1. Introduction
Plocamium cartilagineum is a broadly distributed red alga that contributes to the structure of algal-dominated coastal benthic ecosystems of the Western Antarctic Peninsula [
1]. Although there is no currently accepted alternate species name,
P. cartilagineum in Antarctica is known to be a distinct species from what is called
P. cartilagineum in other parts of the world [
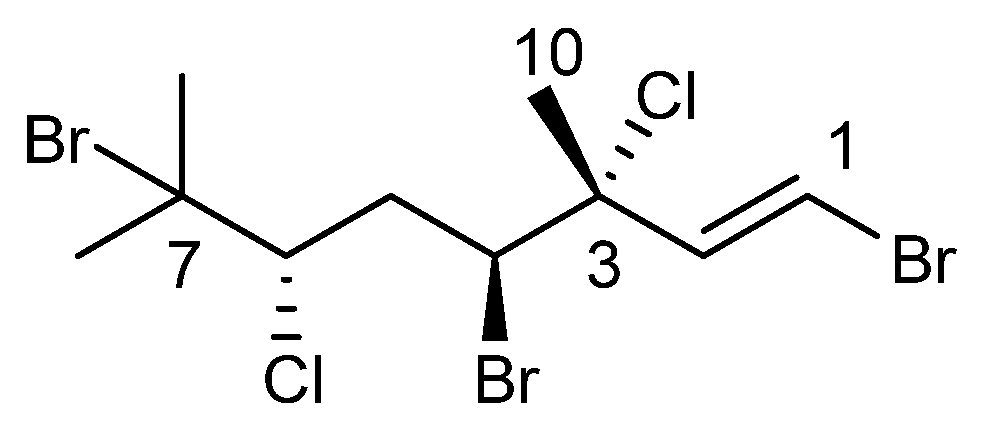
2]. This rhodophyte is known to produce many different polyhalogenated monoterpenes, including anverene (
1,
Scheme 1), which has been shown to convey ecological relevance as a feeding deterrent toward sympatric macroalgal consumers [
3,
4]. Previous studies have shown that chemical diversity among phenotypes of this alga is high, with specific chemotypes (chemogroups [
1]) displaying different halogenated terpenes with variable relative abundances. Patterns of chemogroup variability have been linked through metabolomics with site-specificity, presumably driven by ecological interactions [
1,
3,
5]. In 2013, Young et al. showed that within a collection of 21 individual algal specimens, there existed two distinct genotypes and five distinct chemotypes [
1]. The current study was carried out to identify and describe the major compounds present in two of the chemogroups previously identified, as well as report new structures found during the analysis.
The study began with a bulk collection of
Plocamium cartilagineum from a 2011 Antarctic field season obtained at Gamage Point on Anvers Island. Upon extraction and gas chromatography/mass spectrometry (GC/MS) analysis, this bulk collection was shown to share many metabolites with previously reported chemogroup 4 [
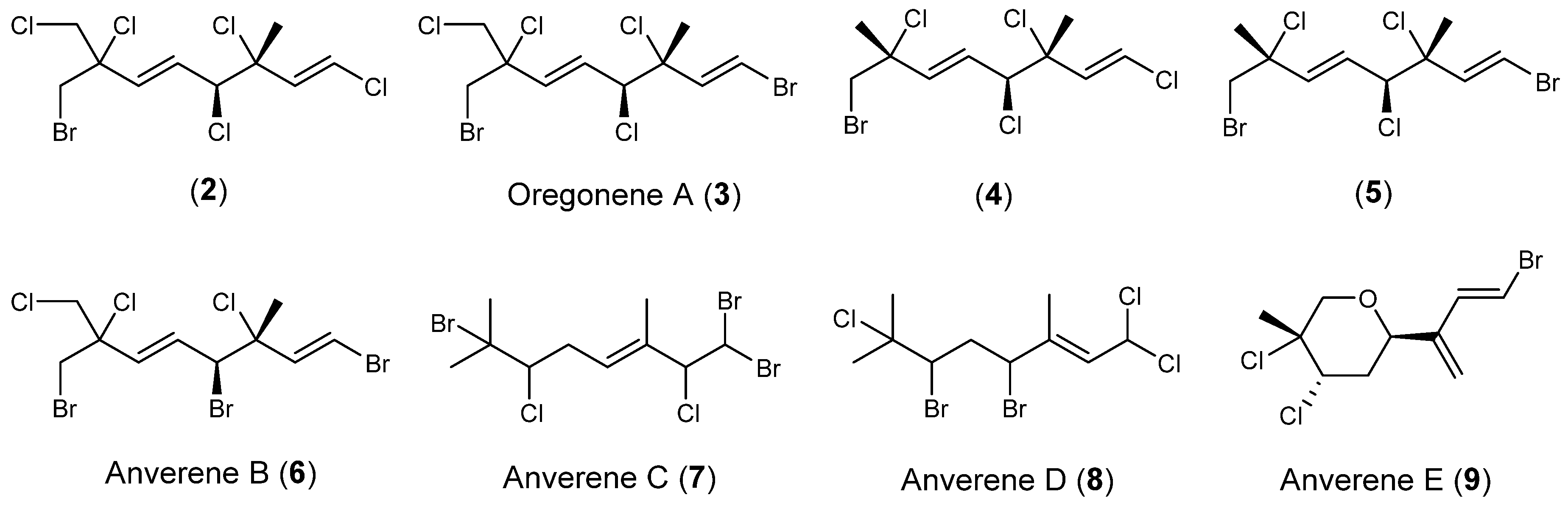
1]. This chemically rich chemogroup produces primarily linear polyhalogenated monoterpenes and yielded known compounds
2–
5 (
Scheme 2), and previously unreported anverene B (
6) as its most abundant metabolites, as well as previously unreported anverenes C and D (
7 and
8) as minor metabolites [
6,
7,
8].
Another field collection was undertaken in the austral summer of 2017 from Norsel Point, which was shown to match the metabolomic profile of the previously reported chemogroup 1, the chemogroup with the least amount of similarity to chemogroup 4 of any of the previously identified chemotypes and one that also belonged to a different genotype [
1]. Upon extraction and fractionation, chemogroup 1 was shown to contain small amounts of
2, however its most abundant metabolite was the previously unreported cyclic monoterpene anverene E (
9), which features a terminal diene not previously reported from this taxon. Fractionation of each algal specimen was guided by GC/MS and nuclear magnetic resonance (NMR) spectroscopy resulting in purified compounds that are traceable to the crude extracts, leading us to the conclusion that no compounds are artifacts of the isolation. Herein we report the characterization of the three new compounds from chemogroup 1, anverenes B–D (
6–
8) and one from chemogroup 4, anverene E (
9) as well as HeLa cytotoxicity for compounds
1–
9.
2. Results and Discussion
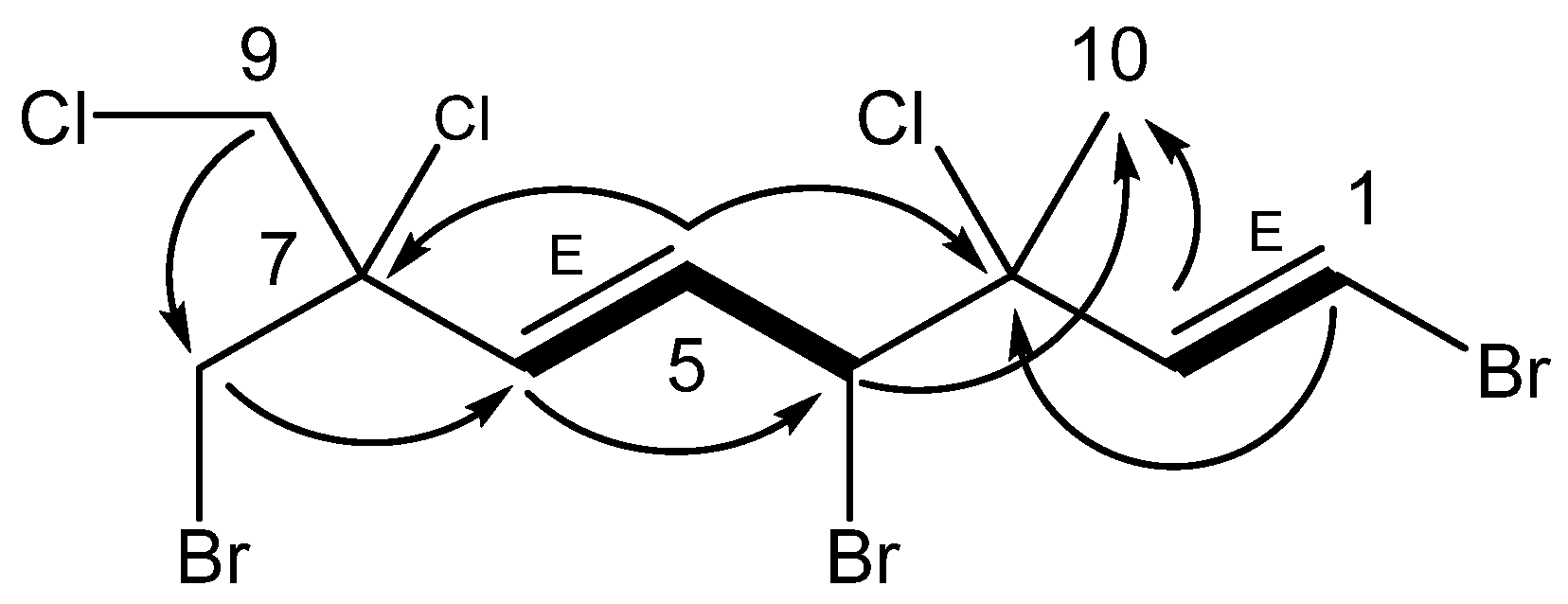
Anverene B (
6) was obtained as a clear oil with spectral data similar to that of
2 and
3. A molecular formula of C
10H
12Br
3Cl
3 was established from high resolution negative chemical ionization mass spectrometry (HRNCIMS) ([M − H]
−: m/z 472.7477, calc. 472.7482), corroborated by
1H and
13C NMR spectra (
Table 1). Key
1H NMR signals (
Figure 1) include a vinyl methine doublet, H-1 (δ
H 6.56), coupled in the COSY spectrum to a second vinyl methine doublet, H-2 (δ
H 6.41), the latter of which could be linked through HMBC correlations to a doublet secondary alkyl methine, H-4 (δ
H 4.60). Additional vinyl methine signals H-5 (δ
H 6.22) and H-6 (δ
H 6.00) were established through COSY correlations as part of an isolated spin system bounded by C-4 and C-6. Two isolated doublet methylene groups, H
2-8 (δ
H 3.83, 3.87) and H
2-9 (δ
H 3.91, 3.98), correlated to C-6 and C-7, as well as to each other, completing the western portion of a monoterpene. The last unaccounted
1H NMR signal, a singlet methyl, H
3-10 (δ
H 1.81), was found through HMBC correlations to C-2 through C-4, as occupying quaternary C-3, yielding the planar structure of anverene B (
Figure 1).
Halogen regiochemistry was primarily defined using
13C NMR shifts. The halogen bearing carbons in the eastern portion of anverene B (
6) including C-1 (δc 110.3), C-3 (δc 70.9), and C-4 (δc 59.3) were matched closely to the carbon shifts reported in anverene A (
1) at the same positions, and were assigned the same substituents; alternate halogenation patterns are not supported by comparison of their carbon shifts to previously published
P. cartilagineum compounds nor calculated chemical shifts (
Table S1) [
4,
6]. Similarly, the halogen bearing carbons in the western portion of anverene B including C-7 (δc 68.8), C-8 (δc 37.2), and C-9 (δc 49.6) matched closely to the carbon shifts reported in
2 at the same positions [
6].
The stereochemical configuration of anverene B (
6) was studied using
3JHH and comparison with previous
Plocamium cartilagineum data sets [
4,
8]. The alkenes were both determined as
E based on
3JHH = 13.6 and 15.3 (
Table 1) [
8,
9]. The relative configuration at C-3 and C-4 was determined by comparison of the carbon shift of C-10 (δc 25.9) with anverene (
1) (C-10 δc 25.5) suggesting the same (
R*,
S*) configuration [
4]. The C-10 methyl group was first noted by Crews as an indicator of differentiation between the (
R,
S) and (
R,
R) configurations at positions C-3 and C-4 within compounds of the same scaffold. He reported three (
R,
S) variants with carbon shifts ranging from δc 24.6–25.4 ppm and two (
R,
R) variants with carbon shifts ranging from 28.0–28.4 ppm, specifically making note of the 3 ppm downfield shift difference between a pair of (
R,S) and (
R,R) diastereomers at positions C-3 and C-4 [
7]. The values seen in anverenes A and B are in agreement with those assignments. The configuration of C-7 was recalcitrant to spectroscopic methods.
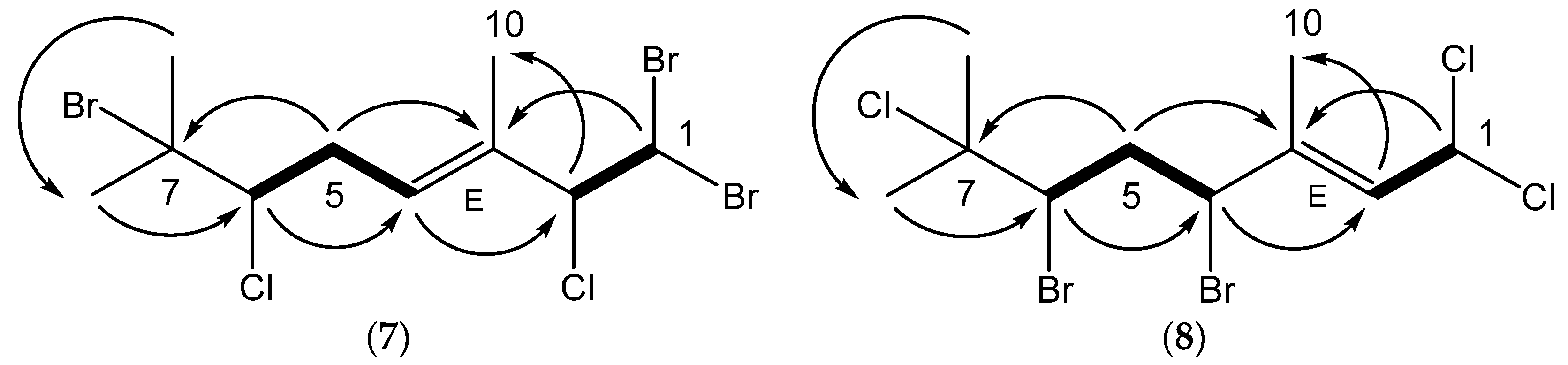
Anverene C (
7) was obtained as a clear oil with spectral data similar to that of (3
E,2
R,6
R,7
S)-1,1,7-tribromo-2,6,8-trichloro-3,7-dimethyloct-3-ene (
10) (
Scheme 3) [
10]. A formula of C
10H
15Br
3Cl
2 was established from HRNCIMS ([M − H]
−: m/z 440.8029, calc. 440.8028). Anverene C departed from the anverene A (
1) and B (
6) motif of the terminal vinyl bromide, displaying a doublet methine, H-1 (δ
H 5.68), on an alkyl-like carbon (δc 45.1) coupled in the COSY spectrum to a similar aliphatic doublet, H-2 (δ
H 4.80) with a carbon shift indicative of an electronegative substituent (δc 73.4). This spin system was extended through HMBC correlation of H-2 to quaternary vinyl carbon C-3 (δc 134.0) and methine C-4 (δc 130.0), the latter bearing a triplet vinyl proton H-4 (δ
H 5.83). A pair of diastereotopic alkyl protons H-5
a (δ
H 2.53) and H-5
b (δ
H 3.15) and methine H-6 (δ
H 4.03) were established through COSY correlations as part of a spin system including C-4 through C-6. H
2-5 could be assigned contiguous to C-4 by HMBC correlations of H-5
a and H-5
b to C-3 and the reciprocal H-4 to C-6. Singlet methyl H
3-10 (δ
H 1.76) displayed HMBC correlations to C-2 through C-4, establishing the eastern portion of anverene C. The remaining two singlet methyl groups, H
3-8 (δ
H 1.80) and H
3-9 (δ
H 1.93) were found on a quaternary alkyl carbon-bearing-heteroatom, C-7 (δ
C 70.5) based on HMBC correlations of their respective protons to C-7, and integrated into the remainder of the monoterpene by virtue of an HMBC correlation to C-6, resulting in the planar structure for anverene C as depicted in
Figure 2. Configuration of the ∆
2 alkene of anverene C was determined as
E based on observation of NOE enhancement of H-2 upon irradiation of H-4.
Halogen regiochemistry was defined, as with anverene B (
6), based on
13C NMR data as well as
1H NMR shifts. The C-1 methine (δc 45.1) of anverene C (
7) requires two halogens; the shift is reminiscent of that for C-1 of
10 (δc 44.2) and incompatible with the value expected for a gem-dichloro (>60 ppm; see anverene D (
8), below). C-2 (δc 73.4) of anverene C is downfield compared to C-2 (δc 64.9) in
10, however a similar proton shift of methine H-2 in
7 and
10 (δ
H 4.80 and 4.73, respectively) argues for the presence of chlorine at C-2 in both [
10]. The two remaining halogens, one chlorine atom and one bromine atom, must fill the remaining open valences at C-6 and C-7. The carbon shifts found in anverene C (C-6, δc 68.8; C-7, δc 70.5) match those found in anverene A (C-6, δc 69.2; C-7, δc 66.3). The proton shifts of the flanking geminal dimethyl group similarly match (anverene C: H
3-8 (δ
H 1.80) and H
3-9 (δ
H 1.93); anverene A: H
3-8 (δ
H 1.81) and H
3-9 (δ
H 1.92)) [
4]. Compare the proton shifts of the gem-dimethyl group of anverene D, where the chloride and bromide have flipped positions, resulting in a 0.1 ppm upfield shift (
Table 2). The rotatable nature of the acyclic scaffold rendered the stereochemical determination at C-2 and C-6 unattainable using spectroscopic methods.
Anverene D (
8) was obtained as a clear oil with spectral data similar to that of anverene C (
7) and certain shifts bearing close resemblance to plocoralide B (
11) (
Scheme 4) [
11]. A formula of C
10H
15Br
2Cl
3 was established from HRNCIMS ([M − H]
−: m/z 396.8535, calc. 396.8533). Anverene D displays a doublet methine, H-1 (δ
H 6.40), on an sp
3 carbon with a shift (δc 66.6) indicative of substitution by electronegative groups but differing from anverene C in that it is coupled in the COSY spectrum to a doublet vinyl proton H-2 (δ
H 6.03) situated on an sp
2 methine carbon (C-2, δc 128.0). This spin system was extended through HMBC correlation of H-1 to quaternary olefinic carbon C-3 (δc 138.2), and further HMBC correlation of H-2 to a methine C-4 (δc 57.8), the latter bearing a proton split as a doublet of doublets (H-4, δ
H 4.81). A pair of diastereotopic alkyl protons H-5
a (δ
H 2.13) and H-5
b (δ
H 2.88) and methine H-6 (δ
H 4.43) were established through COSY correlations as part of a spin system including C-4 through C-6. Further expansion of that partial structure was facilitated by HMBC correlation of a singlet methyl group (H
3-10), (δ
H 1.94) to C-2 through C-4. The remaining two singlet methyl groups, H
3-8 (δ
H 1.71) and H
3-9 (δ
H 1.84) displayed HMBC correlation to a quaternary aliphatic carbon, C-7 (δc 70.8) as well as C-6, completing the planar structure for anverene D as depicted in
Figure 2. Configuration of the ∆
2 alkene of anverene D was determined as
E based on observation of NOE enhancement of H-4 upon irradiation of H-2.
Halogen regiochemistry was defined, as with anverenes B (
6) and C (
7), based on
13C NMR as well as
1H NMR shifts. The halogen bearing carbon C-1 of anverene D (
8) was much farther downfield in both carbon shift (δc 66.6) and proton shift of the methine H-1 (δ
H 6.40) in relation to the analogous carbon shift (δc 45.1) and proton shift of the methine H-1 (δ
H 5.68) of anverene C at the same C-1 position, and closely resembled the C-1 (δc 66.5) and H-1 (δ
H 6.36) shifts reported in plocoralide B (
11), leading us to assign a geminal dichloride substituent that accounted for two of the three chlorine atoms in the molecular formula. The halogen bearing carbon C-4 (δc 57.8) of anverene D was similar to the carbon shift reported in anverene A (
1) (δc 59.8) and plocoralide B (
11) (δc 55.9) at the same position C-4, and hence could be assigned the same bromine substituent [
10]. The halogen bearing carbon C-6 (δc 62.7) was upfield in relation to the carbon shift reported in anverene A at the chlorine bearing C-6 (δc 69.2) position, and was assigned a bromine substituent, accounting for the second of the two bromine atoms in the formula. Finally, the halogen bearing carbon C-7 (δc 70.8) was downfield in relation to the carbon shift reported in anverene A at the bromine bearing C-7 (δc 66.3) position and was assigned a chlorine substituent accounting for the final halogen of the formula. This final assignment is supported by the differences seen in the proton shifts of the flanking geminal dimethyl groups of anverene D (H
3-8, δ
H 1.71; H
3-10, δ
H 1.84), compared to that of anverene A at the same positions (H
3-8, δ
H 1.81; H
3-9, δ
H 1.92) reflecting the greater deshielding effects of the bulkier bromine substituent seen in anverene A in relation to protons occupying the flanking geminal groups [
4]. The rotatable nature of the acyclic scaffold rendered the stereochemical determination at C-4 and C-6 impractical using spectroscopic methods.
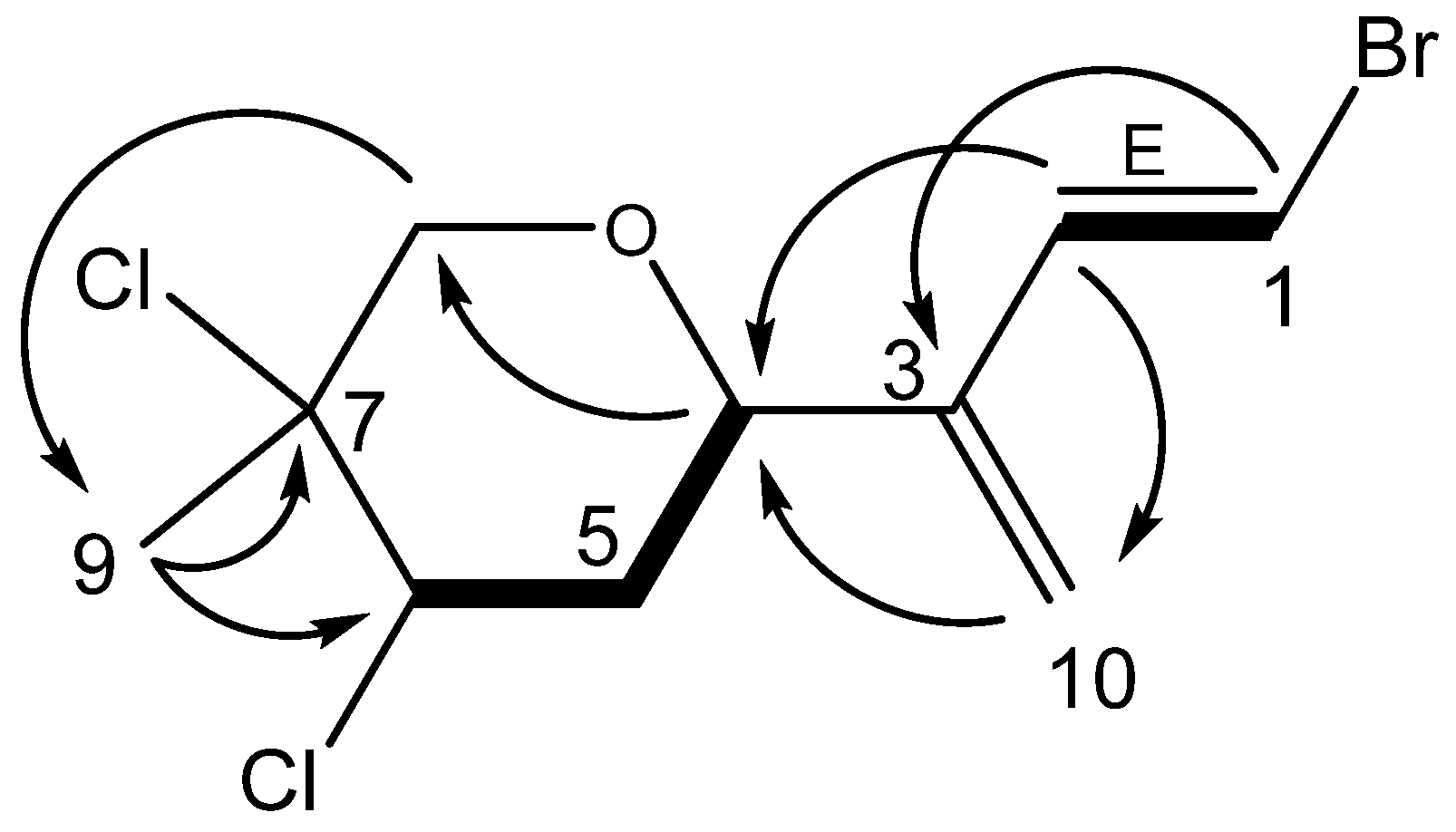
Anverene E (
9) was obtained as a clear oil with a formula of C
10H
13BrCl
2O established from HRNCIMS ([M + Br]-: m/z 376.8705, calc. 376.8716) and corroborated by
1H and
13C NMR spectra (
Table 3). Key
1H NMR signals (
Figure 3) include a vinyl methine doublet, H-1 (δ
H 6.56), coupled in the COSY spectrum to a second vinyl methine doublet, H-2 (δ
H 6.73). This spin system was extended through HMBC correlation of H-1 to quaternary vinyl carbon C-3 (δc 143.2), and further HMBC correlation of H-2 to a methine C-4 (δc 72.0) bearing a proton coupled as a doublet of doublets (H-4, δ
H 4.68). A terminal olefin C-10 (δc 116.8) bearing two singlet vinyl protons H-10
a (δ
H 5.26) and H-10
b (δ
H 5.28) was established as part of a diene residing in the linear eastern portion of a monoterpene through HMBC correlations of H-2 to C-10 as well as H-10
a and H-10
b to both C-3 and C-4. A pair of diastereotopic methylene protons H-5
β (δ
H 2.30) and H-5
α (δ
H 2.42) and a triplet methine H-6 (δ
H 4.52) were established through COSY correlations as part of a spin system including C-4 through C-6. The position of another pair of diastereotopic methylene protons H-8
β (δ
H 3.65) and H-8
α (δ
H 4.05) was assigned through HMBC correlations of H
2-8 to C-4 as well as to C-6, determining its position within a cyclic system comprising the western portion of the monoterpene, with C-4 and C-8 linked through the same electronegative substituent reflected by their similar carbon shifts C-4 (δc 72.0) and C-8 (δc 71.5). Based on the carbon shifts and the oxygen atom in the molecular formula, the cyclic system must be a pyran. The remaining singlet methyl group, H
3-9 (δ
H 1.80) was found on a quaternary alkyl carbon-bearing-heteroatom, C-7 (δc 66.4) based on an HMBC correlation of H
3-9 to C-7, and integrated into the ring system of the western portion by virtue of HMBC correlations to C-6 and C-8, resulting in the planar structure for anverene E as depicted in
Figure 3.
Halogen regiochemistry was defined, as with anverenes B–D (
6–
8), based on
13C NMR as well as
1H NMR shifts. The halogen bearing olefinic carbon of anverene E (
9) (C-1, δc 108.2) as well as its methine proton (H-1, δ
H 6.56) was similar to the carbon (δc 109.7) and proton (δ
H 6.58) shifts reported in anverene A (
1) at the same C-1 position and was assigned analogous bromide substituent accounting for the lone bromine atom in the molecule [
4]. The remaining chlorine atoms were assigned to the other two halogenated carbons on the molecule including C-6 which displays a methine proton shift H-6 (δ
H 4.52) and adjacent methyl group H
3-9 (δ
H 1.80) similar to the analogous moiety reported in
2 (H-4, δ
H 4.48; H
3-9, δ
H 1.75) [
6].
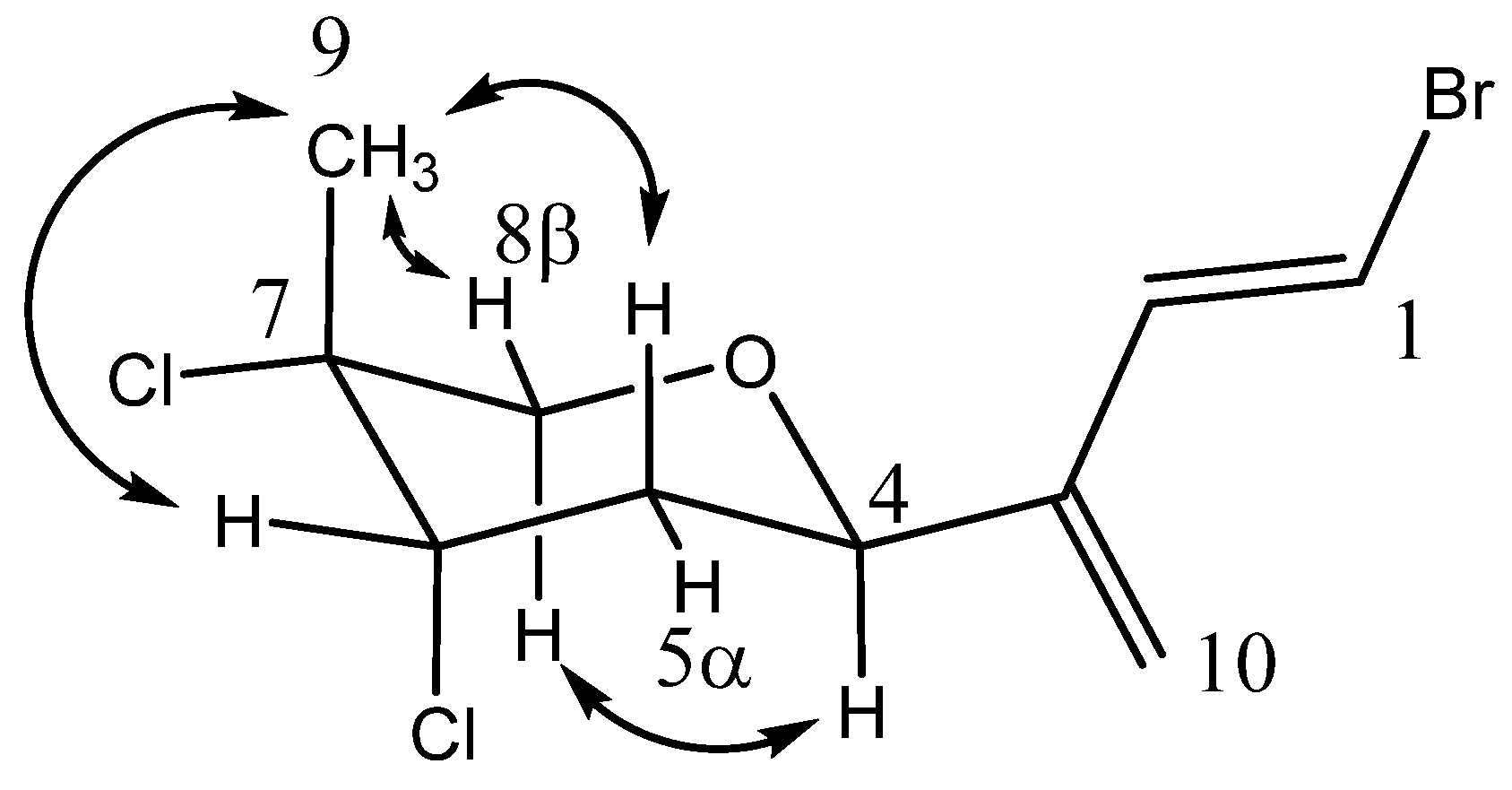
NOESY correlations facilitated the assignment of the relative stereochemical configuration of anverene E (
9) (
Figure 4). A correlation from H-4 (δ
H 4.68) to H-8
α (δ
H 4.05) established the axial orientation of these protons on the α face of the pyran ring and placing the diene chain in the equatorial position. A correlation from H-5
β (δ
H 2.30) to H
3-9 (δ
H 1.80) then informed the placement of the methyl group as axial and occupying the β face of the pyran ring, an assignment further supported by a correlation to from H
3-9 to the adjacent equatorial H-8
β (δ
H 3.65). The equatorial position of H-6 (δ
H 4.52) was derived both by its NOESY correlation to the axial H
3-9 methyl group and through evaluation of the coupling constant (
3J5β,6 = 8.4). The alkene at C-1 and C-2 was determined as
E based on
3J1,2 = 14.1 (
Table 3).
Previous investigations into the therapeutic potential of halogenated monoterpenes have yielded a plethora of biological activities ranging from antimicrobial to antitumor properties. The cytotoxic nature of these compounds is likely derived from their original ecological function as feeding deterrents and antifouling agents [
3,
4,
5]. Compound
4 was previously evaluated for its activity against a human esophageal cancer cell line by Knott et al. (2005) who reported an IC
50 of 9.3 µM. Compounds
1–
9 were consequently evaluated in vitro against a human cervical cancer cell line (HeLa) and all of the compounds showed low micromolar cytotoxicity, with
2,
3, and anverene D (
7) displaying single-digit micromolar activity (
Table 4). Anverene A was on hand from previous investigations [
4].

 ,
, 









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}