5-O-Acetyl-Renieramycin T from Blue Sponge Xestospongia sp. Induces Lung Cancer Stem Cell Apoptosis


 , and
, and 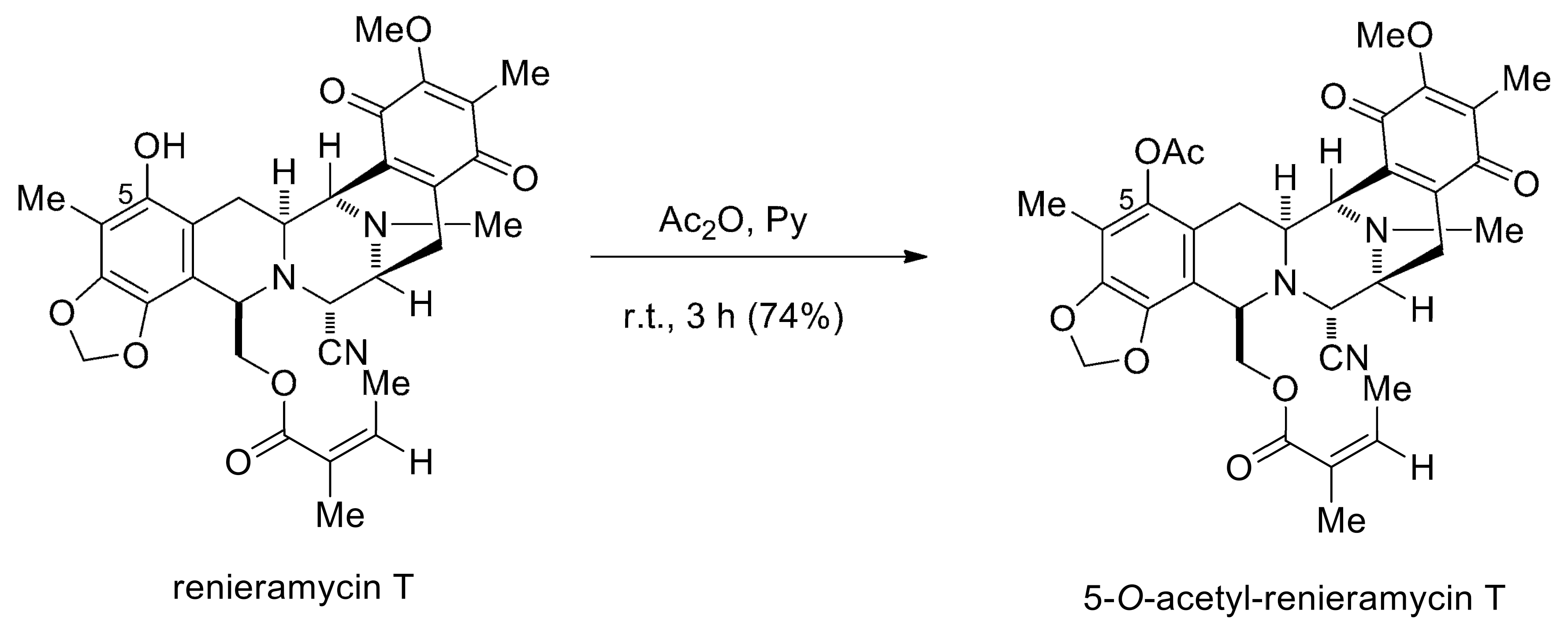{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
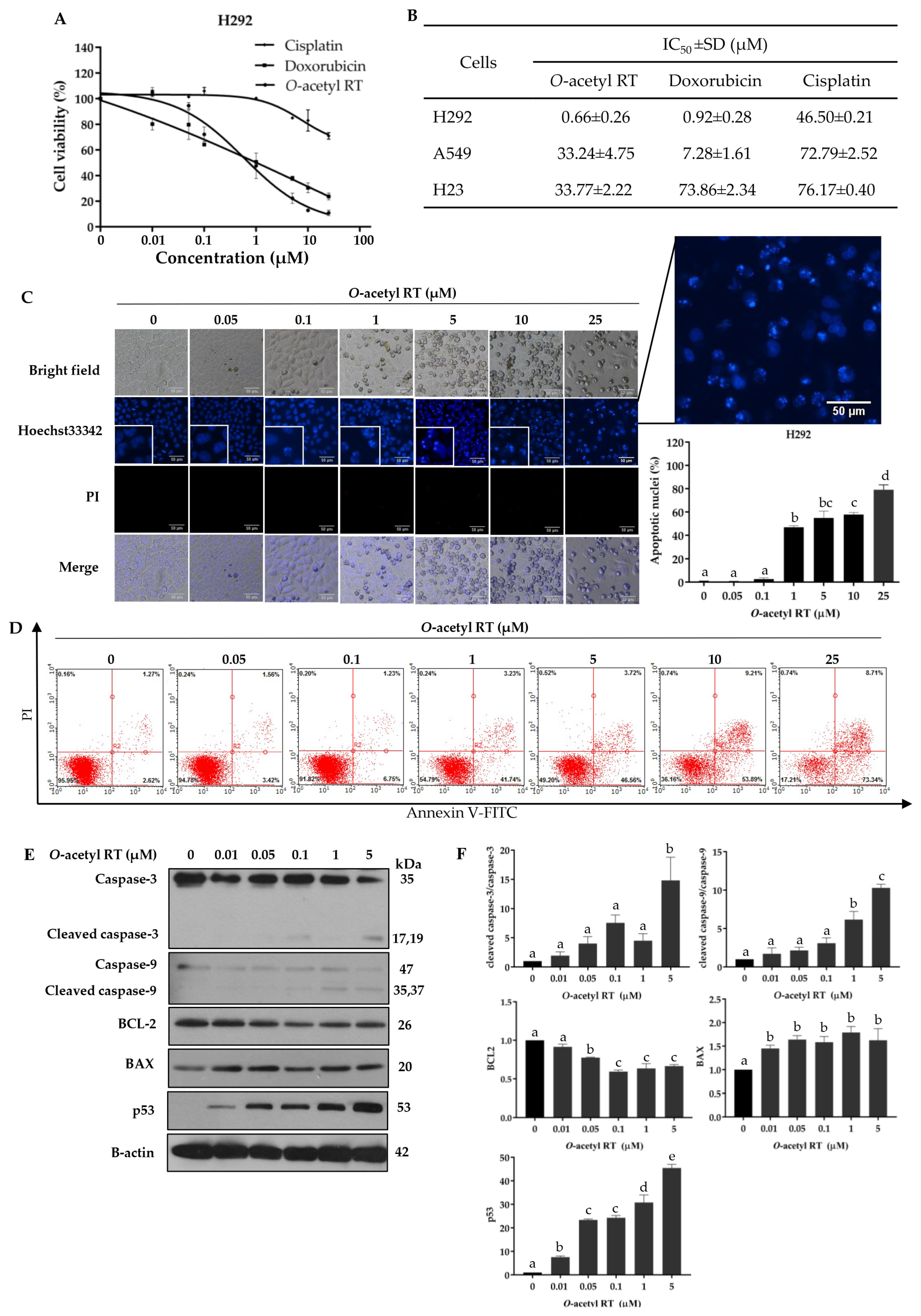
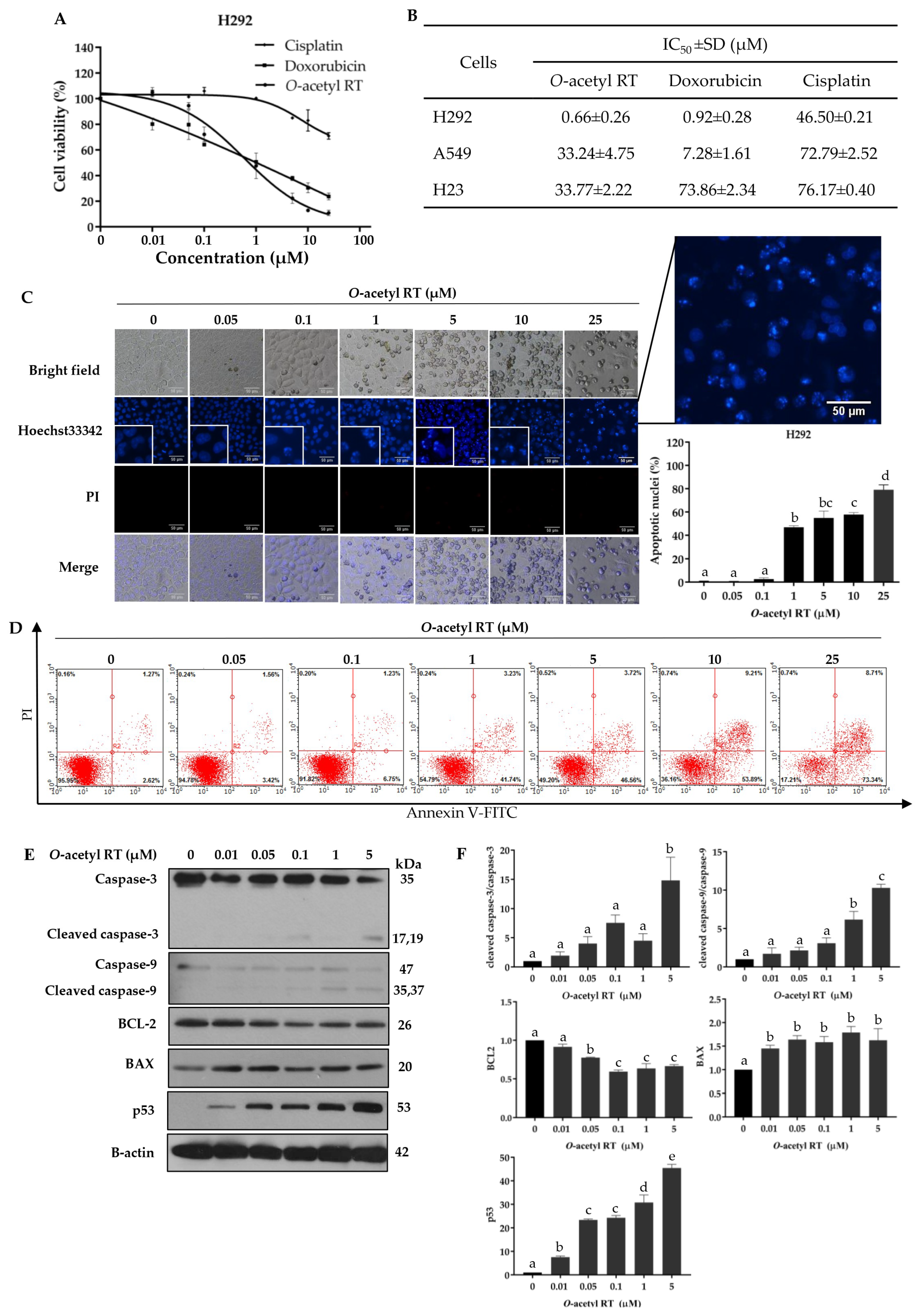
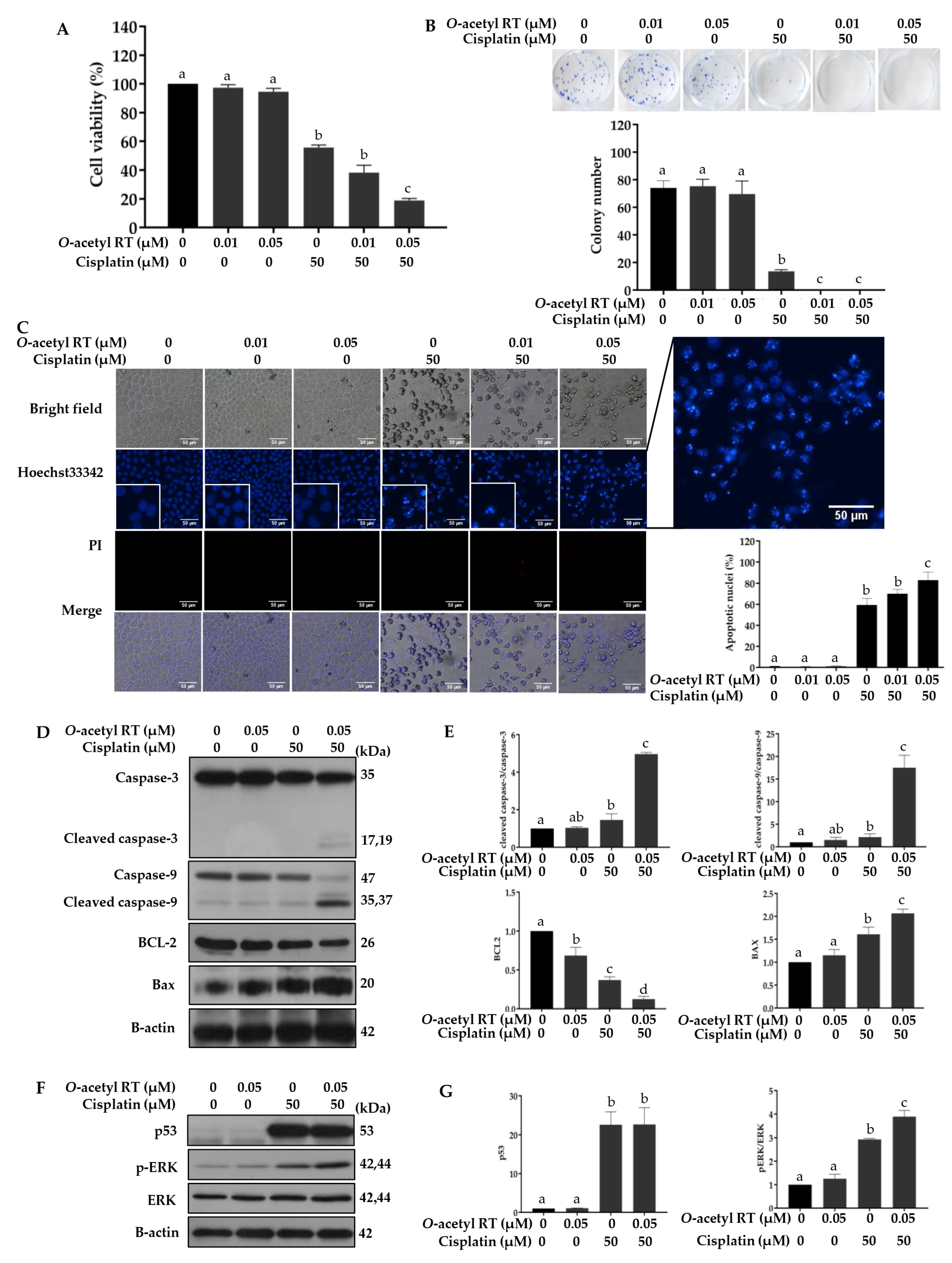
2.1. O-Acetyl RT Reduced the Cell Viability and Induced the Apoptosis of Non-Small-Cell Lung Carcinoma (NSCLC) Cells
2.2. O-Acetyl RT Induced Apoptosis through p53 Activation
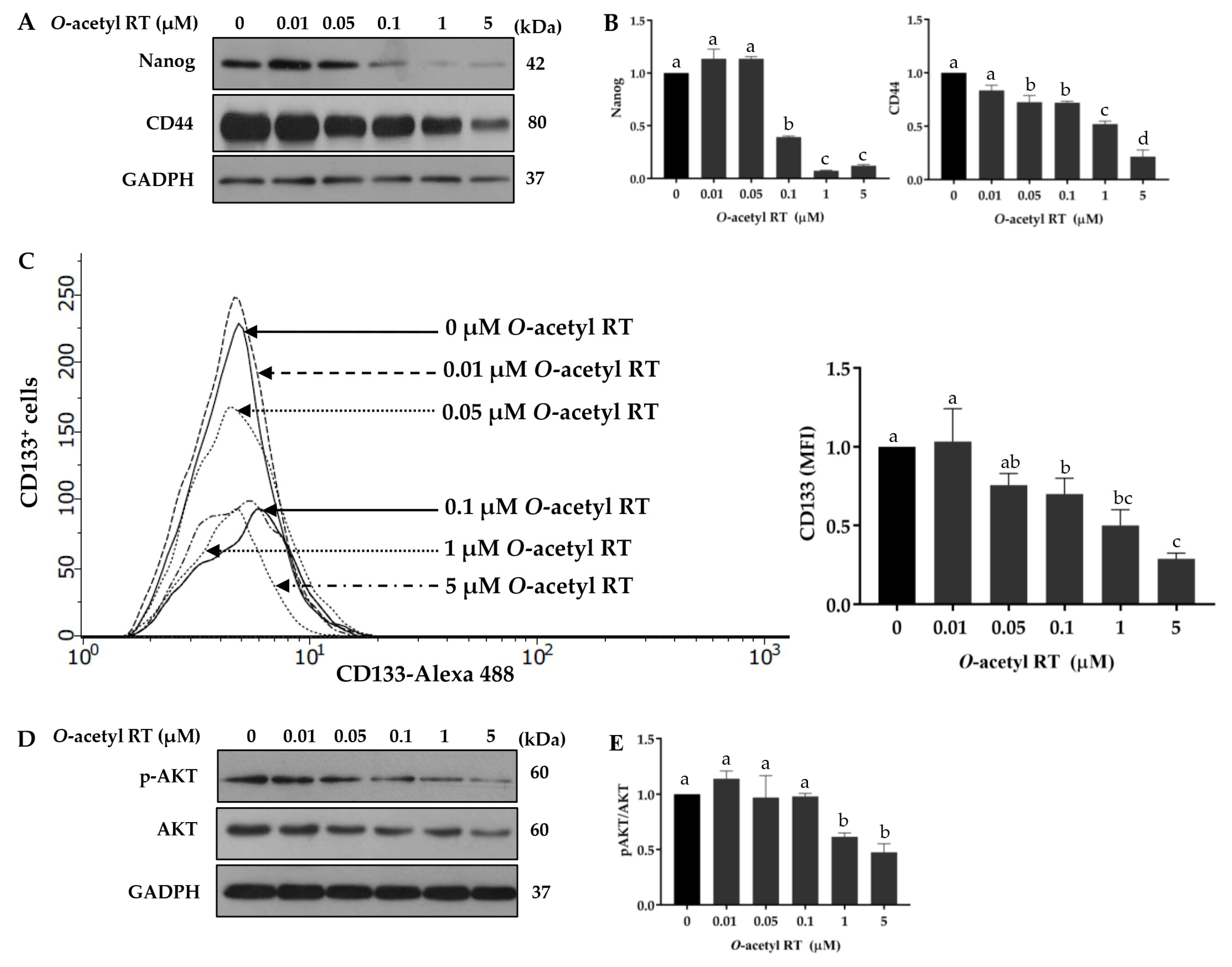
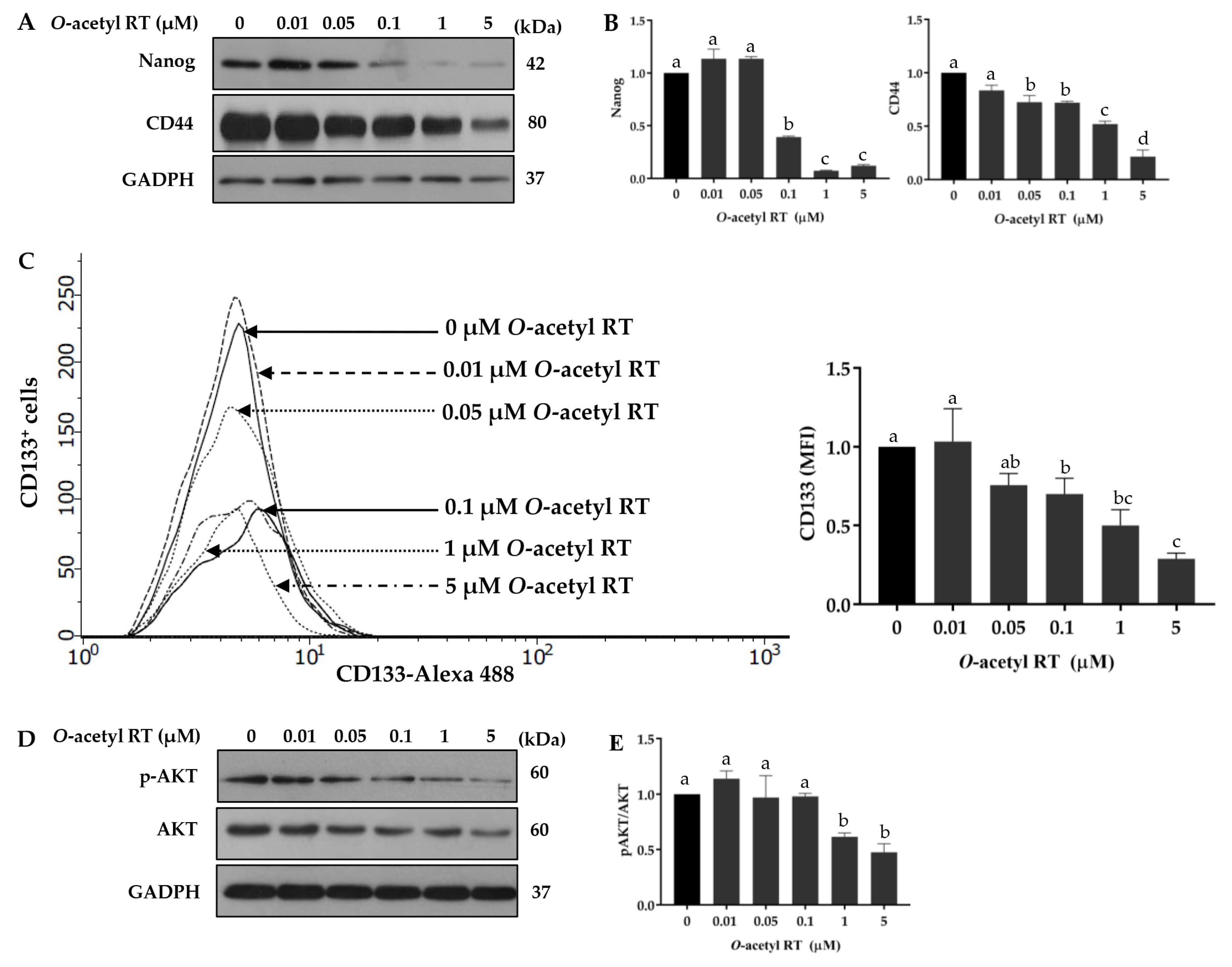
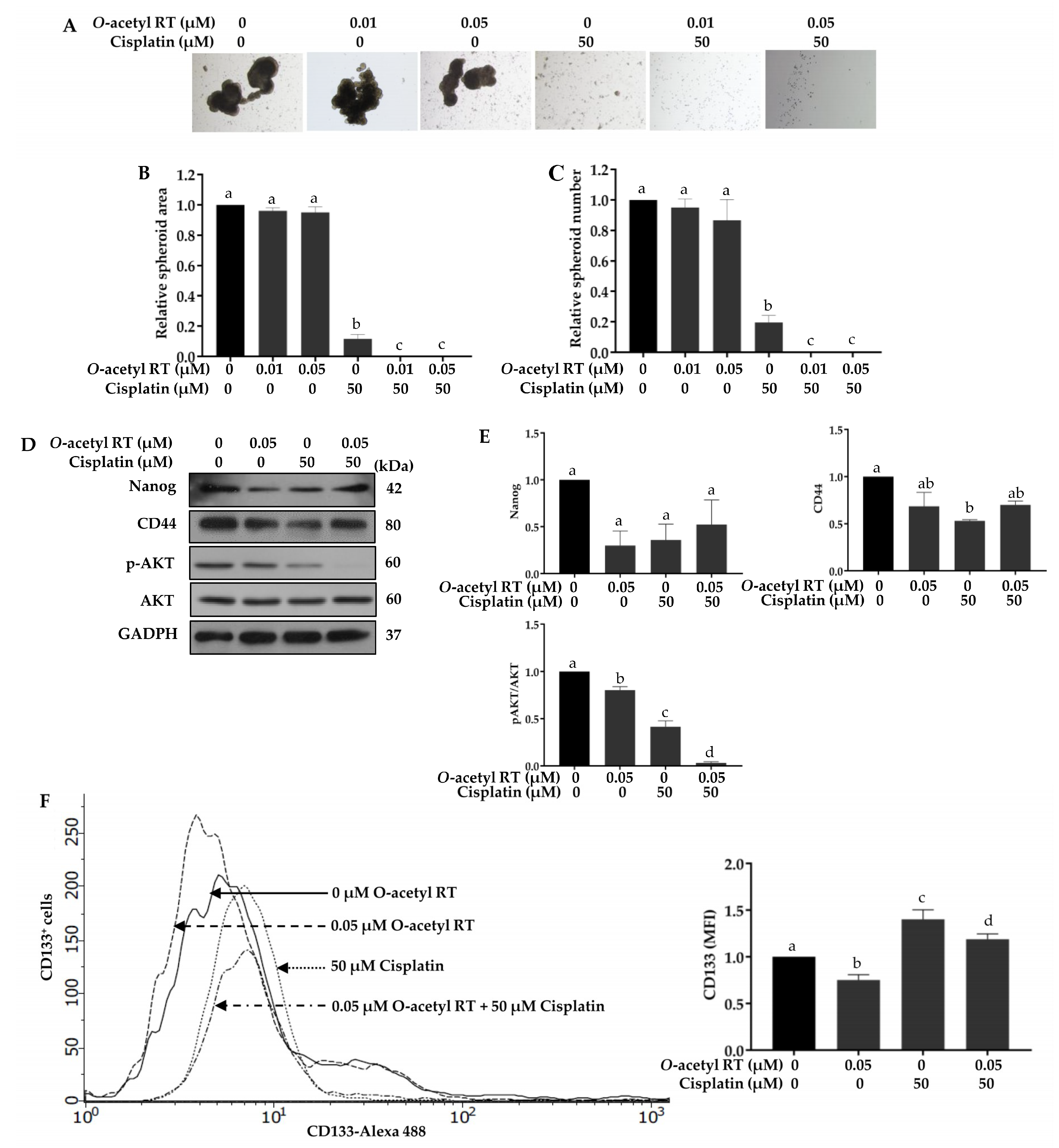
2.3. O-Acetyl RT Suppresses Cancer Stem Cell Signals in H292 Cells
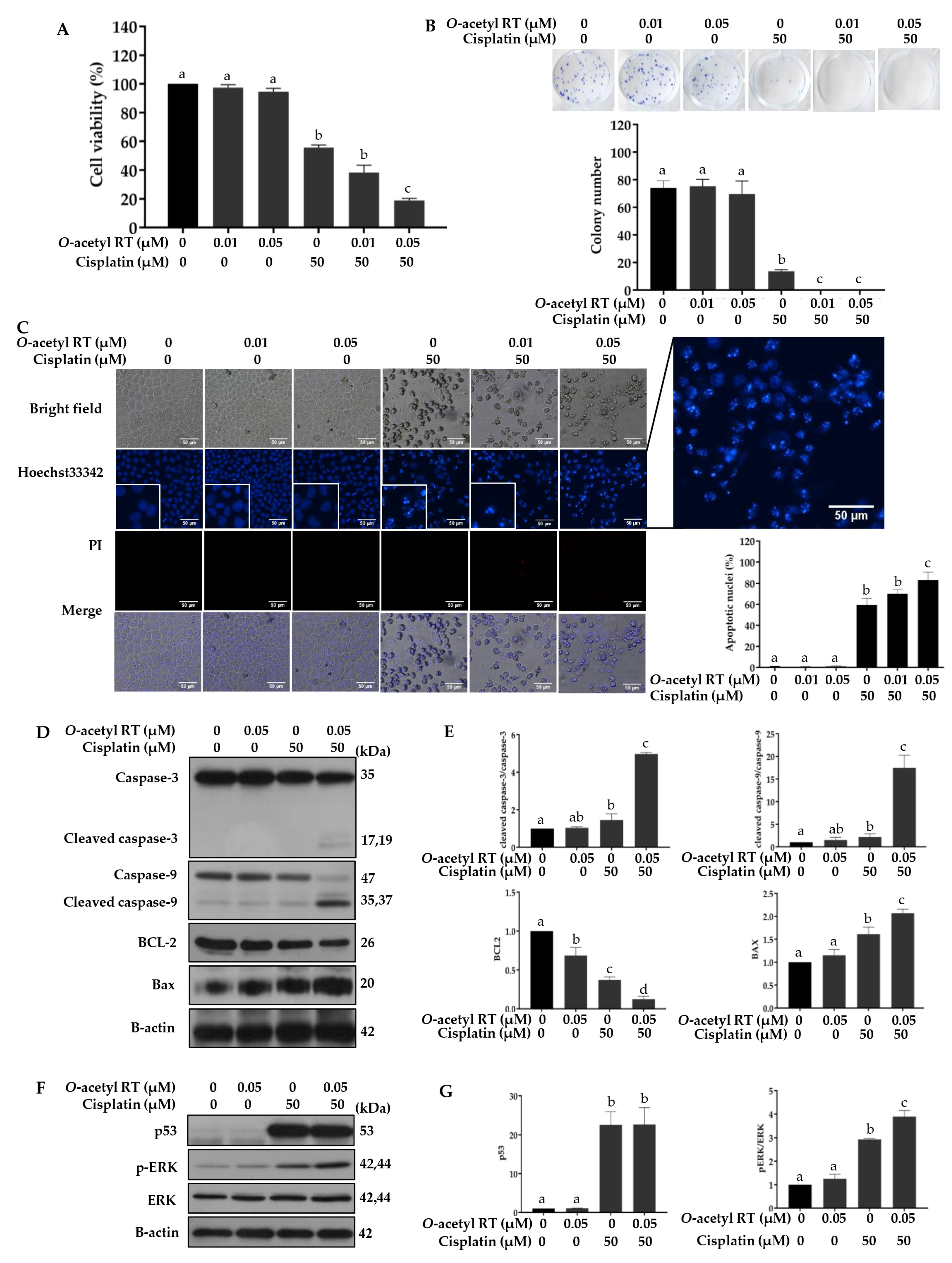
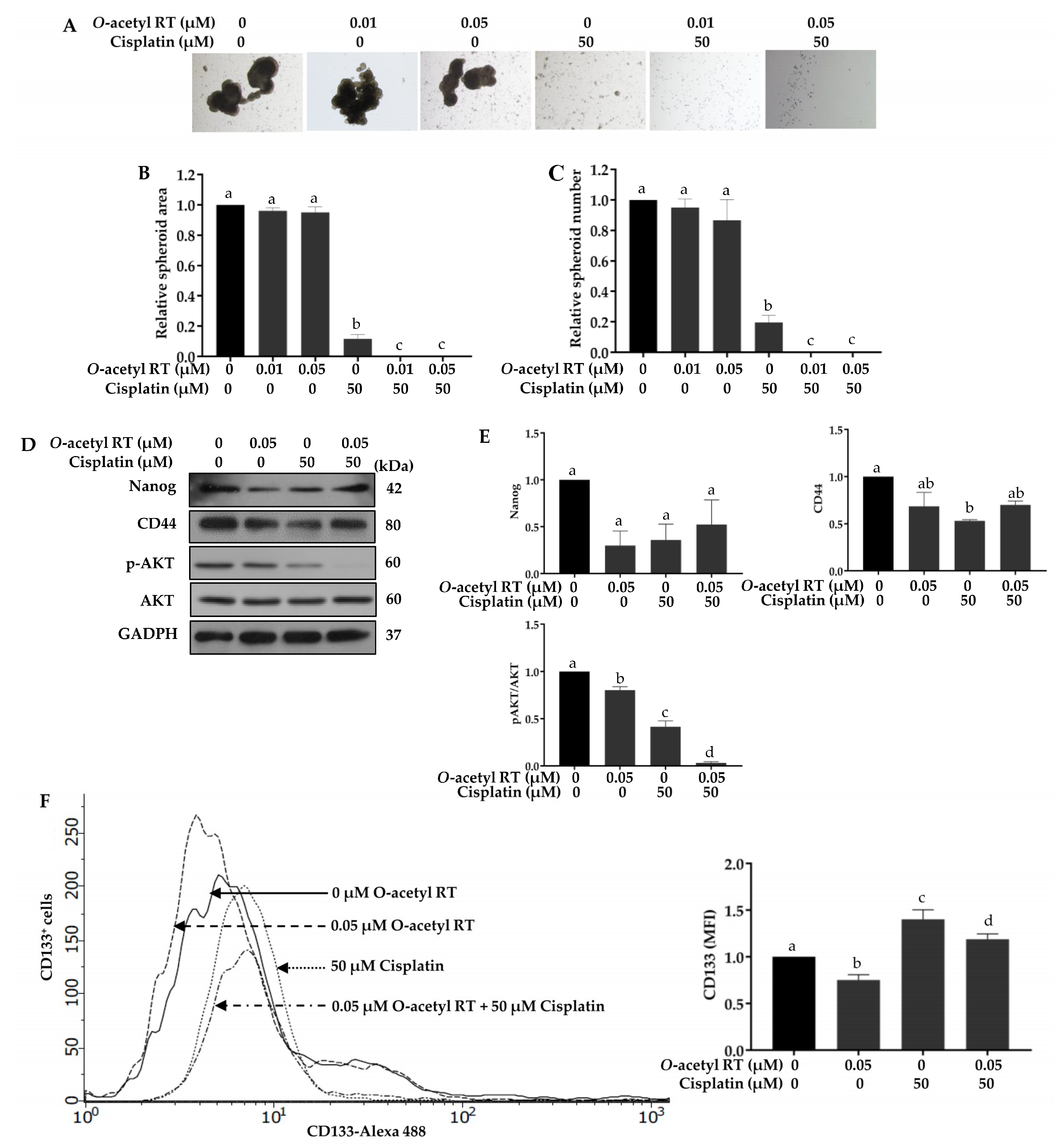
2.4. O-Acetyl RT Increases Sensitivity of H292 Cells to Cisplatin
2.5. O-Acetyl RT Reduces Cisplatin-Induced CD133+ Cells Subpopulation in H292 Cells
3. Discussion
4. Materials and Methods
4.1. Reagents and Antibodies
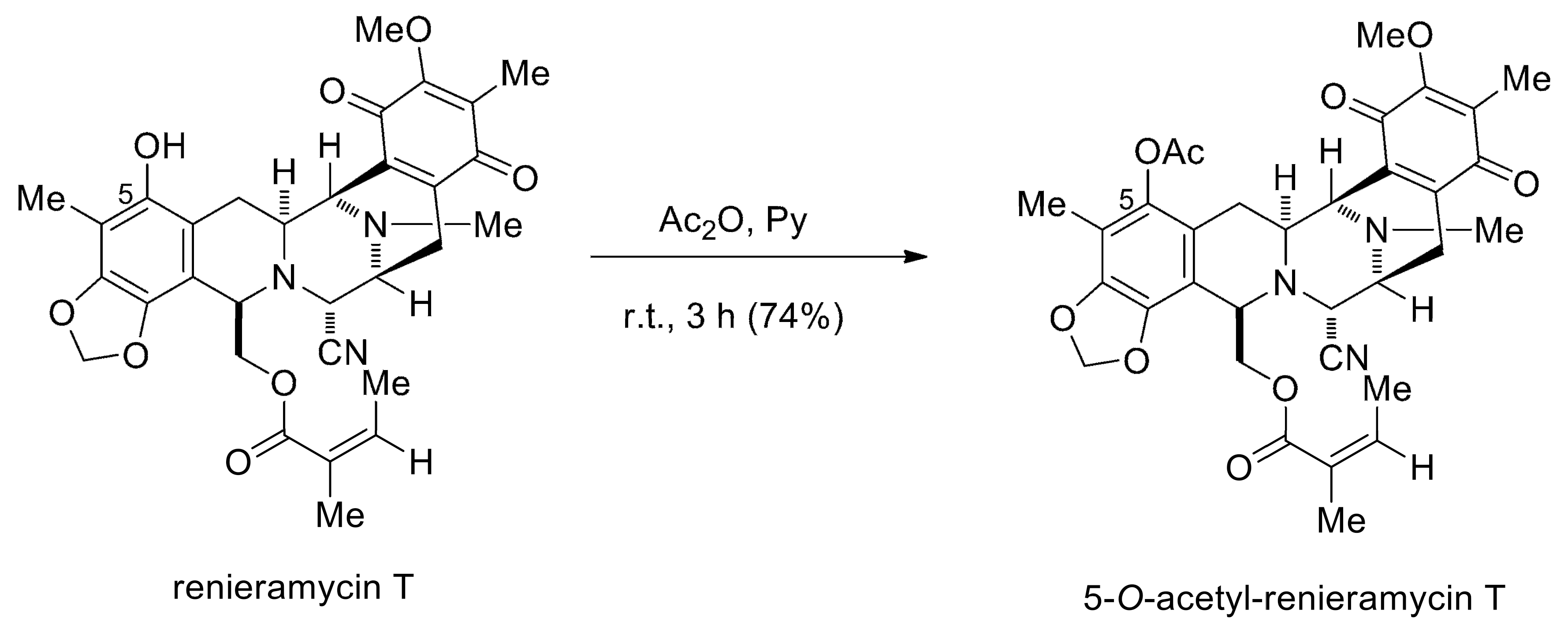
4.2. Preparation of O-Acetyl RT
4.3. Preparation of O-Acetyl RT and Cisplatin Stock Solution
4.4. Cell Lines and Culture
4.5. Cell Viability Assay
4.6. Colony Formation Assay
4.7. Spheroid Formation Assay
4.8. Nuclear Staining Assay
4.9. Annexin V and Propidium Iodide Apoptosis Assay
4.10. Western Blot Analysis
4.11. Flow Cytometry Analysis
4.12. Statistical Analysis
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Molinski, T.F.; Dalisay, D.S.; Lievens, S.L.; Saludes, J.P. Drug development from marine natural products. Nat. Rev. Drug Discov. 2009, 8, 69–85. [Google Scholar] [CrossRef] [PubMed]
- Cragg, G.M.; Grothaus, P.G.; Newman, D.J. Impact of natural products on developing new anti-cancer agents. Chem. Rev. 2009, 109, 3012–3043. [Google Scholar] [CrossRef] [PubMed]
- Moharil, R.B.; Dive, A.; Khandekar, S.; Bodhade, A. Cancer stem cells: An insight. J. Oral Maxillofac. Pathol. 2017, 21, 463. [Google Scholar] [CrossRef] [PubMed]
- Ayob, A.Z.; Ramasamy, T.S. Cancer stem cells as key drivers of tumour progression. J. Biomed. Sci. 2018, 25, 20. [Google Scholar] [CrossRef] [PubMed]
- Koren, A.; Motaln, H.; Cufer, T. Lung cancer stem cells: A biological and clinical perspective. Cell Oncol. (Dordr) 2013, 36, 265–275. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Liu, X.; Ren, Y.; Zhang, J.; Chen, J.; Zhou, W.; Guo, W.; Wang, X.; Chen, H.; Li, M.; et al. Cisplatin-enriching cancer stem cells confer multidrug resistance in non-small cell lung cancer via enhancing TRIB1/HDAC activity. Cell Death Dis. 2017, 8, e2746. [Google Scholar] [CrossRef]
- Levina, V.; Marrangoni, A.M.; DeMarco, R.; Gorelik, E.; Lokshin, A.E. Drug-selected human lung cancer stem cells: Cytokine network, tumorigenic and metastatic properties. PLoS ONE 2008, 3, e3077. [Google Scholar] [CrossRef]
- MacDonagh, L.; Gray, S.G.; Breen, E.; Cuffe, S.; Finn, S.P.; O’Byrne, K.J.; Barr, M.P. Lung cancer stem cells: The root of resistance. Cancer Lett. 2016, 372, 147–156. [Google Scholar] [CrossRef]
- Wang, Y.; Jiang, M.; Du, C.; Yu, Y.; Liu, Y.; Li, M.; Luo, F. Utilization of lung cancer cell lines for the study of lung cancer stem cells (Review). Oncol. Lett. 2018, 15, 6791–6798. [Google Scholar] [CrossRef]
- Salnikov, A.V.; Gladkich, J.; Moldenhauer, G.; Volm, M.; Mattern, J.; Herr, I. CD133 is indicative for a resistance phenotype but does not represent a prognostic marker for survival of non-small cell lung cancer patients. Int. J. Cancer 2010, 126, 950–958. [Google Scholar] [CrossRef]
- Liu, Y.-P.; Yang, C.-J.; Huang, M.-S.; Yeh, C.-T.; Wu, A.T.H.; Lee, Y.-C.; Lai, T.-C.; Lee, C.-H.; Hsiao, Y.-W.; Lu, J.; et al. Cisplatin selects for multidrug-resistant CD133+ cells in lung adenocarcinoma by activating Notch signaling. Cancer Res. 2013, 73, 406–416. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Zhou, B.P. Activation of β-catenin and Akt pathways by Twist are critical for the maintenance of EMT associated cancer stem cell-like characters. BMC Cancer 2011, 11, 49. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.K.; Zhu, Y.L.; Qiu, F.M.; Zhang, T.; Chen, Z.G.; Zheng, S.; Huang, J. Activation of Akt and MAPK pathways enhances the tumorigenicity of CD133+ primary colon cancer cells. Carcinogenesis 2010, 31, 1376–1380. [Google Scholar] [CrossRef] [PubMed]
- Scott, J.D.; Williams, R.M. Chemistry and biology of the tetrahydroisoquinoline antitumor antibiotics. Chem. Rev. 2002, 102, 1669–1730. [Google Scholar] [CrossRef] [PubMed]
- Edrada, R.A.; Proksch, P.; Wray, V.; Christ, R.; Witte, L.; Van Soest, R.W.M. Bioactive isoquinoline quinone from an undescribed Philippine marine sponge of the genus Xestospongia. J. Nat. Prod. 1996, 59, 973–976. [Google Scholar] [CrossRef]
- Suwanborirux, K.; Amnuoypol, S.; Plubrukarn, A.; Pummangura, S.; Kubo, A.; Tanaka, C.; Saito, N. Chemistry of renieramycins. Part 3. Isolation and structure of stabilized renieramycin type derivatives possessing antitumor activity from Thai sponge Xestospongia species, pretreated with potassium cyanide. J. Nat. Prod. 2003, 66, 1441–1446. [Google Scholar] [CrossRef] [PubMed]
- Amnuoypol, S.; Suwanborirux, K.; Pummangura, S.; Kubo, A.; Tanaka, C.; Saito, N. Chemistry of renieramycins. Part 5. Structure elucidation of renieramycin-type derivatives O, Q, R, and S from Thai marine sponge Xestospongia species pretreated with potassium cyanide. J. Nat. Prod. 2004, 67, 1023–1028. [Google Scholar] [CrossRef] [PubMed]
- Chamni, S.; Sirimangkalakitti, N.; Chanvorachote, P.; Saito, N.; Suwanborirux, K. Chemistry of renieramycins. 17. A new generation of renieramycins: Hydroquinone 5-O-monoester analogues of renieramycin M as potential cytotoxic agents against non-small-cell lung cancer cells. J. Nat. Prod. 2017, 80, 1541–1547. [Google Scholar] [CrossRef] [PubMed]
- Tatsukawa, M.; Punzalan, L.L.C.; Magpantay, H.D.S.; Villaseñor, I.M.; Concepcion, G.P.; Suwanborirux, K.; Yokoya, M.; Saito, N. Chemistry of renieramycins. Part 13: Isolation and structure of stabilized renieramycin type derivatives, renieramycins W–Y, from Philippine blue sponge Xestospongia sp., pretreated with potassium cyanide. Tetrahedron 2012, 68, 7422–7428. [Google Scholar] [CrossRef]
- Singh, I.P.; Shah, P. Tetrahydroisoquinolines in therapeutics: A patent review (2010–2015). Expert Opin. Ther. Pat. 2017, 27, 17–36. [Google Scholar] [CrossRef]
- Yokoya, M.; Toyoshima, R.; Suzuki, T.; Le, V.H.; Williams, R.M.; Saito, N. Stereoselective total synthesis of (−)-renieramycin T. J. Org. Chem. 2016, 81, 4039–4047. [Google Scholar] [CrossRef] [PubMed]
- Kimura, S.; Saito, N. Construction of the pentacyclic core and formal total synthesis of (rac)-renieramycin T. ChemistryOpen 2018, 7, 764–771. [Google Scholar] [CrossRef] [PubMed]
- US Food and Drug Administration. FDA Approves New Therapy for Certain Types of Advanced Soft Tissue Sarcoma. Available online: https://www.accessdata.fda.gov/drugsatfda_docs/label/2015/207953s000lbl.pdf (accessed on 28 January 2019).
- Daikuhara, N.; Tada, Y.; Yamaki, S.; Charupant, K.; Amnuoypol, S.; Suwanborirux, K.; Saito, N. Chemistry of renieramycins. Part 7: Renieramycins T and aU, novel renieramycin–ecteinascidin hybrid marine natural products from Thai sponge Xestospongia sp. Tetrahedron Lett. 2009, 50, 4276–4278. [Google Scholar] [CrossRef]
- Charupant, K.; Daikuhara, N.; Saito, E.; Amnuoypol, S.; Suwanborirux, K.; Owa, T.; Saito, N. Chemistry of renieramycins. Part 8: Synthesis and cytotoxicity evaluation of renieramycin M–jorunnamycin A analogues. Bioorg. Med. Chem. 2009, 17, 4548–4558. [Google Scholar] [CrossRef] [PubMed]
- Johnstone, R.W.; Ruefli, A.A.; Lowe, S.W. Apoptosis: A link between cancer genetics and chemotherapy. Cell 2002, 108, 153–164. [Google Scholar] [CrossRef]
- Vazquez, A.; Bond, E.E.; Levine, A.J.; Bond, G.L. The genetics of the p53 pathway, apoptosis and cancer therapy. Nat. Rev. Drug Discov. 2008, 7, 979–987. [Google Scholar] [CrossRef] [PubMed]
- Fiandalo, M.V.; Kyprianou, N. Caspase control: Protagonists of cancer cell apoptosis. Exp. Oncol. 2012, 34, 165–175. [Google Scholar] [PubMed]
- Wesarg, E.; Hoffarth, S.; Wiewrodt, R.; Kröll, M.; Biesterfeld, S.; Huber, C.; Schuler, M. Targeting BCL-2 family proteins to overcome drug resistance in non-small cell lung cancer. Int. J. Cancer 2007, 121, 2387–2394. [Google Scholar] [CrossRef]
- Luanpitpong, S.; Chanvorachote, P. Nitric oxide and aggressive behavior of lung cancer cells. Anticancer Res. 2015, 35, 4585–4592. [Google Scholar]
- Wang, X.; Martindale, J.L.; Holbrook, N.J. Requirement for ERK activation in cisplatin-induced apoptosis. J. Biol. Chem. 2000, 275, 39435–39443. [Google Scholar] [CrossRef]
- Horn, H.F.; Vousden, K.H. Coping with stress: Multiple ways to activate p53. Oncogene 2007, 26, 1306–1316. [Google Scholar] [CrossRef]
- Pflaum, J.; Schlosser, S.; Müller, M. p53 family and cellular stress responses in cancer. Front. Oncol. 2014, 4, 285. [Google Scholar] [CrossRef]
- Dasari, S.; Tchounwou, P.B. Cisplatin in cancer therapy: Molecular mechanisms of action. Eur. J. Pharmacol. 2014, 740, 364–378. [Google Scholar] [CrossRef]
- Wang, G.; Reed, E.; Li, Q.Q. Molecular basis of cellular response to cisplatin chemotherapy in non-small cell lung cancer. Oncol. Rep. 2004, 12, 955–965. [Google Scholar] [CrossRef]
- Galluzzi, L.; Senovilla, L.; Vitale, I.; Michels, J.; Martins, I.; Kepp, O.; Castedo, M.; Kroemer, G. Molecular mechanisms of cisplatin resistance. Oncogene 2012, 31, 1869–1883. [Google Scholar] [CrossRef]
- Guntur, V.P.; Waldrep, J.C.; Guo, J.J.; Selting, K.; Dhand, R. Increasing P53 protein sensitizes non-small cell lung cancer to paclitaxel and cisplatin in vitro. Anticancer Res. 2010, 30, 3557–3564. [Google Scholar]
- Yang, M.; Yuan, F.; Li, P.; Chen, Z.; Chen, A.; Li, S.; Hu, C. Interferon regulatory factor 4 binding protein is a novel p53 target gene and suppresses cisplatin-induced apoptosis of breast cancer cells. Mol. Cancer 2012, 11, 54. [Google Scholar] [CrossRef]
- Liu, R.; Ji, P.; Liu, B.; Qiao, H.; Wang, X.; Zhou, L.; Deng, T.; Ba, Y. Apigenin enhances the cisplatin cytotoxic effect through p53-modulated apoptosis. Oncol. Lett. 2017, 13, 1024–1030. [Google Scholar] [CrossRef]
- Persons, D.L.; Yazlovitskaya, E.M.; Pelling, J.C. Effect of extracellular signal-regulated kinase on p53 accumulation in response to cisplatin. J. Biol. Chem. 2000, 275, 35778–35785. [Google Scholar] [CrossRef]
- Zhang, Z.; Wu, Y.; Liang, H.; Zhang, B.; Zhang, W. MAPK/ERK activation sensitizes MKN-28 cells to cisplatin-induced apoptosis. Cancer Stud. Mol. Med. Open J. 2015, 2, 52–59. [Google Scholar] [CrossRef]
- Miyake, M.; Goodison, S.; Lawton, A.; Gomes-Giacoia, E.; Rosser, C.J. Angiogenin promotes tumoral growth and angiogenesis by regulating matrix metallopeptidase-2 expression via the ERK1/2 pathway. Oncogene 2015, 34, 890–901. [Google Scholar] [CrossRef]
- Kale, J.; Kutuk, O.; Brito, G.C.; Andrews, T.S.; Leber, B.; Letai, A.; Andrews, D.W. Phosphorylation switches Bax from promoting to inhibiting apoptosis thereby increasing drug resistance. EMBO Rep. 2018, 19, e45235. [Google Scholar] [CrossRef]
- Sartorius, U.A.; Krammer, P.H. Upregulation of bcl-2 is involved in the mediation of chemotherapy resistance in human small cell lung cancer cell lines. Int. J. Cancer 2002, 97, 584–592. [Google Scholar] [CrossRef]
- Wongvaranon, P.; Pongrakhananon, V.; Chunhacha, P.; Chanvorachote, P. Acquired resistance to chemotherapy in lung cancer cells mediated by prolonged nitric oxide exposure. Anticancer Res. 2013, 33, 5433–5444. [Google Scholar]
- Jin, J.; Xiong, Y.; Cen, B. Bcl-2 and Bcl-xL mediate resistance to receptor tyrosine kinase targeted therapy in lung and gastric cancer. Anticancer Drugs 2017, 28, 1141–1149. [Google Scholar] [CrossRef]
- Liu, Y.; Yang, Y.; Ye, Y.-C.; Shi, Q.-F.; Chai, K.; Tashiro, S.; Onodera, S.; Ikejima, T. Activation of ERK–p53 and ERK-mediated phosphorylation of Bcl-2 are involved in autophagic cell death induced by the c-Met inhibitor SU11274 in human lung cancer A549 cells. J. Pharmacol. Sci. 2012, 118, 423–432. [Google Scholar] [CrossRef]
- Sun, P.-L.; Sasano, H.; Gao, H. Bcl-2 family in non-small cell lung cancer: Its prognostic and therapeutic implications. Pathol. Int. 2017, 67, 121–130. [Google Scholar] [CrossRef]
- Lobo, N.A.; Shimono, Y.; Qian, D.; Clarke, M.F. The biology of cancer stem cells. Annu. Rev. Cell Dev. Biol. 2007, 23, 675–699. [Google Scholar] [CrossRef]
- Srinual, S.; Chanvorachote, P.; Pongrakhananon, V. Suppression of cancer stem-like phenotypes in NCI-H460 lung cancer cells by vanillin through an Akt-dependent pathway. Int. J. Oncol. 2017, 50, 1341–1351. [Google Scholar] [CrossRef]
- Dubrovska, A.; Kim, S.; Salamone, R.J.; Walker, J.R.; Maira, S.-M.; García-Echeverría, C.; Schultz, P.G.; Reddy, V.A. The role of PTEN/Akt/PI3K signaling in the maintenance and viability of prostate cancer stem-like cell populations. PNAS 2009, 106, 268–273. [Google Scholar] [CrossRef]
- Shan, J.; Shen, J.; Liu, L.; Xia, F.; Xu, C.; Duan, G.; Xu, Y.; Ma, Q.; Yang, Z.; Zhang, Q.; et al. Nanog regulates self-renewal of cancer stem cells through the insulin-like growth factor pathway in human hepatocellular carcinoma. Hepatology 2012, 56, 1004–1014. [Google Scholar] [CrossRef]
- Mizugaki, H.; Sakakibara-Konishi, J.; Kikuchi, J.; Moriya, J.; Hatanaka, K.C.; Kikuchi, E.; Kinoshita, I.; Oizumi, S.; Dosaka-Akita, H.; Matsuno, Y.; et al. CD133 expression: A potential prognostic marker for non-small cell lung cancers. Int. J. Clin. Oncol. 2014, 19, 254–259. [Google Scholar] [CrossRef]
- Leung, E.L.-H.; Fiscus, R.R.; Tung, J.W.; Tin, V.P.-C.; Cheng, L.C.; Sihoe, A.D.-L.; Fink, L.M.; Ma, Y.; Wong, M.P. Non-small cell lung cancer cells expressing CD44 are enriched for stem cell-like properties. PLoS ONE 2010, 5, e14062. [Google Scholar] [CrossRef]
- Maiuthed, A.; Chantarawong, W.; Chanvorachote, P. Lung cancer stem cells and cancer stem cell-targeting natural compounds. Anticancer Res. 2018, 38, 3797–3809. [Google Scholar] [CrossRef]
- Koury, J.; Zhong, L.; Hao, J. Targeting signaling pathways in cancer stem cells for cancer treatment. Stem Cells Int. 2017, 2017, 2925869. [Google Scholar] [CrossRef]
- Matsui, W.H. Cancer stem cell signaling pathways. Medicine (Baltimore) 2016, 95, S8–S19. [Google Scholar] [CrossRef]
- Phi, L.T.H.; Sari, I.N.; Yang, Y.-G.; Lee, S.-H.; Jun, N.; Kim, K.S.; Lee, Y.K.; Kwon, H.Y. Cancer stem cells (CSCs) in drug resistance and their therapeutic implications in cancer treatment. Stem Cells Int. 2018, 2018, 5416923. [Google Scholar] [CrossRef]
- Castellano, E.; Downward, J. RAS Interaction with PI3K. Genes Cancer 2011, 2, 261–274. [Google Scholar] [CrossRef]
- Hu, B.; Ma, Y.; Yang, Y.; Zhang, L.; Han, H.; Chen, J. CD44 promotes cell proliferation in non-small cell lung cancer. Oncol. Lett. 2018, 15, 5627–5633. [Google Scholar] [CrossRef]
- Barr, M.P.; Gray, S.G.; Hoffmann, A.C.; Hilger, R.A.; Thomale, J.; O’Flaherty, J.D.; Fennell, D.A.; Richard, D.; O’Leary, J.J.; O’Byrne, K.J. Generation and characterisation of cisplatin-resistant non-small cell lung cancer cell lines displaying a stem-like signature. PLoS ONE 2013, 8, e54193. [Google Scholar] [CrossRef]
- Bertolini, G.; Roz, L.; Perego, P.; Tortoreto, M.; Fontanella, E.; Gatti, L.; Pratesi, G.; Fabbri, A.; Andriani, F.; Tinelli, S.; et al. Highly tumorigenic lung cancer CD133+ cells display stem-like features and are spared by cisplatin treatment. PNAS 2009, 106, 16281–16286. [Google Scholar] [CrossRef]
- Lan, X.; Wu, Y.-Z.; Wang, Y.; Wu, F.-R.; Zang, C.-B.; Tang, C.; Cao, S.; Li, S.L. CD133 silencing inhibits stemness properties and enhances chemoradiosensitivity in CD133-positive liver cancer stem cells. Int. J. Mol. Med. 2013, 31, 315–324. [Google Scholar] [CrossRef]
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chantarawong, W.; Chamni, S.; Suwanborirux, K.; Saito, N.; Chanvorachote, P. 5-O-Acetyl-Renieramycin T from Blue Sponge Xestospongia sp. Induces Lung Cancer Stem Cell Apoptosis. Mar. Drugs 2019, 17, 109. https://doi.org/10.3390/md17020109
Chantarawong W, Chamni S, Suwanborirux K, Saito N, Chanvorachote P. 5-O-Acetyl-Renieramycin T from Blue Sponge Xestospongia sp. Induces Lung Cancer Stem Cell Apoptosis. Marine Drugs. 2019; 17(2):109. https://doi.org/10.3390/md17020109
Chicago/Turabian StyleChantarawong, Wipa, Supakarn Chamni, Khanit Suwanborirux, Naoki Saito, and Pithi Chanvorachote. 2019. "5-O-Acetyl-Renieramycin T from Blue Sponge Xestospongia sp. Induces Lung Cancer Stem Cell Apoptosis" Marine Drugs 17, no. 2: 109. https://doi.org/10.3390/md17020109
APA StyleChantarawong, W., Chamni, S., Suwanborirux, K., Saito, N., & Chanvorachote, P. (2019). 5-O-Acetyl-Renieramycin T from Blue Sponge Xestospongia sp. Induces Lung Cancer Stem Cell Apoptosis. Marine Drugs, 17(2), 109. https://doi.org/10.3390/md17020109