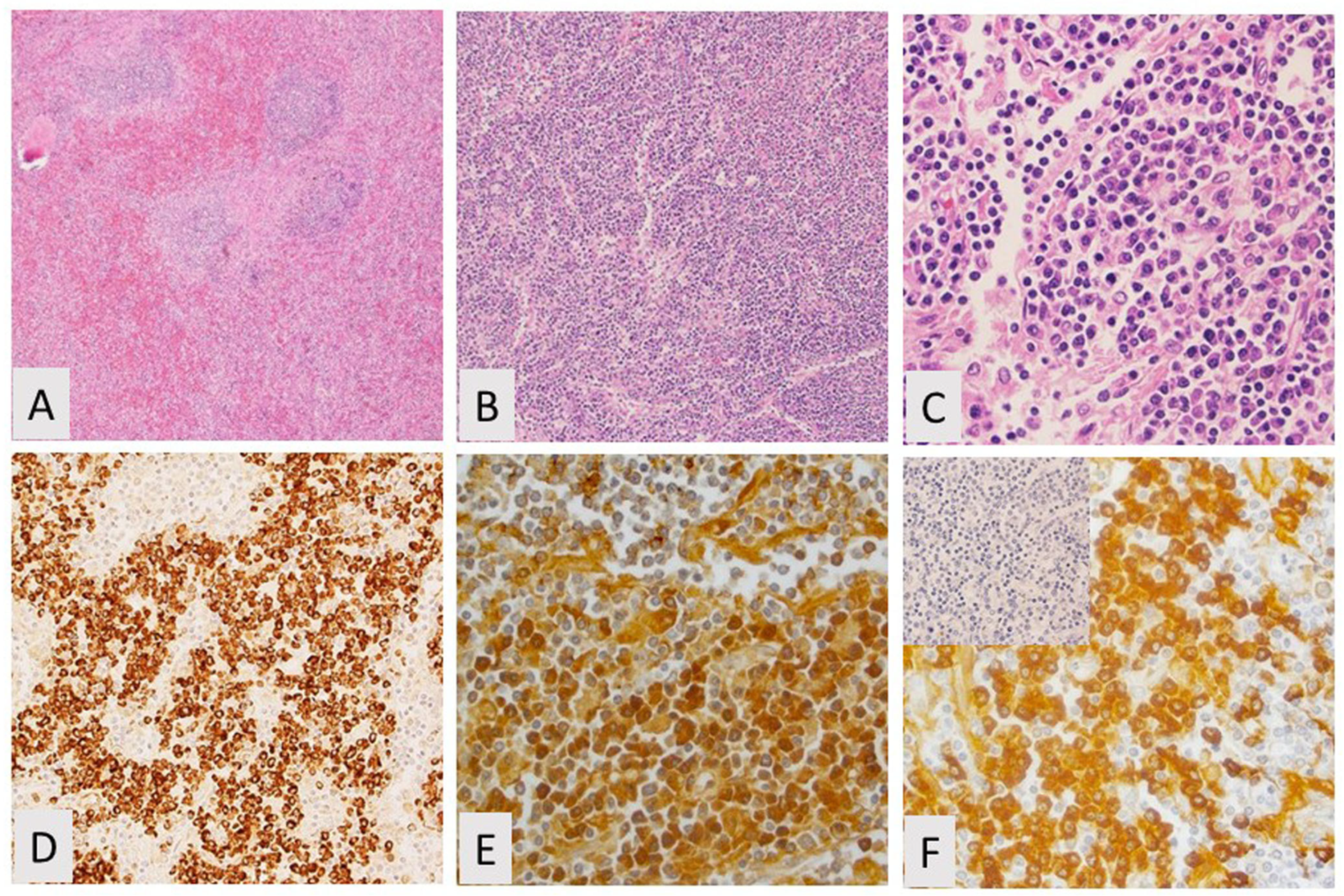
2.1. Spleen and Lymph Node Involvement
2.2. Blood and Bone Marrow Morphology
2.3. Phenotyping by Immunohistochemistry and Flow Cytometry
2.4. Molecular Testing of SMZL
2.5. Treatment and Prognosis
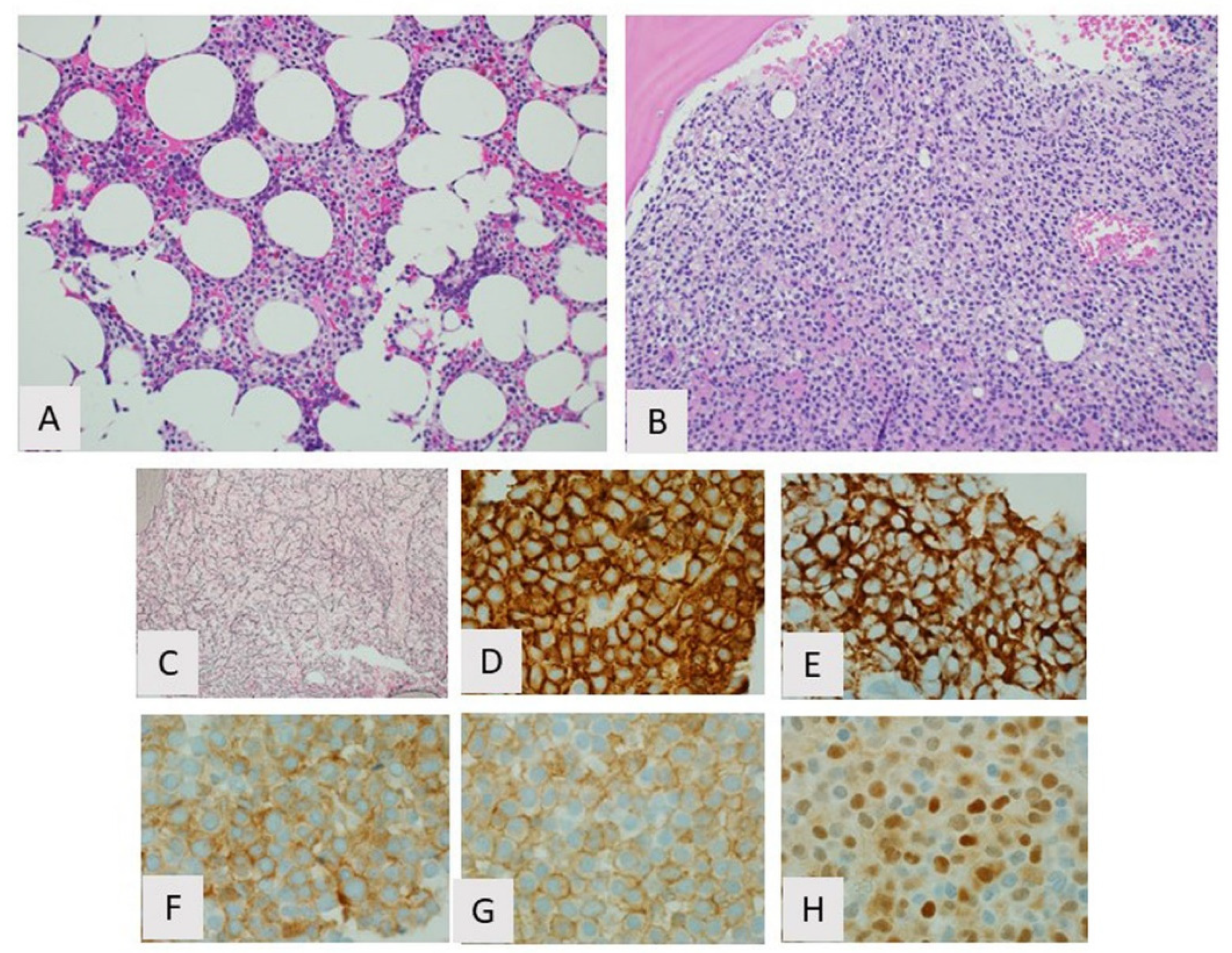
3. Hairy Cell Leukemia
3.1. Introduction
3.2. Biological Features/Pathogenesis
3.3. Clinicopathologic Features and Diagnosis
3.4. Treatment and Prognosis
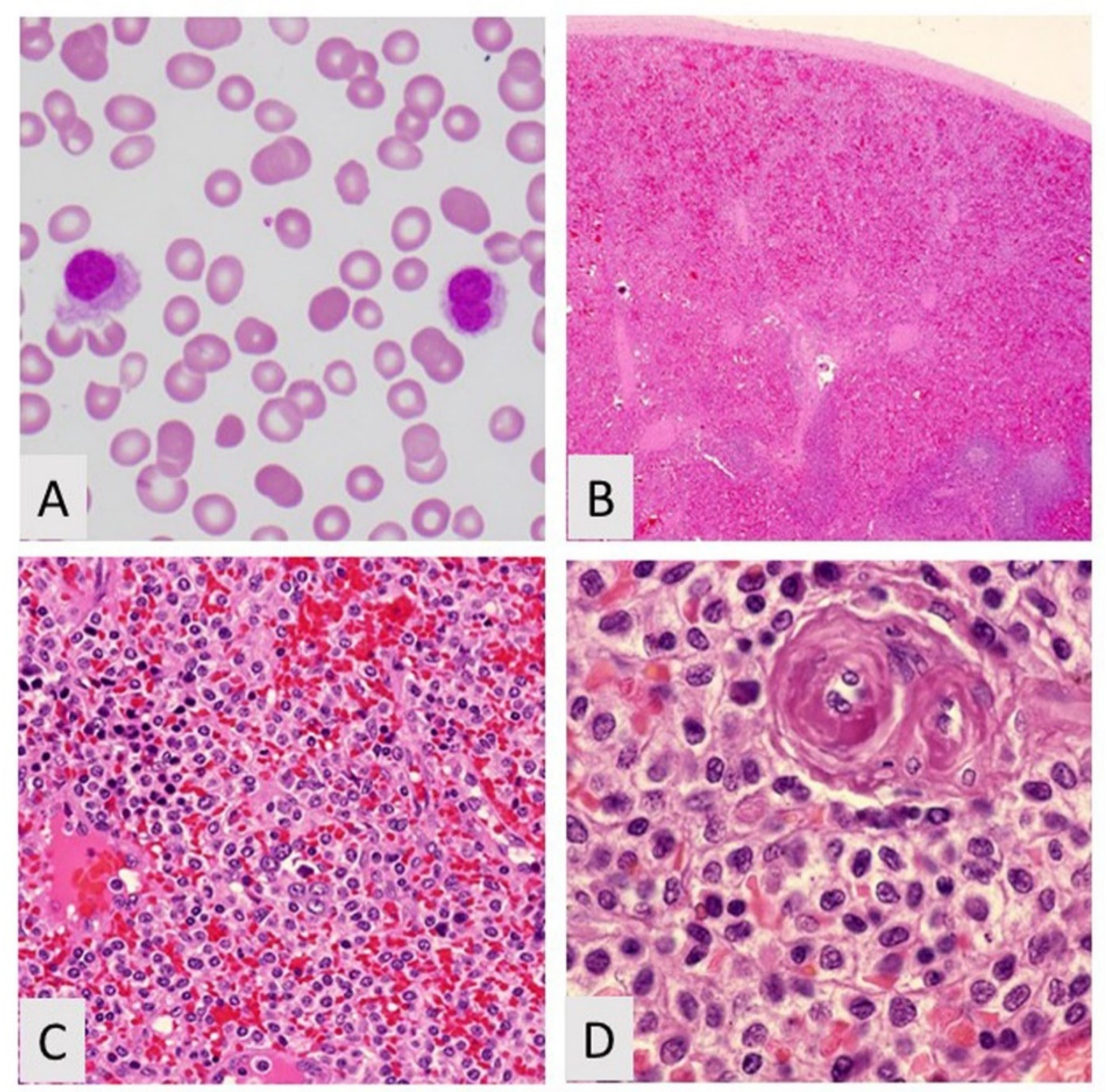
4. Hairy Cell Leukemia Variant
4.1. Introduction
4.2. Biological Features/Pathogenesis
4.3. Clinicopathologic Features and Diagnosis
4.4. Treatment and Prognosis
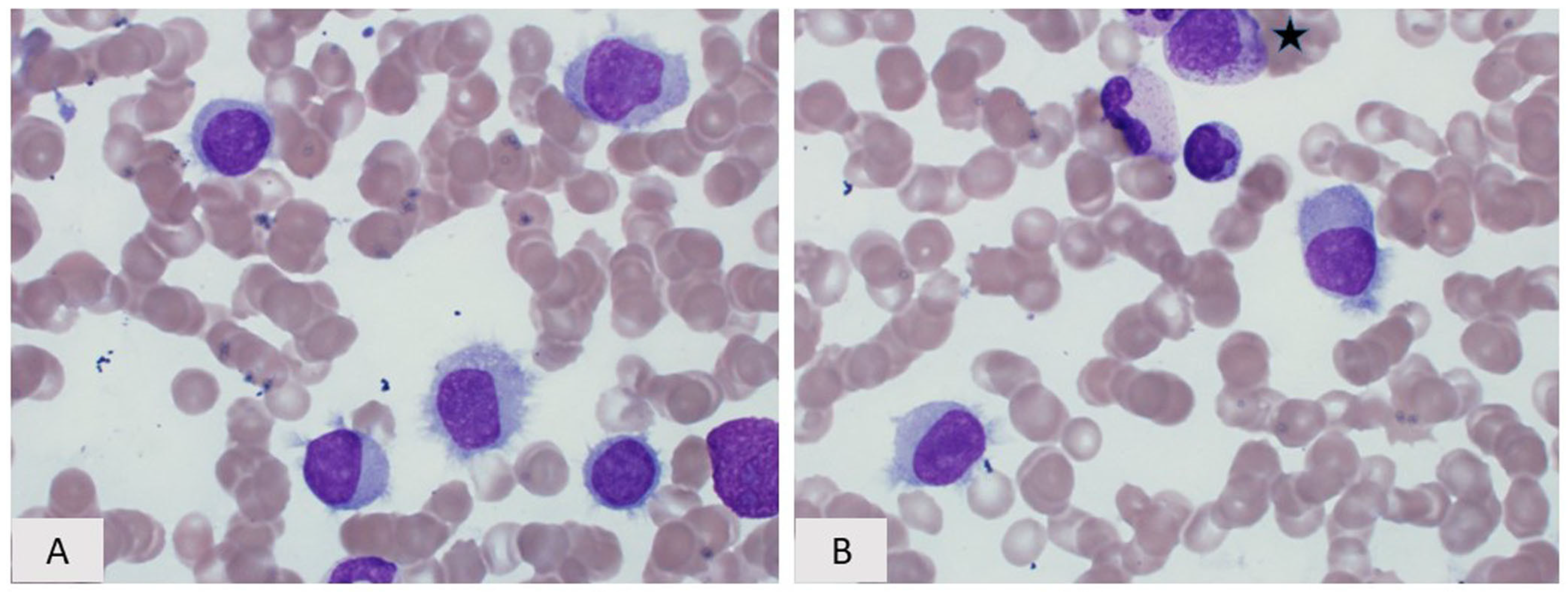
5. Lymphoplasmacytic Lymphoma
5.1. Introduction
5.2. Morphology and Immunophenotype
5.3. Immunophenotype
5.4. Cytogenetic/Molecular Findings
6. Splenic Diffuse Red Pulp Small B-Cell Lymphoma
Funding
Acknowledgments
Conflicts of Interest
References
- Fallah, J.; Olszewski, A.J. Diagnostic and therapeutic splenectomy for splenic lymphomas: Analysis of the National Cancer Data Base. Hematology 2019, 24, 378–386. [Google Scholar] [CrossRef]
- Sohani, A.R.; Zukerberg, L.R. Small B-cell lymphomas of the spleen: How to tell them apart. J. Hematopathol. 2014, 7, 109–121. [Google Scholar] [CrossRef]
- Liu, L.; Wang, H.; Chen, Y.; Rustveld, L.; Liu, G.; Du, X.L. Splenic marginal zone lymphoma: A population-based study on the 2001-2008 incidence and survival in the United States. Leuk. Lymphoma 2013, 54, 1380–1386. [Google Scholar] [CrossRef] [PubMed]
- Fetica, B.; Pop, B.; Blaga, M.; Fulop, A.; Dima, D.; Zdrenghea, M.T.; Vlad, C.I.; Bojan, A.S.; Achimas-Cadariu, P.; Lisencu, C.I.; et al. High prevalence of viral hepatitis in a series of splenic marginal zone lymphomas from Romania. Blood Cancer J. 2016, 6, e498. [Google Scholar] [CrossRef] [PubMed]
- O’Malley, D.P.; Louissaint, A., Jr.; Vasef, M.A.; Auerbach, A.; Miranda, R.; Brynes, R.K.; Fedoriw, Y.; Hudnall, S.D. Recommendations for gross examination and sampling of surgical specimens of the spleen. Ann. Diagn. Pathol. 2015, 19, 288–295. [Google Scholar] [CrossRef]
- Mollejo, M.; Menárguez, J.; Lloret, E.; Sánchez, A.; Campo, E.; Algara, P.; Cristóbal, E.; Sánchez, E. Splenic marginal zone lymphoma: A distinctive type of low-grade B-cell lymphoma. A clinicopathological study of 13 cases. Am. J. Surg. Pathol. 1995, 19, 1146–1157. [Google Scholar] [CrossRef] [PubMed]
- Swerdlow, S.H.; Campo, E.; Harris, N.L.; Jaffe, E.S.; Pileri, S.A.; Stein, H.; Thiele, J.; Arber, D.A.; Hasserjian, R.P.; Le Beau, M.M.; et al. WHO Classification of Tumours of Hematopoietic and Lymphoid Tissues, 4th ed.; IARC Press: Lyon, France, 2017; pp. 223–225, 229–230. [Google Scholar]
- Dufresne, S.D.; Felgar, R.E.; Sargent, R.L.; Surti, U.; Gollin, S.M.; McPhail, E.D.; Cook, J.R.; Swerdlow, S.H. Defining the borders of splenic marginal zone lymphoma: A multiparameter study. Hum. Pathol. 2010, 41, 540–551. [Google Scholar] [CrossRef]
- Molina, T.J.; Lin, P.; Swerdlow, S.H.; Cook, J.R. Marginal zone lymphomas with plasmacytic differentiation and related disorders. Am. J. Clin. Pathol. 2011, 136, 211–225. [Google Scholar] [CrossRef] [PubMed]
- Lloret, E.; Mollejo, M.; Mateo, M.S.; Villuendas, R.; Algara, P.; Martínez, P.; Piris, M.A. Splenic marginal zone lymphoma with increased number of blasts: An aggressive variant? Hum. Pathol. 1999, 30, 1153–1160. [Google Scholar] [CrossRef]
- Camacho, F.I.; Mollejo, M.; Mateo, M.S.; Algara, P.; Navas, C.; Hernández, J.M.; Santoja, C.; Solé, F.; Sánchez-Beato, M.; Piris, M.A. Progression to large B-cell lymphoma in splenic marginal zone lymphoma: A description of a series of 12 cases. Am. J. Surg. Pathol. 2001, 25, 1268–1276. [Google Scholar] [CrossRef] [PubMed]
- Isaacson, P.G.; Matutes, E.; Burke, M.; Catovsky, D. The histopathology of splenic lymphoma with villous lymphocytes. Blood 1994, 84, 3828–3834. [Google Scholar] [CrossRef] [PubMed]
- Mollejo, M.; Lloret, E.; Menárguez, J.; Piris, M.A.; Isaacson, P.G. Lymph node involvement by splenic marginal zone lymphoma: Morphological and immunohistochemical features. Am. J. Surg. Pathol. 1997, 21, 772–780. [Google Scholar] [CrossRef]
- Melo, J.V.; Hedge, U.; Parreira, A.; Thompson, I.; Lampert, I.A.; Catovsky, D. Splenic B cell lymphoma with circulating villous lymphocytes: Differential diagnosis of B cell leukaemias with large spleens. J. Clin. Pathol. 1987, 40, 642–651. [Google Scholar] [CrossRef] [PubMed]
- Troussard, X.; Valensi, F.; Duchayne, E.; Garand, R.; Felman, P.; Tulliez, M.; Henry-Amar, M.; Bryon, P.A.; Flandrin, G. Splenic lymphoma with villous lymphocytes: Clinical presentation, biology and prognostic factors in a series of 100 patients. Br. J. Haematol. 1996, 93, 731–736. [Google Scholar] [CrossRef]
- Chacón, J.I.; Mollejo, M.; Muñoz, E.; Algara, P.; Mateo, M.; Lopez, L.; Andrade, J.; Carbonero, I.G.; Martínez, B.; Piris, M.A.; et al. Splenic marginal zone lymphoma: Clinical characteristics and prognostic factors in a series of 60 patients. Blood 2002, 100, 1648–1654. [Google Scholar] [CrossRef]
- Xochelli, A.; Kalpadakis, C.; Gardiner, A.; Baliakas, P.; Vassilakopoulos, T.P.; Mould, S.; Davis, Z.; Stalika, E.; Kanellis, G.; Angelopoulou, M.K.; et al. Clonal B-cell lymphocytosis exhibiting immunophenotypic features consistent with a marginal-zone origin: Is this a distinct entity? Blood 2014, 123, 1199–1206. [Google Scholar] [CrossRef] [PubMed]
- Catovsky, D.; Matutes, E. Splenic lymphoma with circulating villous lymphocytes/splenic marginal-zone lymphoma. Semin. Hematol. 1999, 36, 148–154. [Google Scholar]
- Challagundla, P.; Medeiros, L.J.; Kanagal-Shamanna, R.; Miranda, R.N.; Jorgensen, J.L. Differential expression of CD200 in B-cell neoplasms by flow cytometry can assist in diagnosis, subclassification, and bone marrow staging. Am. J. Clin. Pathol. 2014, 142, 837–844. [Google Scholar] [CrossRef]
- Baseggio, L.; Traverse-Glehen, A.; Petinataud, F.; Callet-Bauchu, E.; Berger, F.; Ffrench, M.; Couris, C.M.; Thieblemont, C.; Morel, D.; Coiffier, B.; et al. CD5 expression identifies a subset of splenic marginal zone lymphomas with higher lymphocytosis: A clinico-pathological, cytogenetic and molecular study of 24 cases. Haematologica 2010, 95, 604–612. [Google Scholar] [CrossRef]
- Salido, M.; Baró, C.; Oscier, D.; Stamatopoulos, K.; Dierlamm, J.; Matutes, E.; Traverse-Glehen, A.; Berger, F.; Felman, P.; Thieblemont, C.; et al. Cytogenetic aberrations and their prognostic value in a series of 330 splenic marginal zone B-cell lymphomas: A multicenter study of the Splenic B-Cell Lymphoma Group. Blood 2010, 116, 1479–1488. [Google Scholar] [CrossRef] [PubMed]
- Rinaldi, A.; Mian, M.; Chigrinova, E.; Arcaini, L.; Bhagat, G.; Novak, U.; Rancoita, P.M.; De Campos, C.P.; Forconi, F.; Gascoyne, R.D.; et al. Genome-wide DNA profiling of marginal zone lymphomas identifies subtype-specific lesions with an impact on the clinical outcome. Blood 2011, 117, 1595–1604. [Google Scholar] [CrossRef]
- Baliakas, P.; Strefford, J.C.; Bikos, V.; Parry, M.; Stamatopoulos, K.; Oscier, D. Splenic marginal-zone lymphoma: Ontogeny and genetics. Leuk. Lymphoma 2015, 56, 301–310. [Google Scholar] [CrossRef]
- Rossi, D.; Trifonov, V.; Fangazio, M.; Bruscaggin, A.; Rasi, S.; Spina, V.; Monti, S.; Vaisitti, T.; Arruga, F.; Famà, R.; et al. The coding genome of splenic marginal zone lymphoma: Activation of NOTCH2 and other pathways regulating marginal zone development. J. Exp. Med. 2012, 209, 1537–1551. [Google Scholar] [CrossRef]
- Kiel, M.J.; Velusamy, T.; Betz, B.L.; Zhao, L.; Weigelin, H.G.; Chiang, M.Y.; Huebner-Chan, D.R.; Bailey, N.G.; Yang, D.T.; Bhagat, G.; et al. Whole-genome sequencing identifies recurrent somatic NOTCH2 mutations in splenic marginal zone lymphoma. J. Exp. Med. 2012, 209, 1553–1565. [Google Scholar] [CrossRef]
- Lee, S.Y.; Kumano, K.; Nakazaki, K.; Sanada, M.; Matsumoto, A.; Yamamoto, G.; Nannya, Y.; Suzuki, R.; Ota, S.; Ota, Y.; et al. Gain-of-function mutations and copy number increases of Notch2 in diffuse large B-cell lymphoma. Cancer Sci. 2009, 100, 920–926. [Google Scholar] [CrossRef] [PubMed]
- Piris, M.A.; Onaindía, A.; Mollejo, M. Splenic marginal zone lymphoma. Best Pract. Res. Clin. Haematol. 2017, 30, 56–64. [Google Scholar] [CrossRef] [PubMed]
- Jaramillo Oquendo, C.; Parker, H.; Oscier, D.; Ennis, S.; Gibson, J.; Strefford, J.C. Systematic review of somatic mutations in splenic marginal zone lymphoma. Sci. Rep. 2019, 9, 10444. [Google Scholar] [CrossRef] [PubMed]
- Bennett, M.; Schechter, G.P. Treatment of splenic marginal zone lymphoma: Splenectomy versus rituximab. Semin. Hematol. 2010, 47, 143–147. [Google Scholar] [CrossRef]
- Lenglet, J.; Traullé, C.; Mounier, N.; Benet, C.; Munoz-Bongrand, N.; Amorin, S.; Noguera, M.E.; Traverse-Glehen, A.; Ffrench, M.; Baseggio, L.; et al. Long-term follow-up analysis of 100 patients with splenic marginal zone lymphoma treated with splenectomy as first-line treatment. Leuk. Lymphoma 2014, 55, 1854–1860. [Google Scholar] [CrossRef] [PubMed]
- Kalpadakis, C.; Pangalis, G.A.; Vassilakopoulos, T.P.; Sachanas, S.; Angelopoulou, M.K. Treatment of splenic marginal zone lymphoma: Should splenectomy be abandoned? Leuk. Lymphoma 2014, 55, 1463–1470. [Google Scholar] [CrossRef] [PubMed]
- Lumish, M.; Falchi, L.; Imber, B.S.; Scordo, M.; von Keudell, G.; Joffe, E. How we treat mature B-cell neoplasms (indolent B-cell lymphomas). J. Hematol. Oncol. 2021, 14, 5. [Google Scholar] [CrossRef] [PubMed]
- Florindez, J.A.; Alderuccio, J.P.; Reis, I.M.; Lossos, I.S. Splenic marginal zone lymphoma: A US population-based survival analysis (1999-2016). Cancer 2020, 126, 4706–4716. [Google Scholar] [CrossRef]
- Bouroncle, B.A.; Wiseman, B.K.; Doan, C.A. Leukemic reticuloendotheliosis. Blood 1958, 13, 609–630. [Google Scholar] [CrossRef] [PubMed]
- Cawley, J.C. The pathophysiology of the hairy cell. Hematol. Oncol. Clin. N. Am. 2006, 20, 1011–1021. [Google Scholar] [CrossRef] [PubMed]
- Tiacci, E.; Liso, A.; Piris, M.; Falini, B. Evolving Concepts in the pathogenesis of hairy-cell leukaemia. Nat. Rev. Cancer 2006, 6, 437–448. [Google Scholar] [CrossRef] [PubMed]
- Arons, E.; Sunshine, J.; Suntum, T.; Kreitman, R.J. Somatic hypermutation and VH gene usage in hairy cell leukaemia. Br. J. Haematol. 2006, 133, 504–512. [Google Scholar] [CrossRef]
- Basso, K.; Liso, A.; Tiacci, E.; Benedetti, R.; Pulsoni, A.; Foa, R.; Di Raimondo, F.; Ambrosetti, A.; Califano, A.; Klein, U.; et al. Gene expression profiling in hairy cell leukemia reveals a phenotype related to memory B cells with altered expression of chemokine and adhesion receptors. J. Exp. Med. 2004, 199, 59–68. [Google Scholar] [CrossRef]
- Falini, B.; Tiacci, E.; Liso, A.; Basso, K.; Sabattini, E.; Pacini, R.; Foa, R.; Pulsoni, A.; Dalla Favera, R.; Pileri, S. Simple diagnostic assay for hairy cell leukaemia by immunocytochemical detection of annexin A1 (ANXA1). Lancet 2004, 363, 1869–1870. [Google Scholar] [CrossRef]
- Tiacci, E.; Trifonov, V.; Schiavoni, G.; Holmes, A.; Kern, W.; Martelli, M.P.; Pucciarini, A.; Bigerna, B.; Pacini, R.; Wells, V.A.; et al. BRAF mutations in hairy-cell leukemia. N. Engl. J. Med. 2011, 364, 2305–2315. [Google Scholar] [CrossRef]
- Arcaini, L.; Zibellini, S.; Boveri, E.; Riboni, R.; Rattotti, S.; Varettoni, M.; Guerrera, M.L.; Lucioni, M.; Tenore, A.; Merli, M.; et al. The BRAF V600E mutation in hairy cell leukemia and other mature B-cell neoplasms. Blood 2012, 119, 188–191. [Google Scholar] [CrossRef]
- Tiacci, E.; Schiavoni, G.; Forconi, F.; Santi, A.; Trentin, L.; Ambrosetti, A.; Cecchini, D.; Sozzi, E.; Francia di Celle, P.; Di Bello, C.; et al. Simple genetic diagnosis of hairy cell leukemia by sensitive detection of the BRAF-V600E mutation. Blood 2012, 119, 192–195. [Google Scholar] [CrossRef] [PubMed]
- Pettirossi, V.; Santi, A.; Imperi, E.; Russo, G.; Pucciarini, A.; Bigerna, B.; Schiavoni, G.; Fortini, E.; Spanhol-Rosseto, A.; Sportoletti, P.; et al. BRAF inhibitors reverse the unique molecular signature and phenotype of hairy cell leukemia and exert potent antileukemic activity. Blood 2015, 125, 1207–1216. [Google Scholar] [CrossRef] [PubMed]
- Dores, G.M.; Curtis, R.E.; Anderson, W.F.; Demierre, M.F. Hairy cell leukaemia: A heterogeneous disease? Br. J. Haematol. 2008, 142, 45–51. [Google Scholar] [CrossRef] [PubMed]
- Bethel, K.J.; Sharpe, R.W. Pathology of hairy-cell leukaemia. Best Pract. Res. Clin. Haematol. 2003, 16, 15–31. [Google Scholar] [CrossRef]
- Shao, H.; Calvo, K.R.; Grönborg, M.; Tembhare, P.R.; Kreitman, R.J.; Stetler-Stevenson, M.; Yuan, C.M. Distinguishing hairy cell leukemia variant from hairy cell leukemia: Development and validation of diagnostic criteria. Leuk. Res. 2013, 37, 401–409. [Google Scholar] [CrossRef] [PubMed]
- Sharpe, R.W.; Bethel, K.J. Hairy cell leukemia: Diagnostic pathology. Hematol. Oncol. Clin. N. Am. 2006, 20, 1023–1049. [Google Scholar] [CrossRef] [PubMed]
- Garnache Ottou, F.; Chandesris, M.O.; Lhermitte, L.; Callens, C.; Beldjord, K.; Garrido, M.; Bedin, A.S.; Brouzes, C.; Villemant, S.; Rubio, M.T.; et al. Peripheral blood 8 colour flow cytometry monitoring of hairy cell leukaemia allows detection of high-risk patients. Br. J. Haematol. 2014, 166, 50–59. [Google Scholar] [CrossRef]
- Jöhrens, K.; Stein, H.; Anagnostopoulos, I. T-bet transcription factor detection facilitates the diagnosis of minimal hairy cell leukemia infiltrates in bone marrow trephines. Am. J. Surg. Pathol. 2007, 31, 1181–1185. [Google Scholar] [CrossRef]
- Morgan, E.A.; Yu, H.; Pinkus, J.L.; Pinkus, G.S. Immunohistochemical detection of hairy cell leukemia in paraffin sections using a highly effective CD103 rabbit monoclonal antibody. Am. J. Clin. Pathol. 2013, 139, 220–230. [Google Scholar] [CrossRef] [PubMed]
- Pillai, V.; Pozdnyakova, O.; Charest, K.; Li, B.; Shahsafaei, A.; Dorfman, D.M. CD200 flow cytometric assessment and semiquantitative immunohistochemical staining distinguishes hairy cell leukemia from hairy cell leukemia-variant and other B-cell lymphoproliferative disorders. Am. J. Clin. Pathol. 2013, 140, 536–543. [Google Scholar] [CrossRef] [PubMed]
- Sherman, M.J.; Hanson, C.A.; Hoyer, J.D. An assessment of the usefulness of immunohistochemical stains in the diagnosis of hairy cell leukemia. Am. J. Clin. Pathol. 2011, 136, 390–399. [Google Scholar] [CrossRef]
- Chen, Y.H.; Tallman, M.S.; Goolsby, C.; Peterson, L. Immunophenotypic variations in hairy cell leukemia. Am. J. Clin. Pathol. 2006, 125, 251–259. [Google Scholar] [CrossRef]
- Dong, H.Y.; Weisberger, J.; Liu, Z.; Tugulea, S. Immunophenotypic analysis of CD103+ B-lymphoproliferative disorders: Hairy cell leukemia and its mimics. Am. J. Clin. Pathol. 2009, 131, 586–595. [Google Scholar] [CrossRef]
- Jasionowski, T.M.; Hartung, L.; Greenwood, J.H.; Perkins, S.L.; Bahler, D.W. Analysis of CD10+ hairy cell leukemia. Am. J. Clin. Pathol. 2003, 120, 228–235. [Google Scholar] [CrossRef] [PubMed]
- Waterfall, J.J.; Arons, E.; Walker, R.L.; Pineda, M.; Roth, L.; Killian, J.K.; Abaan, O.D.; Davis, S.R.; Kreitman, R.J.; Meltzer, P.S. High prevalence of MAP2K1 mutations in variant and IGHV4-34-expressing hairy-cell leukemias. Nat. Genet. 2014, 46, 8–10. [Google Scholar] [CrossRef]
- Xi, L.; Arons, E.; Navarro, W.; Calvo, K.R.; Stetler-Stevenson, M.; Raffeld, M.; Kreitman, R.J. Both variant and IGHV4-34-expressing hairy cell leukemia lack the BRAF V600E mutation. Blood 2012, 119, 3330–3332. [Google Scholar] [CrossRef] [PubMed]
- Robak, T. Current treatment options in hairy cell leukemia and hairy cell leukemia variant. Cancer Treat. Rev. 2006, 32, 365–376. [Google Scholar] [CrossRef] [PubMed]
- Grever, M.R. How I treat hairy cell leukemia. Blood 2010, 115, 21–28. [Google Scholar] [CrossRef] [PubMed]
- Turakhia, S.; Lanigan, C.; Hamadeh, F.; Swerdlow, S.H.; Tubbs, R.R.; Cook, J.R. Immunohistochemistry for BRAF V600E in the differential diagnosis of hairy cell leukemia vs other splenic B-cell lymphomas. Am. J. Clin. Pathol. 2015, 144, 87–93. [Google Scholar] [CrossRef] [PubMed]
- Tiacci, E.; Park, J.H.; De Carolis, L.; Chung, S.S.; Broccoli, A.; Scott, S.; Zaja, F.; Devlin, S.; Pulsoni, A.; Chung, Y.R.; et al. Targeting mutant BRAF in relapsed or refractory hairy-cell leukemia. N. Engl. J. Med. 2015, 373, 1733–1747. [Google Scholar] [CrossRef]
- Matutes, E.; Wotherspoon, A.; Catovsky, D. The variant form of hairy-cell leukaemia. Best Pract. Res. Clin. Haematol. 2003, 16, 41–56. [Google Scholar] [CrossRef]
- Cessna, M.H.; Hartung, L.; Tripp, S.; Perkins, S.L.; Bahler, D.W. Hairy cell leukemia variant: Fact or fiction. Am. J. Clin. Pathol. 2005, 123, 132–138. [Google Scholar] [CrossRef] [PubMed]
- Hockley, S.L.; Giannouli, S.; Morilla, A.; Wotherspoon, A.; Morgan, G.J.; Matutes, E.; Gonzalez, D. Insight into the molecular pathogenesis of hairy cell leukaemia, hairy cell leukaemia variant and splenic marginal zone lymphoma, provided by the analysis of their IGH rearrangements and somatic hypermutation patterns. Br. J. Haematol. 2010, 148, 666–669. [Google Scholar] [CrossRef]
- Hockley, S.L.; Morgan, G.J.; Leone, P.E.; Walker, B.A.; Morilla, A.; Else, M.; Wotherspoon, A.; Dearden, C.; Catovsky, D.; Gonzalez, D.; et al. High-resolution genomic profiling in hairy cell leukemia-variant compared with typical hairy cell leukemia. Leukemia 2011, 25, 1189–1192. [Google Scholar] [CrossRef][Green Version]
- Raess, P.W.; Mintzer, D.; Husson, M.; Nakashima, M.O.; Morrissette, J.J.; Daber, R.; Bagg, A. BRAF V600E is also seen in unclassifiable splenic B-cell lymphoma/leukemia, a potential mimic of hairy cell leukemia. Blood 2013, 122, 3084–3085. [Google Scholar] [CrossRef][Green Version]
- Hockley, S.L.; Else, M.; Morilla, A.; Wotherspoon, A.; Dearden, C.; Catovsky, D.; Gonzalez, D.; Matutes, E. The prognostic impact of clinical and molecular features in hairy cell leukaemia variant and splenic marginal zone lymphoma. Br. J. Haematol. 2012, 158, 347–354. [Google Scholar] [CrossRef]
- Kanellis, G.; Garcia-Alonso, L.; Camacho, F.I.; Garcia, J.F.; Mollejo, M.; Montes-Moreno, S.; Garcia-Vela, J.A.; Piris, M.A. Hairy cell leukemia, blastic type: Description of spleen morphology and immunophenotype of a distinctive case. Leuk. Lymphoma 2011, 52, 1589–1592. [Google Scholar] [CrossRef] [PubMed]
- Matutes, E. Immunophenotyping and differential diagnosis of hairy cell leukemia. Hematol. Oncol. Clin. N. Am. 2006, 20, 1051–1063. [Google Scholar] [CrossRef]
- Alapat, D.; Coviello-Malle, J.; Owens, R.; Qu, P.; Barlogie, B.; Shaughnessy, J.D.; Lorsbach, R.B. Diagnostic usefulness and prognostic impact of CD200 expression in lymphoid malignancies and plasma cell myeloma. Am. J. Clin. Pathol. 2012, 137, 93–100. [Google Scholar] [CrossRef] [PubMed]
- Kreitman, R.J.; Wilson, W.; Calvo, K.R.; Arons, E.; Roth, L.; Sapolsky, J.; Zhou, H.; Raffeld, M.; Stetler-Stevenson, M. Cladribine with immediate rituximab for the treatment of patients with variant hairy cell leukemia. Clin. Cancer Res. 2013, 19, 6873–6881. [Google Scholar] [CrossRef]
- Fang, H.; Kapoor, P.; Gonsalves, W.I.; Frederick, L.A.; Viswanatha, D.; Howard, M.T.; He, R.; Morice, W.G., II; McPhail, E.D.; Greipp, P.T. Defining Lymphoplasmacytic Lymphoma: Does MYD88L265P define a pathologically distinct entity among patients with an IgM paraprotein and bone marrow-based low-grade b-cell lymphomas with plasmacytic differentiation? Am. J. Clin. Pathol. 2018, 150, 168–176. [Google Scholar] [CrossRef] [PubMed]
- Maqbool, M.G.; Tam, C.S.; Morison, I.M.; Simpson, D.; Mollee, P.; Schneider, H.; Chan, H.; Juneja, S.; Yasmin Harvey, Y.; Nath, L.; et al. A practical guide to laboratory investigations at diagnosis and follow up in Waldenström macroglobulinaemia: Recommendations from the Medical and Scientific Advisory Group, Myeloma Australia, the Pathology Sub-committee of the Lymphoma and Related Diseases Registry and the Australasian Association of Clinical Biochemists Monoclonal Gammopathy Working Group. Pathology 2020, 52, 167–178. [Google Scholar] [CrossRef]
- Rosado, F.; Morice, W.; He, R.; Howard, M.T.; Timm, M.; McPhail, E.D. Immunophenotypic features by multiparameter flow cytometry can help distinguish low grade B-cell lymphomas with plasmacytic differentiation from plasma cell proliferative disorders with an unrelated clonal B-cell process. Br. J. Haematol. 2015, 169, 368–376. [Google Scholar] [CrossRef]
- Gertz, M.A. Waldenström macroglobulinemia: 2019 update on diagnosis, risk stratification, and management. Am. J. Hematol. 2019, 94, 266–276. [Google Scholar] [CrossRef]
- Wang, W.; Lin, P. Lymphoplasmacytic lymphoma and Waldenström macroglobulinaemia: Clinicopathological features and differential diagnosis. Pathology 2020, 52, 6–14. [Google Scholar] [CrossRef]
- de Groen, R.A.L.; Schrader, A.M.R.; Kersten, M.J.; Pals, S.T.; Vermaat, J.S.P. MYD88 in the driver’s seat of B-cell lymphomagenesis: From molecular mechanisms to clinical implications. Haematologica 2019, 104, 2337–2348. [Google Scholar] [CrossRef] [PubMed]
- Kaiser, L.M.; Hunter, Z.R.; Treon, S.P.; Buske, C. CXCR4 in Waldenström’s Macroglobulinema: Chances and challenges. Leukemia 2021, 35, 333–345. [Google Scholar] [CrossRef]
- Schmidt, J.; Federmann, B.; Schindler, N.; Steinhilber, J.; Bonzheim, I.; Fend, F.; Quintanilla-Martinez, L. MYD88 L265P and CXCR4 mutations in lymphoplasmacytic lymphoma identify cases with high disease activity. Br. J. Haematol. 2015, 169, 795–803. [Google Scholar] [CrossRef] [PubMed]
- Ohno, H.; Ueda, C.; Akasaka, T. The t(9;14)(p13;q32) translocation in B-cell non-Hodgkin’s lymphoma. Leuk. Lymphoma. 2000, 36, 435–445. [Google Scholar] [CrossRef]
- Yun, S.; Johnson, A.C.; Okolo, O.N.; Arnold, S.J.; McBride, A.; Zhang, L.; Baz, R.C.; Anwer, F. Waldenström macroglobulinemia: Review of pathogenesis and management. Clin. Lymphoma Myeloma Leuk. 2017, 17, 252–262. [Google Scholar] [CrossRef]
- Nguyen-Khac, F.; Lambert, J.; Chapiro, E.; Grelier, A.; Mould, S.; Barin, C.; Daudignon, A.; Gachard, N.; Struski, S.; Henry, C.; et al. Chromosomal aberrations and their prognostic value in a series of 174 untreated patients with Waldenström’s macroglobulinemia. Haematologica 2013, 98, 649–654. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Low-Grade CD5(−)CD10(−) B-Cell LPDs | Other Neoplasms | |
|---|---|---|
| Diffuse miliary small white nodules | Splenic marginal zone lymphoma | Follicular lymphoma Mantle cell lymphoma CLL/SLL Classic Hodgkin lymphoma, rarely |
| Large mass-like nodules, solitary or multiple | Diffuse large B-cell lymphoma, follicular lymphoma, usually higher grade, classic Hodgkin lymphoma; Vascular neoplasms including littoral cell angioma, hemangioma, and splenic hamartoma | |
| Homogeneous, beefy red cut surface | Hairy cell leukemia, hairy cell leukemia variant, splenic diffuse red pulp small B-cell lymphoma | Hepatosplenic T-cell lymphoma, T-cell prolymphocytic leukemia |
| Antibody | Immunoreactivity | Staining Pattern | Description | Also Detected in |
|---|---|---|---|---|
| Annexin A1 | No | Cell membrane | HCL, specific marker | HCL |
| BRAF | No | Nuclear | None | HCL |
| Cyclin D1 | No | Nuclear | None | MCL, PCM |
| BCL2 | Yes | Cell membrane and cytoplasm | (+) in neoplastic B- cells, (−) in residual germinal centers | Most other small B-cell lymphomas |
| BCL6 | No | Nuclear | (−) in neoplastic cells, (+) in residual germinal centers, may be (+) in transformed SMZL | BL, DLBCL, FL |
| CD5 | Yes | Cell membrane | (+) in ~20% of cases, often dim reactivity | CLL/SLL, MCL |
| CD10 | No | Cell membrane | (+) only in residual germinal centers | BL, DLBCL, FL |
| CD11c | Yes | Cell membrane | Flow marker, (+) in 50% of cases | HCL |
| CD20 | Yes | Cell membrane | Pan-B-cell marker, neoplastic B-cells (+) | Most other B-cell lymphomas |
| CD21 | Yes | Dendritic pattern | (+) in follicular dendritic cell meshworks | FL |
| CD23 | No | Dendritic pattern | (+) in follicular dendritic cell meshworks | CLL/SLL, FL |
| CD25 | Yes | Cell membrane | Flow and immunohistochemistry marker, (+) in subset of cases | HCL |
| CD103 | Yes | Cell membrane | Flow and immunohistochemistry marker, (+) in small % of cases | HCL |
| DBA44 | Yes | Cell membrane | (+) in subset of cases | HCL |
| IgD | Yes | Cell membrane | (+) in subset of cases | Reactive mantle zones |
| IgM | Yes | Cell membrane | (+), most cases | |
| Ki-67 | Yes | Nuclear | Positive staining in both the germinal center and marginal zone which has been described as the so-called targetoid pattern. | Differing proliferation index in each lymphoma |
| Neoplasm | Peripheral Blood | Bone Marrow | Spleen |
|---|---|---|---|
| SRDPL | Mature lymphocytes with villous projections | Intrasinusoidal pattern of involvement. No plasmacytic differentiation | Cord and sinusoidal expansion +/− blood lakes |
| HCL | Lymphocytes (less chromatin clumping than normal lymphocytes) with villous projections | Subtle to extensive interstitial infiltrates of lymphocytes. Often extensive reticulin fibrosis | Red pulp cord expansion; atrophic white pulp; blood lakes common. |
| HCL-v | Small lymphocytes with visible nucleoli and villous projections | Intrasinusoidal pattern of involvement; atrophic white pulp | Fill dilated sinusoids +/− blood lakes; atrophic white pulp |
| LPL/WM | Spectrum of lymphocytes, plasmacytoid lymphocytes, and true plasma cells. | Diffuse, interstitial, and nodular involvement by a spectrum of lymphocytes, plasmacytoid lymphocytes, and true plasma cells | Red pulp cord and sinus expansion; atrophic white pulp. |
| Antibody | Immunoreactivity in SDRPL | Immunoreactivity in HCL | Immunoreactivity in HCL-V | Immunoreactivity in LPL/WM | Staining Pattern |
|---|---|---|---|---|---|
| CD20 | Yes | Yes | Yes | Yes | Cell membrane |
| CD138 | No | No | No | + in plasma cells | Cell membrane |
| DBA44 | Yes | Yes | Yes | No | Cell membrane |
| IgG | Yes | Yes | −/+ | Cell membrane | |
| Cyclin D3 | Yes | No | No | No | Nuclear |
| TRAP | No | Yes | No | No | Granular cytoplasmic |
| CD11c | −/+ | Yes | Yes | No | Cell membrane |
| CD5 | No | No | No | No | Cell membrane |
| CD123 | No | Yes | +/− | No | Cell membrane |
| Annexin A1 | No | Yes | No | No | Cell membrane |
| CD103 | No | Yes | Variable | No | Cell membrane |
| CD25 | No | Yes | No | +/- | Cell membrane |
| BRAF | No | Yes | No | No | Nuclear |
| Abnormality | SDRPL | HCL | HCL-V | LPL |
|---|---|---|---|---|
| CCND3 mutation | 26% | Negative | 13% | Negative |
| BRAF mutation | Negative | >95% | Negative | Negative |
| MYD88 mutation | Negative | Negative | Negative | >90% |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Schmieg, J.J.; Muir, J.M.; Aguilera, N.S.; Auerbach, A. CD5-Negative, CD10-Negative Low-Grade B-Cell Lymphoproliferative Disorders of the Spleen. Curr. Oncol. 2021, 28, 5124-5147. https://doi.org/10.3390/curroncol28060430
Schmieg JJ, Muir JM, Aguilera NS, Auerbach A. CD5-Negative, CD10-Negative Low-Grade B-Cell Lymphoproliferative Disorders of the Spleen. Current Oncology. 2021; 28(6):5124-5147. https://doi.org/10.3390/curroncol28060430
Chicago/Turabian StyleSchmieg, John J., Jeannie M. Muir, Nadine S. Aguilera, and Aaron Auerbach. 2021. "CD5-Negative, CD10-Negative Low-Grade B-Cell Lymphoproliferative Disorders of the Spleen" Current Oncology 28, no. 6: 5124-5147. https://doi.org/10.3390/curroncol28060430
APA StyleSchmieg, J. J., Muir, J. M., Aguilera, N. S., & Auerbach, A. (2021). CD5-Negative, CD10-Negative Low-Grade B-Cell Lymphoproliferative Disorders of the Spleen. Current Oncology, 28(6), 5124-5147. https://doi.org/10.3390/curroncol28060430