
Diapause-like Drug-Tolerant Persister State: The Key to Nirvana Rebirth



Abstract
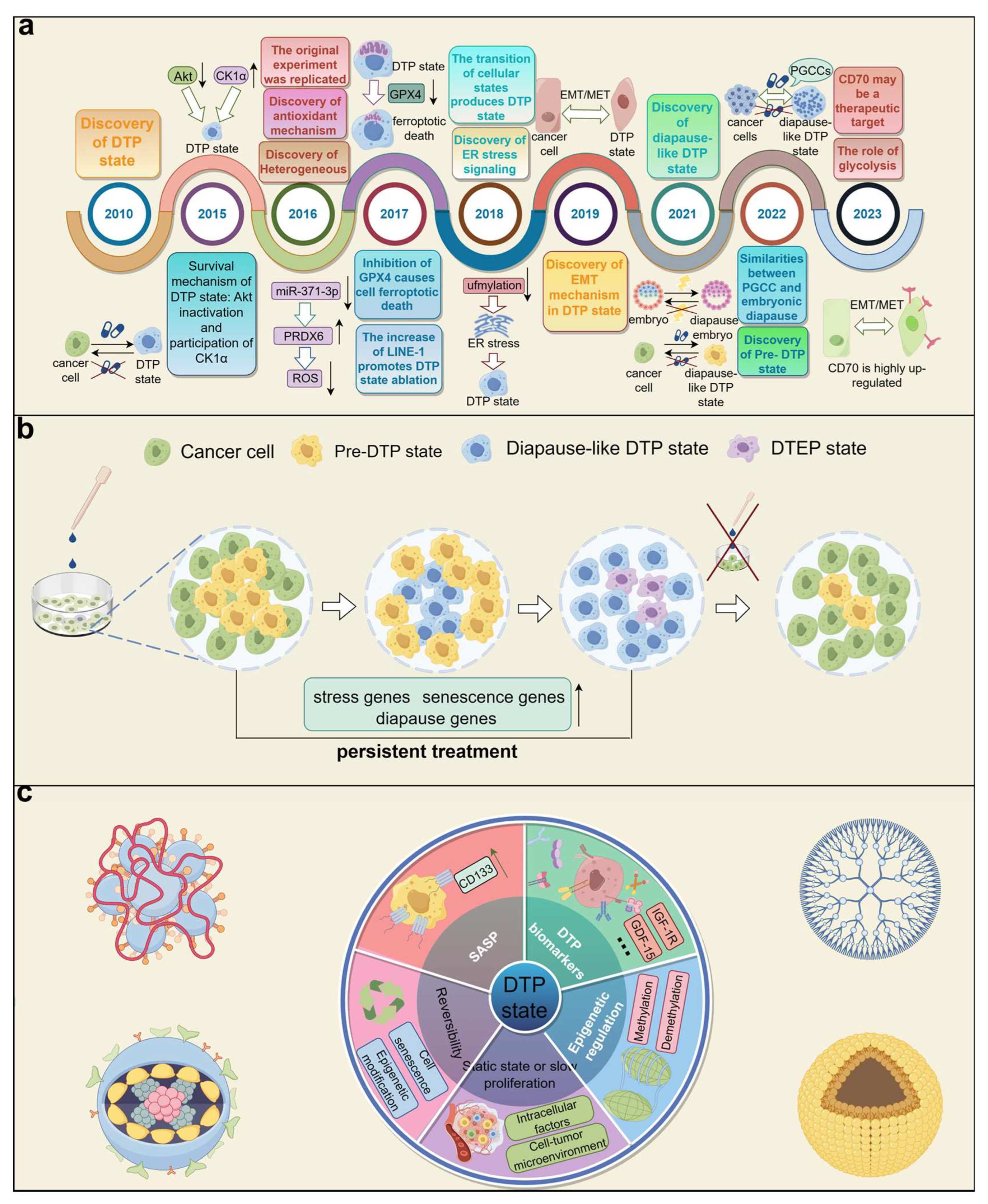
1. Introduction
2. Characteristics of the DTP State
2.1. Static State or Slow Proliferation
2.2. Reversibility
2.3. DTP Biomarkers
2.4. Senescence-Associated Secretory Phenotype (SASP)
2.5. Epigenetic Regulation
3. Similarity between Diapause-like DTP State and Embryonic Diapause
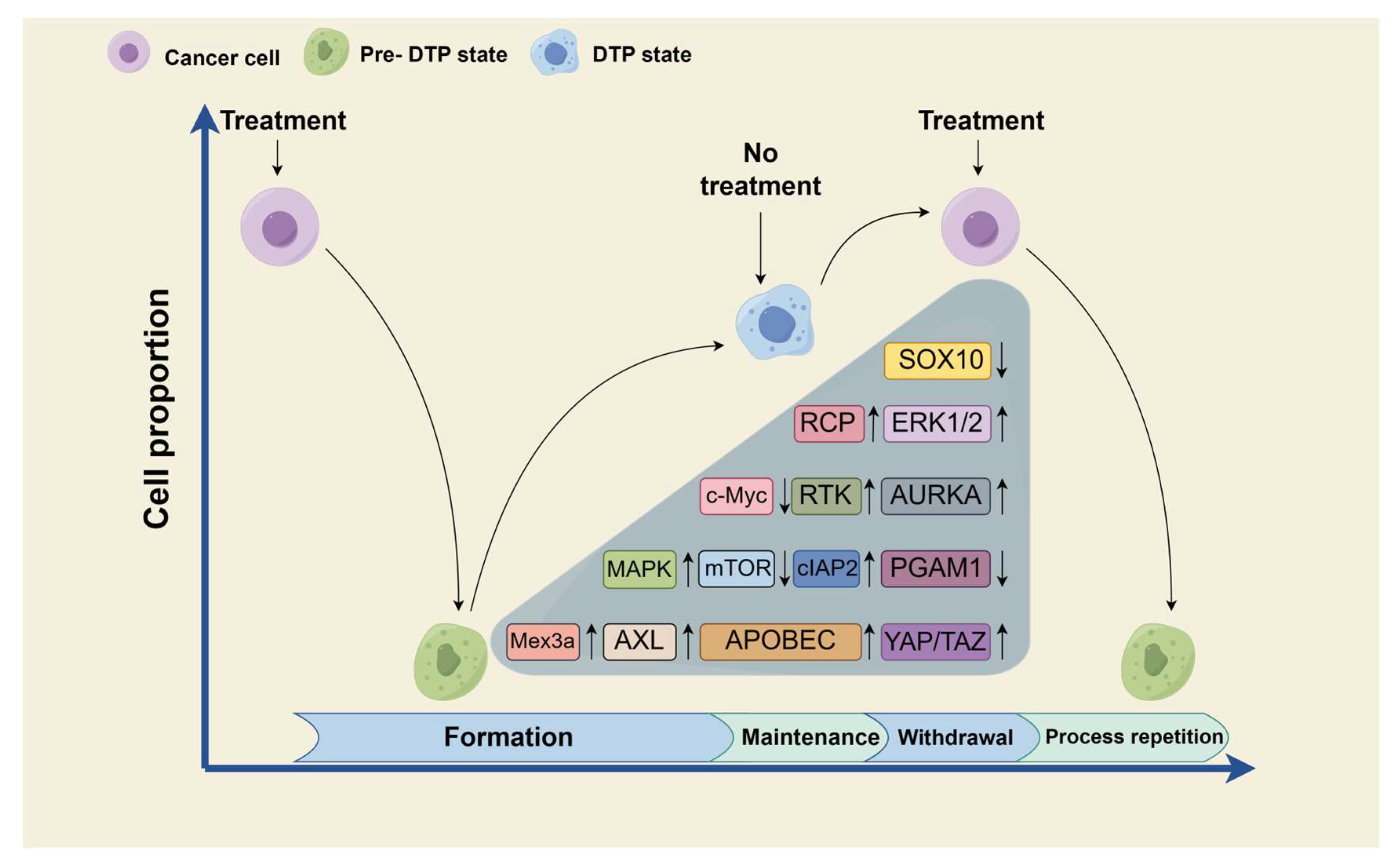
4. Treatment Strategy of Diapause-like DTP State
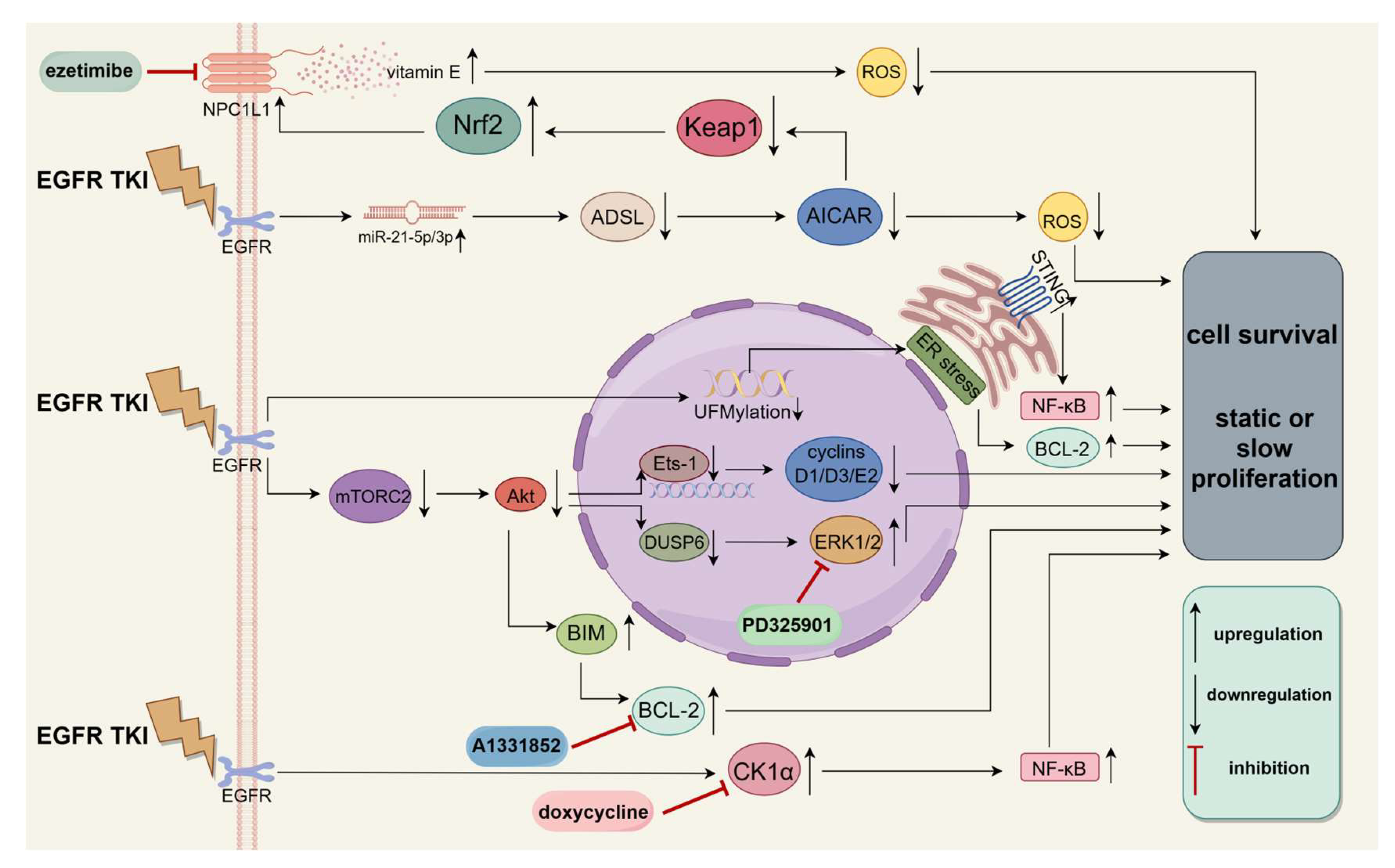
4.1. Prevention of Diapause-like DTP State Formation
4.2. Maintenance of Diapause-like DTP State
4.3. Withdrawal of Diapause-like State
5. Perspectives and Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Chen, S.M.; Cao, Z.; Prettner, K.; Kuhn, M.; Yang, J.T.; Jiao, L.R.; Wang, Z.R.; Li, W.M.; Geldsetzer, P.; Baernighausen, T.; et al. Estimates and Projections of the Global Economic Cost of 29 Cancers in 204 Countries and Territories from 2020 to 2050. JAMA Oncol. 2023, 9, 465–472. [Google Scholar] [CrossRef] [PubMed]
- Siegel, R.L.; Miller, K.D.; Wagle, N.S.; Jemal, A. Cancer statistics, 2023. CA-Cancer J. Clin. 2023, 73, 17–48. [Google Scholar] [CrossRef] [PubMed]
- Balayan, V.; Guddati, A.K. Tumor Dormancy: Biologic and Therapeutic Implications. World J. Oncol. 2022, 13, 8–19. [Google Scholar] [CrossRef] [PubMed]
- Gomatou, G.; Syrigos, N.; Vathiotis, I.A.; Kotteas, E.A. Tumor Dormancy: Implications for Invasion and Metastasis. Int. J. Mol. Sci. 2021, 22, 4862. [Google Scholar] [CrossRef] [PubMed]
- Butturini, E.; de Prati, A.C.; Boriero, D.; Mariotto, S. Tumor Dormancy and Interplay with Hypoxic Tumor Microenvironment. Int. J. Mol. Sci. 2019, 20, 4305. [Google Scholar] [CrossRef] [PubMed]
- Sharma, S.V.; Lee, D.Y.; Li, B.H.; Quinlan, M.P.; Takahashi, F.; Maheswaran, S.; McDermott, U.; Azizian, N.; Zou, L.; Fischbach, M.A.; et al. A Chromatin-Mediated Reversible Drug-Tolerant State in Cancer Cell Subpopulations. Cell 2010, 141, 69–80. [Google Scholar] [CrossRef] [PubMed]
- You, B.; Xia, T.; Gu, M.; Zhang, Z.X.; Zhang, Q.C.; Shen, J.H.; Fan, Y.; Yao, H.; Pan, S.; Lu, Y.N.; et al. AMPK-mTOR-Mediated Activation of Autophagy Promotes Formation of Dormant Polyploid Giant Cancer Cells. Cancer Res. 2022, 82, 846–858. [Google Scholar] [CrossRef]
- Rehman, S.K.; Haynes, J.; Collignon, E.; Brown, K.R.; Wang, Y.D.; Nixon, A.M.L.; Bruce, J.P.; Wintersinger, J.A.; Mer, A.S.; Lo, E.B.L.; et al. Colorectal Cancer Cells Enter a Diapause-like DTP State to Survive Chemotherapy. Cell 2021, 184, 226–242.e21. [Google Scholar] [CrossRef]
- Guo, S.; Tian, Z.; Wu, Q.W.; King-Jones, K.; Liu, W.; Zhu, F.; Wang, X.P. Steroid hormone ecdysone deficiency stimulates preparation for photoperiodic reproductive diapause. PLoS Genet. 2021, 17, 28. [Google Scholar] [CrossRef]
- Hutfilz, C. Endocrine Regulation of Lifespan in Insect Diapause. Front. Physiol. 2022, 13, 18. [Google Scholar] [CrossRef]
- Duy, C.; Li, M.; Teater, M.; Meydan, C.; Garrett-Bakelman, F.E.; Lee, T.C.; Chin, C.R.; Durmaz, C.; Kawabata, K.C.; Dhimolea, E.; et al. Chemotherapy Induces Senescence-Like Resilient Cells Capable of Initiating AML Recurrence. Cancer Discov. 2021, 11, 1542–1561. [Google Scholar] [CrossRef] [PubMed]
- Chen, T.M.; Huang, C.M.; Setiawan, S.A.; Hsieh, M.S.; Sheen, C.C.; Yeh, C.T. KDM5D Histone Demethylase Identifies Platinum-Tolerant Head and Neck Cancer Cells Vulnerable to Mitotic Catastrophe. Int. J. Mol. Sci. 2023, 24, 5310. [Google Scholar] [CrossRef] [PubMed]
- Glasheen, M.Q.; Caksa, S.; Young, A.G.; Wilski, N.A.; Ott, C.A.; Chervoneva, I.; Flaherty, K.T.; Herlyn, M.; Xu, X.; Aplin, A.E.; et al. Targeting Upregulated cIAP2 in SOX10-Deficient Drug Tolerant Melanoma. Mol. Cancer Ther. 2023, 22, 1087–1099. [Google Scholar] [CrossRef] [PubMed]
- Huang, F.; Cai, F.Y.; Dahabieh, M.S.; Gunawardena, K.; Talebi, A.; Dehairs, J.; El-Turk, F.; Park, J.Y.; Li, M.Q.; Goncalves, C.; et al. Peroxisome disruption alters lipid metabolism and potentiates antitumor response with MAPK-targeted therapy in melanoma. J. Clin. Investig. 2023, 133, 20. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.; Carvajal, R.; Kim, M.; Yang, H.W. Kinetics of RTK activation determine ERK reactivation and resistance to dual BRAF/MEK inhibition in melanoma. Cell Rep. 2023, 42, 20. [Google Scholar] [CrossRef] [PubMed]
- Martin, M.J.; Floc’h, N.; Pfeifer, M.; Criscione, S.; Delpuech, O.; Gagrica, S.; Yao, Y.; McDermott, U.; Smith, P.D. Pharmaceutical Reactivation of Attenuated Apoptotic Pathways Leads to Elimination of Osimertinib Drug-Tolerant Cells. Cancer Res. Commun. 2022, 2, 1312–1325. [Google Scholar] [CrossRef] [PubMed]
- Szebényi, K.; Füredi, A.; Bajtai, E.; Sama, S.N.; Csiszar, A.; Gombos, B.; Szabó, P.; Grusch, M.; Szakács, G. Effective targeting of breast cancer by the inhibition of P-glycoprotein mediated removal of toxic lipid peroxidation byproducts from drug tolerant persister cells. Drug Resist. Updates 2023, 71, 12. [Google Scholar] [CrossRef]
- Wang, H.; Liang, Y.K.; Zhang, T.; Yu, X.N.; Song, X.M.; Chen, Y.Z.; Mao, Q.X.; Xia, W.J.; Chen, B.; Xu, L.; et al. C-IGF1R encoded by cIGF1R acts as a molecular switch to restrict mitophagy of drug-tolerant persister tumour cells in non-small cell lung cancer. Cell Death Differ. 2023, 30, 2365–2381. [Google Scholar] [CrossRef]
- Zhou, X.; An, J.Y.; Kurilov, R.; Brors, B.; Hu, K.; Peccerella, T.; Roessler, S.; Pfuetze, K.; Schulz, A.; Wolf, S.; et al. Persister cell phenotypes contribute to poor patient outcomes after neoadjuvant chemotherapy in PDAC. Nat. Cancer 2023, 4, 1362–1381. [Google Scholar] [CrossRef]
- Jayappa, K.D.; Tran, B.; Gordon, V.L.; Morris, C.; Saha, S.; Farrington, C.C.; O’Connor, C.M.; Zawacki, K.P.; Isaac, K.M.; Kester, M.; et al. PP2A modulation overcomes multidrug resistance in chronic lymphocytic leukemia via mPTP-dependent apoptosis. J. Clin. Investig. 2023, 133, e155938. [Google Scholar] [CrossRef]
- Lin, Q.Y.; Chen, J.Q.; Gu, L.F.; Dan, X.G.; Zhang, C.; Yang, Y.Z. New insights into mitophagy and stem cells. Stem Cell Res. Ther. 2021, 12, 14. [Google Scholar] [CrossRef]
- Gan, Z.Y.; Callegari, S.; Cobbold, S.A.; Cotton, T.R.; Mlodzianoski, M.J.; Schubert, A.F.; Geoghegan, N.D.; Rogers, K.L.; Leis, A.; Dewson, G.; et al. Activation mechanism of PINK1. Nature 2022, 602, 328–335. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Chen, H.X.; Xie, X.; Yang, B.; Wang, X.J.; Zhang, J.Y.; Qiao, T.; Guan, J.; Qiu, Y.T.; Huang, Y.X.; et al. PINK1-Mediated Mitophagy Promotes Oxidative Phosphorylation and Redox Homeostasis to Induce. Cancer Res. 2023, 83, 398–413. [Google Scholar] [CrossRef] [PubMed]
- Shen, S.S.; Vagner, S.; Robert, C. Persistent Cancer Cells: The Deadly Survivors. Cell 2020, 183, 860–874. [Google Scholar] [CrossRef] [PubMed]
- Kim, D.; An, L.Y.; Moon, J.; Maymi, V.I.; McGurk, A.I.; Rudd, B.D.; Fowell, D.J.; White, A.C. Ccr2± Monocyte-Derived Macrophages Influence Trajectories of Acquired Therapy Resistance in Braf-Mutant Melanoma. Cancer Res. 2023, 83, 2328–2344. [Google Scholar] [CrossRef] [PubMed]
- Fane, M.E.; Chhabra, Y.; Alicea, G.M.; Maranto, D.A.; Douglass, S.M.; Webster, M.R.; Rebecca, V.W.; Marino, G.E.; Almeida, F.; Ecker, B.L.; et al. Stromal changes in the aged lung induce an emergence from melanoma dormancy. Nature 2022, 606, 396–405. [Google Scholar] [CrossRef] [PubMed]
- Oren, Y.; Tsabar, M.; Cuoco, M.S.; Amir-Zilberstein, L.; Cabanos, H.F.; Hutter, J.C.; Hu, B.M.; Thakore, P.I.; Tabaka, M.; Fulco, C.P.; et al. Cycling cancer persister cells arise from lineages with distinct programs. Nature 2021, 596, 576–582. [Google Scholar] [CrossRef] [PubMed]
- Moghal, N.; Li, Q.; Stewart, E.L.; Navab, R.; Mikubo, M.; D’Arcangelo, E.; Martins, S.N.; Raghavan, V.; Pham, N.A.; Li, M.; et al. Single-Cell Analysis Reveals Transcriptomic Features of Drug-Tolerant Persisters and Stromal Adaptation in a Patient-Derived EGFR-Mutated Lung Adenocarcinoma Xenograft Model. J. Thorac. Oncol. 2023, 18, 499–515. [Google Scholar] [CrossRef] [PubMed]
- Sun, X.; He, L.; Liu, H.; Thorne, R.F.; Zeng, T.; Liu, L.; Zhang, B.; He, M.; Huang, Y.; Li, M.; et al. The diapause-like colorectal cancer cells induced by SMC4 attenuation are characterized by low proliferation and chemotherapy insensitivity. Cell Metab. 2023, 35, 1563–1579.e8. [Google Scholar] [CrossRef]
- Mikubo, M.; Inoue, Y.; Liu, G.; Tsao, M.S. Mechanism of Drug Tolerant Persister Cancer Cells: The Landscape and Clinical Implication for Therapy. J. Thorac. Oncol. 2021, 16, 1798–1809. [Google Scholar] [CrossRef]
- Delahaye, C.; Figarol, S.; Pradines, A.; Favre, G.; Mazieres, J.; Calvayrac, O. Early Steps of Resistance to Targeted Therapies in Non-Small-Cell Lung Cancer. Cancers 2022, 14, 2613. [Google Scholar] [CrossRef]
- Demaria, M.; O’Leary, M.N.; Chang, J.H.; Shao, L.J.; Liu, S.; Alimirah, F.; Koenig, K.; Le, C.; Mitin, N.; Deal, A.M.; et al. Cellular Senescence Promotes Adverse Effects of Chemotherapy and Cancer Relapse. Cancer Discov. 2017, 7, 165–176. [Google Scholar] [CrossRef]
- Saleh, T.; Tyutyunyk-Massey, L.; Gewirtz, D.A. Tumor Cell Escape from Therapy-Induced Senescence as a Model of Disease Recurrence after Dormancy. Cancer Res. 2019, 79, 1044–1046. [Google Scholar] [CrossRef]
- Rochette, L.; Zeller, M.; Cottin, Y.; Vergely, C. Insights Into Mechanisms of GDF15 and Receptor GFRAL: Therapeutic Targets. Trends Endocrinol. Metab. 2020, 31, 939–951. [Google Scholar] [CrossRef] [PubMed]
- Bellio, C.; Emperador, M.; Castellano, P.; Gris-Oliver, A.; Canals, F.; Sanchez-Pla, A.; Zamora, E.; Arribas, J.; Saura, C.; Serra, V.; et al. GDF15 Is an Eribulin Response Biomarker also Required for Survival of DTP Breast Cancer Cells. Cancers 2022, 14, 2562. [Google Scholar] [CrossRef] [PubMed]
- Chang, C.A.; Jen, J.; Jiang, S.W.; Sayad, A.; Mer, A.S.; Brown, K.R.; Nixon, A.M.L.; Dhabaria, A.; Tang, K.H.; Venet, D.; et al. Ontogeny and Vulnerabilities of Drug-Tolerant Persisters in HER2(+) Breast Cancer. Cancer Discov. 2022, 12, 1022–1045. [Google Scholar] [CrossRef] [PubMed]
- Noronha, A.; Nataraj, N.B.; Lee, J.S.; Zhitomirsky, B.; Oren, Y.; Oster, S.; Lindzen, M.; Mukherjee, S.; Will, R.; Ghosh, S.; et al. AXL and Error-Prone DNA Replication Confer Drug Resistance and Offer Strategies to Treat EGFR-Mutant Lung Cancer. Cancer Discov. 2022, 12, 2666–2683. [Google Scholar] [CrossRef] [PubMed]
- Cox, M.A.; Bassi, C.; Saunders, M.E.; Nechanitzky, R.; Morgado-Palacin, I.; Zheng, C.; Mak, T.W. Beyond neurotransmission: Acetylcholine in immunity and inflammation. J. Intern. Med. 2020, 287, 120–133. [Google Scholar] [CrossRef] [PubMed]
- Nie, M.; Chen, N.; Pang, H.H.; Jiang, T.; Jiang, W.; Tian, P.W.; Yao, L.; Chen, Y.Z.; DeBerardinis, R.J.; Li, W.M.; et al. Targeting acetylcholine signaling modulates persistent drug tolerance in EGFR-mutant lung cancer and impedes tumor relapse. J. Clin. Investig. 2022, 132, 17. [Google Scholar] [CrossRef]
- du Manoir, S.; Delpech, H.; Orsetti, B.; Jacot, W.; Pirot, N.; Noel, J.; Colombo, P.E.; Sardet, C.; Theillet, C. In high-grade ovarian carcinoma, platinum-sensitive tumor recurrence and acquired-resistance derive from quiescent residual cancer cells that overexpress CRYAB, CEACAM6, and SOX2. J. Pathol. 2022, 257, 367–378. [Google Scholar] [CrossRef]
- Dhanyamraju, P.K.; Schell, T.D.; Amin, S.; Robertson, G.P. Drug-Tolerant Persister Cells in Cancer Therapy Resistance. Cancer Res. 2022, 82, 2503–2514. [Google Scholar] [CrossRef]
- Cuesta-Borràs, E.; Salvans, C.; Arqués, O.; Chicote, I.; Ramírez, L.; Cabellos, L.; Martínez-Quintanilla, J.; Mur-Espinosa, A.; García-Alvarez, A.; Hernando, J.; et al. DPPA3-HIF1a axis controls colorectal cancer chemoresistance by imposing a slow cell-cycle phenotype. Cell Rep. 2023, 42, 33. [Google Scholar] [CrossRef] [PubMed]
- Wiley, C.D.; Campisi, J. The metabolic roots of senescence: Mechanisms and opportunities for intervention. Nat. Metab. 2021, 3, 1290–1301. [Google Scholar] [CrossRef] [PubMed]
- Saleh, T.; Gewirtz, D.A. Considering therapy-induced senescence as a mechanism of tumour dormancy contributing to disease recurrence. Br. J. Cancer 2022, 126, 1363–1365. [Google Scholar] [CrossRef] [PubMed]
- Hernandez-Segura, A.; de Jong, T.V.; Melov, S.; Guryev, V.; Campisi, J.; Demaria, M. Unmasking Transcriptional Heterogeneity in Senescent Cells. Curr. Biol. 2017, 27, 2652–2660. [Google Scholar] [CrossRef] [PubMed]
- Kunimasa, K.; Nagano, T.; Shimono, Y.; Dokuni, R.; Kiriu, T.; Tokunaga, S.; Tamura, D.; Yamamoto, M.; Tachihara, M.; Kobayashi, K.; et al. Glucose metabolism-targeted therapy and withaferin A are effective for epidermal growth factor receptor tyrosine kinase inhibitor-induced drug-tolerant persisters. Cancer Sci. 2017, 108, 1368–1377. [Google Scholar] [CrossRef]
- Mingay, M.; Chaturvedi, A.; Bilenky, M.; Cao, Q.; Jackson, L.; Hui, T.; Moksa, M.; Heravi-Moussavi, A.; Humphries, R.K.; Heuser, M.; et al. Vitamin C-induced epigenomic remodelling in IDH1 mutant acute myeloid leukaemia. Leukemia 2018, 32, 11–20. [Google Scholar] [CrossRef]
- Sun, H.Y.; Huang, B.H.; Cao, J.; Yan, Q.; Yin, M.Z. Editorial: Epigenetic Regulation and Tumor Immunotherapy. Front. Oncol. 2022, 12, 4. [Google Scholar] [CrossRef]
- Hatch, S.B.; Yapp, C.; Montenegro, R.C.; Savitsky, P.; Gamble, V.; Tumber, A.; Ruda, G.F.; Bavetsias, V.; Fedorov, O.; Atrash, B.; et al. Assessing histone demethylase inhibitors in cells: Lessons learned. Epigenetics Chromatin 2017, 10, 17. [Google Scholar] [CrossRef]
- Vinogradova, M.; Gehling, V.S.; Gustafson, A.; Arora, S.; Tindell, C.A.; Wilson, C.; Williamson, K.E.; Guler, G.D.; Gangurde, P.; Manieri, W.; et al. An inhibitor of KDM5 demethylases reduces survival of drug-tolerant cancer cells. Nat. Chem. Biol. 2016, 12, 531–538. [Google Scholar] [CrossRef]
- Staberg, M.; Rasmussen, R.D.; Michaelsen, S.R.; Pedersen, H.; Jensen, K.E.; Villingshoj, M.; Skjoth-Rasmussen, J.; Brennum, J.; Vitting-Seerup, K.; Poulsen, H.S.; et al. Targeting glioma stem-like cell survival and chemoresistance through inhibition of lysine-specific histone demethylase KDM2B. Mol. Oncol. 2018, 12, 406–420. [Google Scholar] [CrossRef] [PubMed]
- Gale, M.; Sayegh, J.; Cao, J.; Norcia, M.; Gareiss, P.; Hoyer, D.; Merkel, J.S.; Yan, Q. Screen-identified selective inhibitor of lysine demethylase 5A blocks cancer cell growth and drug resistance. Oncotarget 2016, 7, 39931–39944. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.N.; Zhang, S.M.; McGeary, M.K.; Krykbaeva, I.; Lai, L.; Jansen, D.J.; Kales, S.C.; Simeonov, A.; Hall, M.D.; Kelly, D.P.; et al. KDM5B Promotes Drug Resistance by Regulating Melanoma-Propagating Cell Subpopulations. Mol. Cancer Ther. 2019, 18, 706–717. [Google Scholar] [CrossRef] [PubMed]
- Montavon, T.; Shukeir, N.; Erikson, G.; Engist, B.; Onishi-Seebacher, M.; Ryan, D.; Musa, Y.; Mittler, G.; Meyer, A.G.; Genoud, C.; et al. Complete loss of H3K9 methylation dissolves mouse heterochromatin organization. Nat. Commun. 2021, 12, 16. [Google Scholar] [CrossRef] [PubMed]
- Wiles, E.T.; McNaught, K.J.; Kaur, G.; Selker, J.M.L.; Ormsby, T.; Aravind, L.; Selker, E.U. Evolutionarily ancient BAH-PHD protein mediates Polycomb silencing. Proc. Natl. Acad. Sci. USA 2020, 117, 11614–11623. [Google Scholar] [CrossRef] [PubMed]
- Guler, G.D.; Tindell, C.A.; Pitti, R.; Wilson, C.; Nichols, K.; Cheung, T.K.; Kim, H.J.; Wongchenko, M.; Yan, Y.B.; Haley, B.; et al. Repression of Stress-Induced LINE-1 Expression Protects Cancer Cell Subpopulations from Lethal Drug Exposure. Cancer Cell 2017, 32, 221–237.e13. [Google Scholar] [CrossRef] [PubMed]
- Menon, D.R.; Hammerlindl, H.; Gimenez, G.; Hammerlindl, S.; Zuegner, E.; Torrano, J.; Bordag, N.; Al Emran, A.; Giam, M.; Denil, S.; et al. H3K4me3 remodeling induced acquired resistance through O-GlcNAc transferase. Drug Resist. Updates 2023, 71, 100993. [Google Scholar] [CrossRef] [PubMed]
- Bulut-Karslioglu, A.; Biechele, S.; Jin, H.; Macrae, T.A.; Hejna, M.; Gertsenstein, M.; Song, J.S.; Ramalho-Santos, M. Inhibition of mTOR induces a paused pluripotent state. Nature 2016, 540, 119–123. [Google Scholar] [CrossRef]
- Scognamiglio, R.; Cabezas-Wallscheid, N.; Thier, M.C.; Altamura, S.; Reyes, A.; Prendergast, A.M.; Baumgartner, D.; Carnevalli, L.S.; Atzberger, A.; Haas, S.; et al. Myc Depletion Induces a Pluripotent Dormant State Mimicking Diapause. Cell 2016, 164, 668–680. [Google Scholar] [CrossRef]
- Lin, X.W.; Xu, W.H. Hexokinase is a key regulator of energy metabolism and ROS activity in insect lifespan extension. Aging 2016, 8, 245–259. [Google Scholar] [CrossRef]
- Dhimolea, E.; Simoes, R.D.; Kansara, D.; Al’Khafaji, A.; Bouyssou, J.; Weng, X.; Sharma, S.; Raja, J.; Awate, P.; Shirasaki, R.; et al. An Embryonic Diapause-like Adaptation with Suppressed Myc Activity Enables Tumor Treatment Persistence. Cancer Cell 2021, 39, 240–256.e11. [Google Scholar] [CrossRef] [PubMed]
- Hussein, A.M.; Wang, Y.L.; Mathieu, J.; Margaretha, L.; Song, C.Z.; Jones, D.C.; Cavanaugh, C.; Miklas, J.W.; Mahen, E.; Showalter, M.R.; et al. Metabolic Control over mTOR-Dependent Diapause-like State. Dev. Cell 2020, 52, 236–250.e7. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.H.; Azizian, N.G.; Sullivan, D.K.; Li, Y.L. mTOR inhibition attenuates chemosensitivity through the induction of chemotherapy resistant persisters. Nat. Commun. 2022, 13, 13. [Google Scholar] [CrossRef] [PubMed]
- Hussein, A.M. Metabolic and Epigenetic Regulation in Development and in Embryonic Diapause. Ph.D. Thesis, University of Washington, Seattle, WA, USA, 2021. [Google Scholar]
- Vaupel, P.; Multhoff, G. Revisiting the Warburg effect: Historical dogma versus current understanding. J. Physiol. 2021, 599, 1745–1757. [Google Scholar] [CrossRef]
- Marcucci, F.; Rumio, C. Glycolysis-induced drug resistance in tumors-A response to danger signals? Neoplasia 2021, 23, 234–245. [Google Scholar] [CrossRef] [PubMed]
- Peng, J.H.; Cui, Y.Y.; Xu, S.P.; Wu, X.W.; Huang, Y.; Zhou, W.B.; Wang, S.; Fu, Z.Y.; Xie, H. Altered glycolysis results in drug-resistant in clinical tumor therapy. Oncol. Lett. 2021, 21, 14. [Google Scholar] [CrossRef] [PubMed]
- Du, D.Y.; Liu, C.; Qin, M.Y.; Zhang, X.; Xi, T.; Yuan, S.T.; Hao, H.P.; Xiong, J. Metabolic dysregulation and emerging therapeutical targets for hepatocellular carcinoma. Acta Pharm. Sin. B 2022, 12, 558–580. [Google Scholar] [CrossRef] [PubMed]
- Cao, Y.H. Adipocyte and lipid metabolism in cancer drug resistance. J. Clin. Investig. 2019, 129, 3006–3017. [Google Scholar] [CrossRef]
- Ichimiya, T.; Yamakawa, T.; Hirano, T.; Yokoyama, Y.; Hayashi, Y.; Hirayama, D.; Wagatsuma, K.; Itoi, T.; Nakase, H. Autophagy and Autophagy-Related Diseases: A Review. Int. J. Mol. Sci. 2020, 21, 8974. [Google Scholar] [CrossRef]
- Lee, J.E.; Oh, H.A.; Song, H.; Jun, J.H.; Roh, C.R.; Xie, H.; Dey, S.K.; Lim, H.J. Autophagy Regulates Embryonic Survival during Delayed Implantation. Endocrinology 2011, 152, 2067–2075. [Google Scholar] [CrossRef]
- Li, X.H.; He, S.K.; Ma, B.Y. Autophagy and autophagy-related proteins in cancer. Mol. Cancer 2020, 19, 16. [Google Scholar] [CrossRef] [PubMed]
- Debnath, J.; Gammoh, N.; Ryan, K.M. Autophagy and autophagy-related pathways in cancer. Nat. Rev. Mol. Cell Biol. 2023, 24, 560–575. [Google Scholar] [CrossRef]
- Kalluri, R.; Weinberg, R.A. The basics of epithelial-mesenchymal transition. J. Clin. Investig. 2009, 119, 1420–1428. [Google Scholar] [CrossRef] [PubMed]
- Ribatti, D.; Tamma, R.; Annese, T. Epithelial-Mesenchymal Transition in Cancer: A Historical Overview. Transl. Oncol. 2020, 13, 9. [Google Scholar] [CrossRef] [PubMed]
- Alhasan, B.A.; Gordeev, S.A.; Knyazeva, A.R.; Aleksandrova, K.V.; Margulis, B.A.; Guzhova, I.V.; Suvorova, I.I. The mTOR Pathway in Pluripotent Stem Cells: Lessons for Understanding Cancer Cell Dormancy. Membranes 2021, 11, 858. [Google Scholar] [CrossRef] [PubMed]
- Zou, L.; Liao, M.R.; Zhen, Y.Q.; Zhu, S.; Chen, X.Y.; Zhang, J.; Hao, Y.; Liu, B. Autophagy and beyond: Unraveling the complexity of UNC-51-like kinase 1 (ULK1) from biological functions to therapeutic implications. Acta Pharm. Sin. B 2022, 12, 3743–3782. [Google Scholar] [CrossRef]
- Fenton, S.E.; Zannikou, M.; Ilut, L.; Fischietti, M.; Ji, C.N.; Oku, C.V.; Horvath, C.M.; Le Poople, I.C.; Bosenberg, M.; Bartom, E.T.; et al. Targeting ULK1 Decreases IFN gamma-Mediated Resistance to Immune Checkpoint Inhibitors. Mol. Cancer Res. 2023, 21, 332–344. [Google Scholar] [CrossRef] [PubMed]
- Bhattacharya, S.; Piya, S.; Ma, H.X.; Sharma, P.; Zhang, Q.; Baran, N.; Ruvolo, V.R.; McQueen, T.; Davis, R.E.; Pourebrahim, R.; et al. Targeting Unc51-like Autophagy Activating Kinase 1 (ULK1) Overcomes Adaptive Drug Resistance in Acute Myelogenous Leukemia. Mol. Cancer Res. 2023, 21, 548–563. [Google Scholar] [CrossRef]
- Risom, T.; Langer, E.M.; Chapman, M.P.; Rantala, J.; Fields, A.J.; Boniface, C.; Alvarez, M.J.; Kendsersky, N.D.; Pelz, C.R.; Johnson-Camacho, K.; et al. Differentiation-state plasticity is a targetable resistance mechanism in basal-like breast cancer. Nat. Commun. 2018, 9, 17. [Google Scholar] [CrossRef]
- Shi, K.X.; Lu, H.J.; Zhang, Z.F.; Fu, Y.J.; Wu, J.; Zhou, S.C.; Ma, P.F.; Ye, K.Y.; Zhang, S.Z.; Shi, H.L.; et al. Transient targeting of BIM-dependent adaptive MCL1 preservation enhances tumor response to molecular therapeutics in non-small cell lung cancer. Cell Death Differ. 2023, 30, 195–207. [Google Scholar] [CrossRef]
- Kashima, Y.; Shibahara, D.; Suzuki, A.; Muto, K.; Kobayashi, I.S.; Plotnick, D.; Udagawa, H.; Izumi, H.; Shibata, Y.; Tanaka, K.; et al. Single-Cell Analyses Reveal Diverse Mechanisms of Resistance to EGFR Tyrosine Kinase Inhibitors in Lung Cancer. Cancer Res. 2021, 81, 4835–4848. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, H.; Sakakibara-Konishi, J.; Furuta, M.; Shoji, T.; Tsuji, K.; Morinaga, D.; Kikuchi, E.; Kikuchi, J.; Noguchi, T.; Hatanaka, K.C.; et al. Notch pathway regulates osimertinib drug-tolerant persistence in EGFR-mutated non-small-cell lung cancer. Cancer Sci. 2023, 114, 1635–1650. [Google Scholar] [CrossRef] [PubMed]
- Fan, Q.W.; Aksoy, O.; Wong, R.A.; Ilkhanizadeh, S.; Novotny, C.J.; Gustafson, W.C.; Truong, A.Y.Q.; Cayanan, G.; Simonds, E.F.; Haas-Kogan, D.; et al. A Kinase Inhibitor Targeted to mTORC1 Drives Regression in Glioblastoma. Cancer Cell 2017, 31, 424–435. [Google Scholar] [CrossRef] [PubMed]
- Rodrik-Outmezguine, V.S.; Okaniwa, M.; Yao, Z.; Novotny, C.J.; McWhirter, C.; Banaji, A.; Won, H.; Wong, W.; Berger, M.; de Stanchina, E.; et al. Overcoming mTOR resistance mutations with a new-generation mTOR inhibitor. Nature 2016, 534, 272–276. [Google Scholar] [CrossRef] [PubMed]
- Egan, D.F.; Chun, M.G.H.; Vamos, M.; Zou, H.X.; Rong, J.; Miller, C.J.; Lou, H.J.; Raveendra-Panickar, D.; Yang, C.C.; Sheffler, D.J.; et al. Small Molecule Inhibition of the Autophagy Kinase ULK1 and Identification of ULK1 Substrates. Mol. Cell 2015, 59, 285–297. [Google Scholar] [CrossRef]
- Appiah, C.O.; Singh, M.; May, L.; Bakshi, I.; Vaidyanathan, A.; Dent, P.; Ginder, G.; Grant, S.; Bear, H.; Landry, J. The epigenetic regulation of cancer cell recovery from therapy exposure and its implications as a novel therapeutic strategy for preventing disease recurrence. Adv. Cancer Res. 2023, 158, 337–385. [Google Scholar] [CrossRef] [PubMed]
- Cambron-Kopco, L.D.; Yocum, G.D.; Yeater, K.M.; Greenlee, K.J. Timing of Diapause Initiation and Overwintering Conditions Alter Gene Expression Profiles in Megachile rotundata. Front. Physiol. 2022, 13, 10. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.S.E.; Guanizo, A.C.; Jakasekara, W.S.N.; Inampudi, C.; Luong, Q.; Garama, D.J.; Alamgeer, M.; Thakur, N.; DeVeer, M.; Ganju, V.; et al. MYC drives platinum resistant SCLC that is overcome by the dual PI3K-HDAC inhibitor fimepinostat. J. Exp. Clin. Cancer Res. 2023, 42, 14. [Google Scholar] [CrossRef]
- Bai, P.Y.; Fan, T.J.; Wang, X.; Zhao, L.J.; Zhong, R.G.; Sun, G.H. Modulating MGMT expression through interfering with cell signaling pathways. Biochem. Pharmacol. 2023, 215, 18. [Google Scholar] [CrossRef]
- Alvarez-Varela, A.; Novellasdemunt, L.; Barriga, F.M.; Hernando-Momblona, X.; Cañellas-Socias, A.; Cano-Crespo, S.; Sevillano, M.; Cortina, C.; Stork, D.; Morral, C.; et al. Mex3a marks drug-tolerant persister colorectal cancer cells that mediate relapse after chemotherapy. Nat. Cancer 2022, 3, 1052–1070. [Google Scholar] [CrossRef]
- Liu, J.S. Giant cells: Linking McClintock’s heredity to early embryogenesis and tumor origin throughout millennia of evolution on Earth. Semin. Cancer Biol. 2022, 81, 176–192. [Google Scholar] [CrossRef]
- Kostal, V. Eco-physiological phases of insect diapause. J. Insect Physiol. 2006, 52, 113–127. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.S.; Niu, N.; Li, X.R.; Zhang, X.D.; Sood, A.K. The life cycle of polyploid giant cancer cells and dormancy in cancer: Opportunities for novel therapeutic interventions. Semin. Cancer Biol. 2022, 81, 132–144. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.S. The “life code”: A theory that unifies the human life cycle and the origin of human tumors. Semin. Cancer Biol. 2020, 60, 380–397. [Google Scholar] [CrossRef] [PubMed]
- Revesz, L.; Norman, U. Chromosome ploidy and radiosensitivity of tumours. Nature 1960, 187, 861–862. [Google Scholar] [CrossRef] [PubMed]
- Moein, S.; Adibi, R.; Meirelles, L.D.; Nardi, N.B.; Gheisari, Y. Cancer regeneration: Polyploid cells are the key drivers of tumor progression. Biochim. Biophys. Acta-Rev. Cancer 2020, 1874, 11. [Google Scholar] [CrossRef] [PubMed]
- Amend, S.R.; Torga, G.; Lin, K.C.; Kostecka, L.G.; de Marzo, A.; Austin, R.H.; Pienta, K.J. Polyploid giant cancer cells: Unrecognized actuators of tumorigenesis, metastasis, and resistance. Prostate 2019, 79, 1489–1497. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.P.; Jiang, S.T.; He, F.H.; Tian, Y.Y.; Hu, H.Y.; Gao, L.; Zhang, L.; Chen, A.L.; Hu, Y.X.; Fan, L.Y.; et al. Single-cell transcriptomics reveals multiple chemoresistant properties in leukemic stem and progenitor cells in pediatric AML. Genome Biol. 2023, 24, 41. [Google Scholar] [CrossRef]
- Goyal, Y.; Busch, G.T.; Pillai, M.; Li, J.X.; Boe, R.H.; Grody, E.I.; Chelvanambi, M.; Dardani, I.P.; Emert, B.; Bodkin, N.; et al. Diverse clonal fates emerge upon drug treatment of homogeneous cancer cells. Nature 2023, 620, 651–659. [Google Scholar] [CrossRef]
- Cotton, J.L.; Estrada Diez, J.; Sagar, V.; Chen, J.; Piquet, M.; Alford, J.; Song, Y.; Li, X.; Riester, M.; DiMare, M.T.; et al. Expressed Barcoding Enables High-Resolution Tracking of the Evolution of Drug Tolerance. Cancer Res. 2023, 83, 3611–3623. [Google Scholar] [CrossRef]
- Wang, K.L.; Kumar, T.; Wang, J.K.; Minussi, D.C.; Sei, E.; Li, J.Z.; Tran, T.M.; Thennavan, A.; Hu, M.; Casasent, A.K.; et al. Archival single-cell genomics reveals persistent subclones during DCIS progression. Cell 2023, 186, 3968–3982.e15. [Google Scholar] [CrossRef] [PubMed]
- Bian, B.J.M.; Paquet, A.; Arguel, M.J.; Meyer, M.; Peyre, L.; Chalabi, A.; Péré, M.; Lebrigand, K.; Waldmann, R.; Barbry, P.; et al. Coupling live-cell imaging and in situ isolation of the same single cell to profile the transient states of predicted drug-tolerant cells. STAR Protoc. 2022, 3, 32. [Google Scholar] [CrossRef] [PubMed]
- Williams, A.F.; Gervasio, D.; Turkal, C.; Stuhlfire, A.; Wang, M.; Mauch, B.; West, A.; Paw, M.; Hairani, M.; Lathrop, C.; et al. Apoptotic DNase DFFB mediates cancer persister cell mutagenesis and acquired drug resistance. Cancer Res. 2023, 83. [Google Scholar] [CrossRef]
- Antoszczak, M.; Müller, S.; Cañeque, T.; Colombeau, L.; Dusetti, N.; Santofimia-Castaño, P.; Gaillet, C.; Puisieux, A.; Iovanna, J.L.; Rodriguez, R. Iron-Sensitive Prodrugs That Trigger Active Ferroptosis in Drug-Tolerant Pancreatic Cancer Cells. J. Am. Chem. Soc. 2022, 144, 11536–11545. [Google Scholar] [CrossRef]
- Gerisch, B.; Tharyan, R.G.; Mak, J.; Denzel, S.I.; Popkes-van Oepen, T.; Henn, N.; Antebi, A. HLH-30/TFEB Is a Master Regulator of Reproductive Quiescence. Dev. Cell 2020, 53, 316–329.e5. [Google Scholar] [CrossRef] [PubMed]
- Lin, X.X.; Sen, I.; Janssens, G.E.; Zhou, X.; Fonslow, B.R.; Edgar, D.; Stroustrup, N.; Swoboda, P.; Yates, J.R., III; Ruvkun, G.; et al. DAF-16/FOXO and HLH-30/TFEB function as combinatorial transcription factors to promote stress resistance and longevity. Nat. Commun. 2018, 9, 15. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
{kind=link}
| Tumor Types | Tolerant Drugs | Key Pathways/ Genes | Changes in Key Pathways/Genes in a Diapause-like DTP State | Drugs to Improve Treatment | References |
|---|---|---|---|---|---|
| HNSCC | cisplatin | KDM5D/ AURKB | upregulation | barasertib | [45] |
| Melanoma | BRAF inhibitor and MEK inhibitor | cIAP2 | upregulation | birinapant | [73] |
| Melanoma | BRAF inhibitor and MEK inhibitor | Peroxisomal and UGCG | upregulation | inhibitor of the PEX3–PEX19 interaction and UGCG inhibitor | [74] |
| Melanoma | BRAF inhibitor and MEK inhibitor | RTK | upregulation | RMC-4550 | [75] |
| NSCLC | EGFR TKI | BIM | upregulation | BH3 mimetics | [39] |
| TNBC | doxorubicin | P-gp | upregulation | tariquidar | [76] |
| NSCLC | EGFR TKI | IGF1R | upregulation | cIGF1R | [77] |
| PDAC | irinotecan | CYP3A | upregulation | ketoconazole | [78] |
| CLL | venetoclax | Bax/Bak | downregulation | DT-061 | [79] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chen, H.-L.; Jin, W.-L. Diapause-like Drug-Tolerant Persister State: The Key to Nirvana Rebirth. Medicina 2024, 60, 228. https://doi.org/10.3390/medicina60020228
Chen H-L, Jin W-L. Diapause-like Drug-Tolerant Persister State: The Key to Nirvana Rebirth. Medicina. 2024; 60(2):228. https://doi.org/10.3390/medicina60020228
Chicago/Turabian StyleChen, Han-Lin, and Wei-Lin Jin. 2024. "Diapause-like Drug-Tolerant Persister State: The Key to Nirvana Rebirth" Medicina 60, no. 2: 228. https://doi.org/10.3390/medicina60020228
APA StyleChen, H.-L., & Jin, W.-L. (2024). Diapause-like Drug-Tolerant Persister State: The Key to Nirvana Rebirth. Medicina, 60(2), 228. https://doi.org/10.3390/medicina60020228