
Discovery of New 3-(Benzo[b]Thiophen-2-yl)Pyrrolidine-2,5-Dione Derivatives as Potent Antiseizure and Antinociceptive Agents—In Vitro and In Vivo Evaluation

 , , , , , ,
, , , , , ,
Abstract
1. Introduction
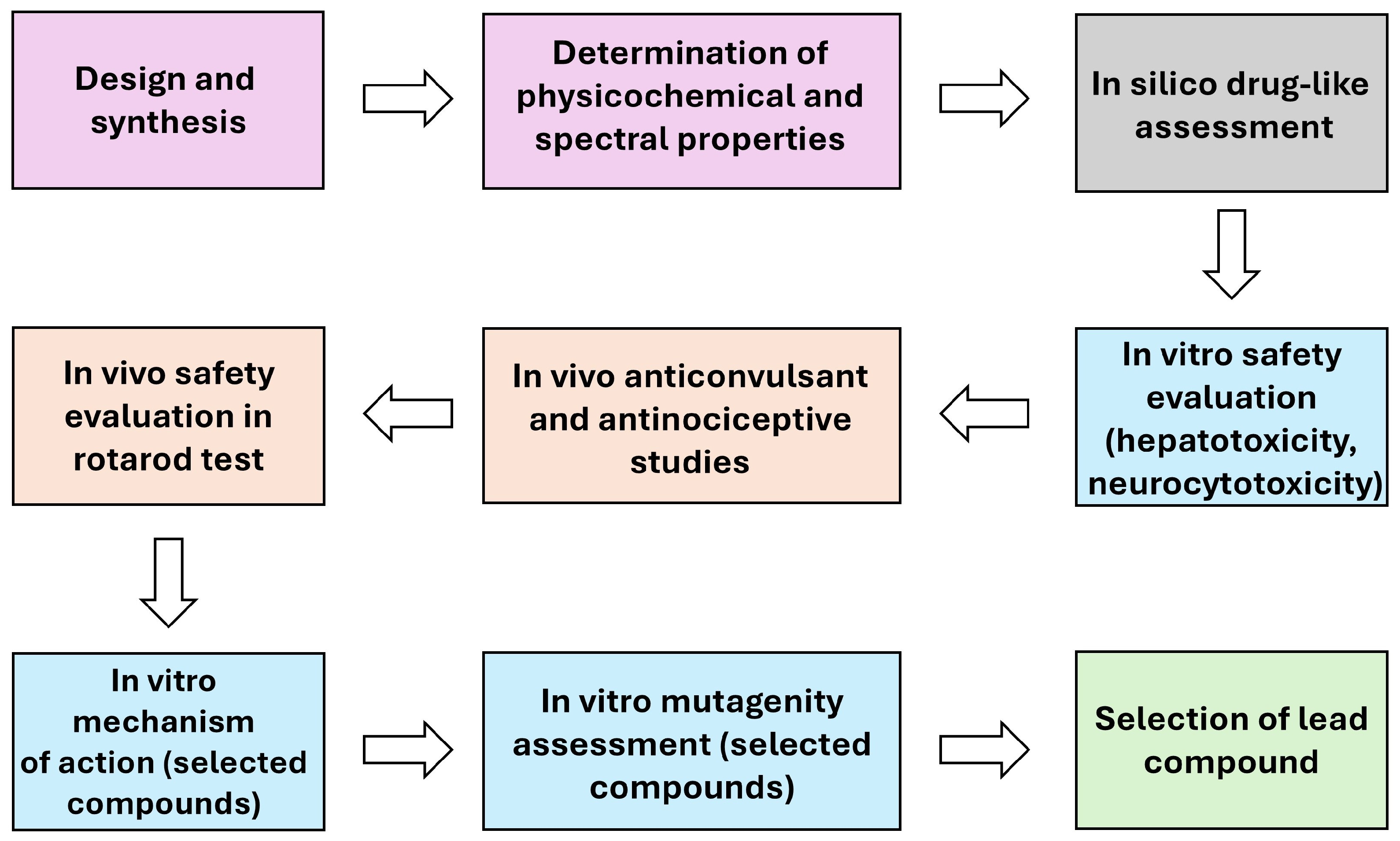
2. Results and Discussion
2.1. Chemistry
2.2. In Silico Studies
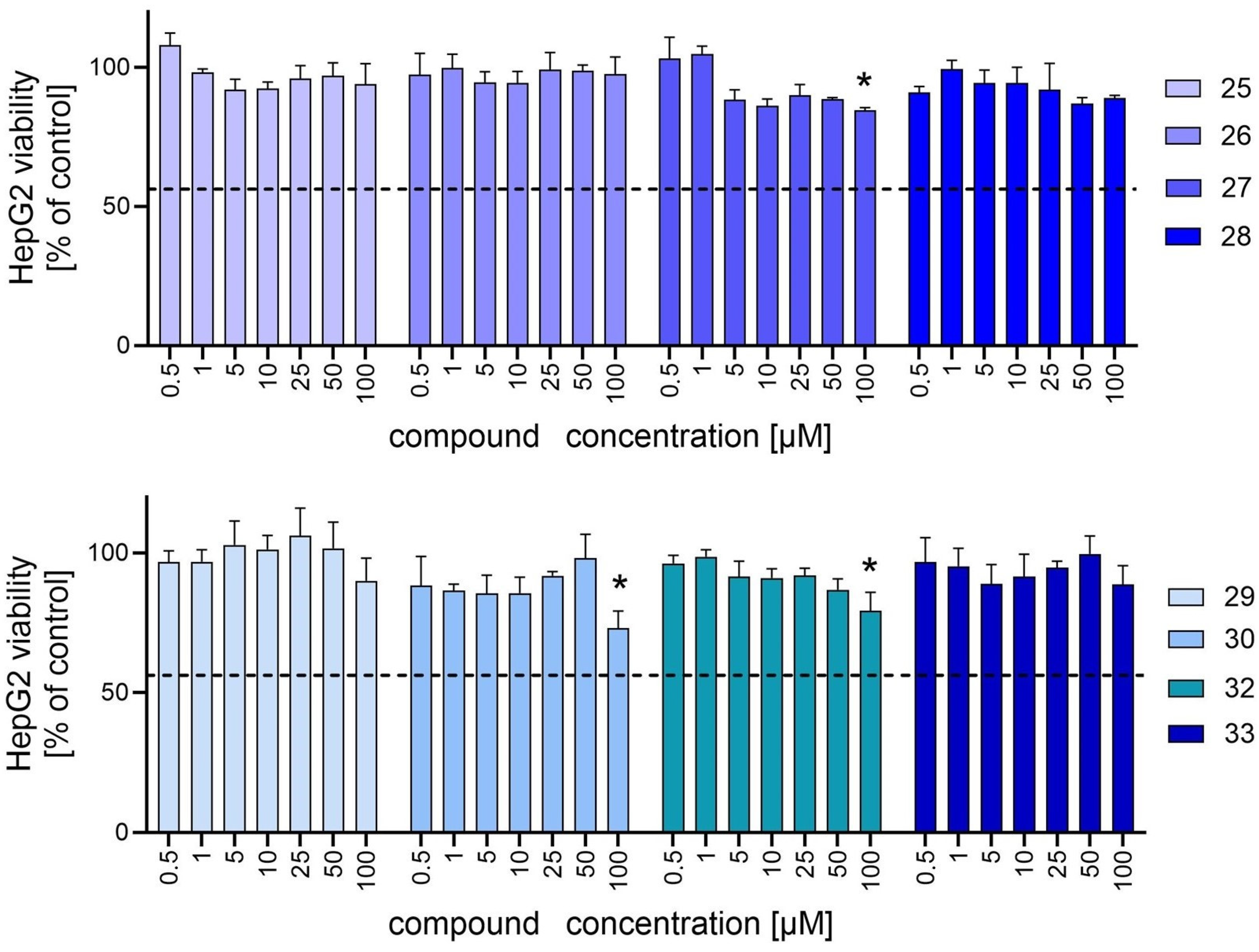
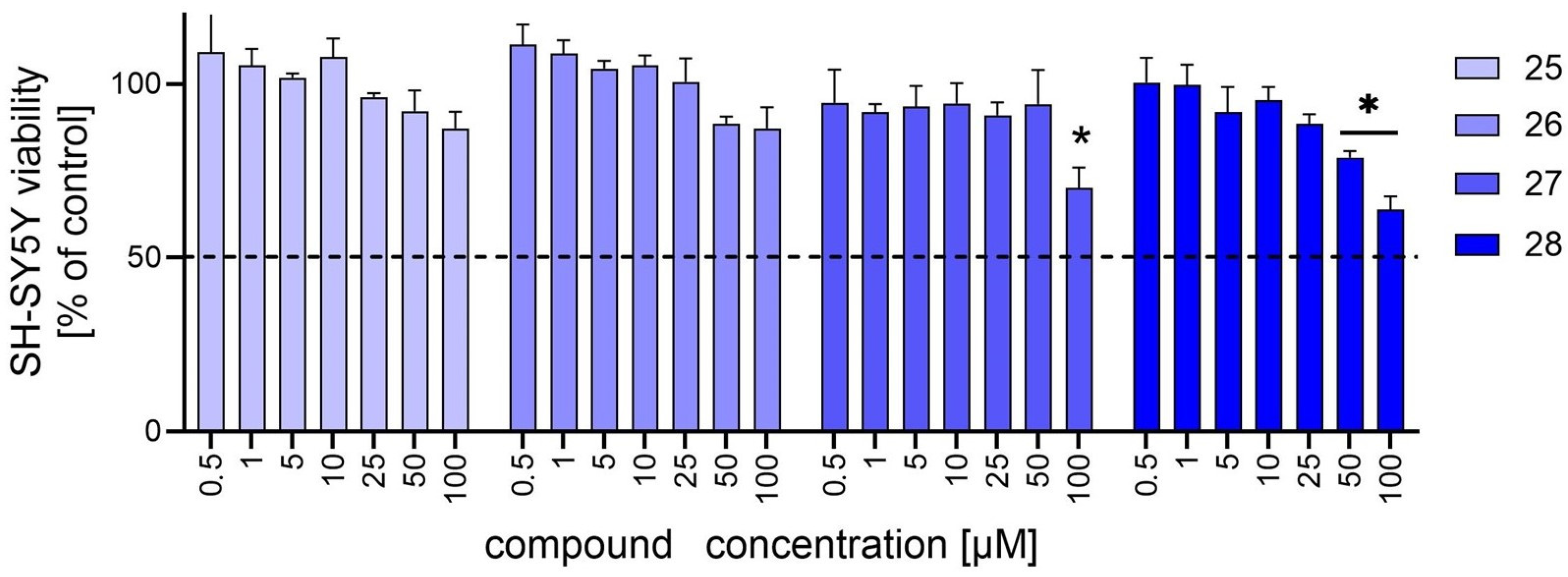
2.3. In Vitro Hepatocytotoxicity and Neurocytotoxicity Assays
2.4. Anticonvulsant Activity
2.5. Antinociceptive Activity
2.5.1. Formalin Test
2.5.2. Oxaliplatin-Induced Neuropathic Pain
2.6. In Vitro Sodium and Calcium Channel Binding Studies
2.7. Analysis of Mutagenicity
3. Materials and Methods
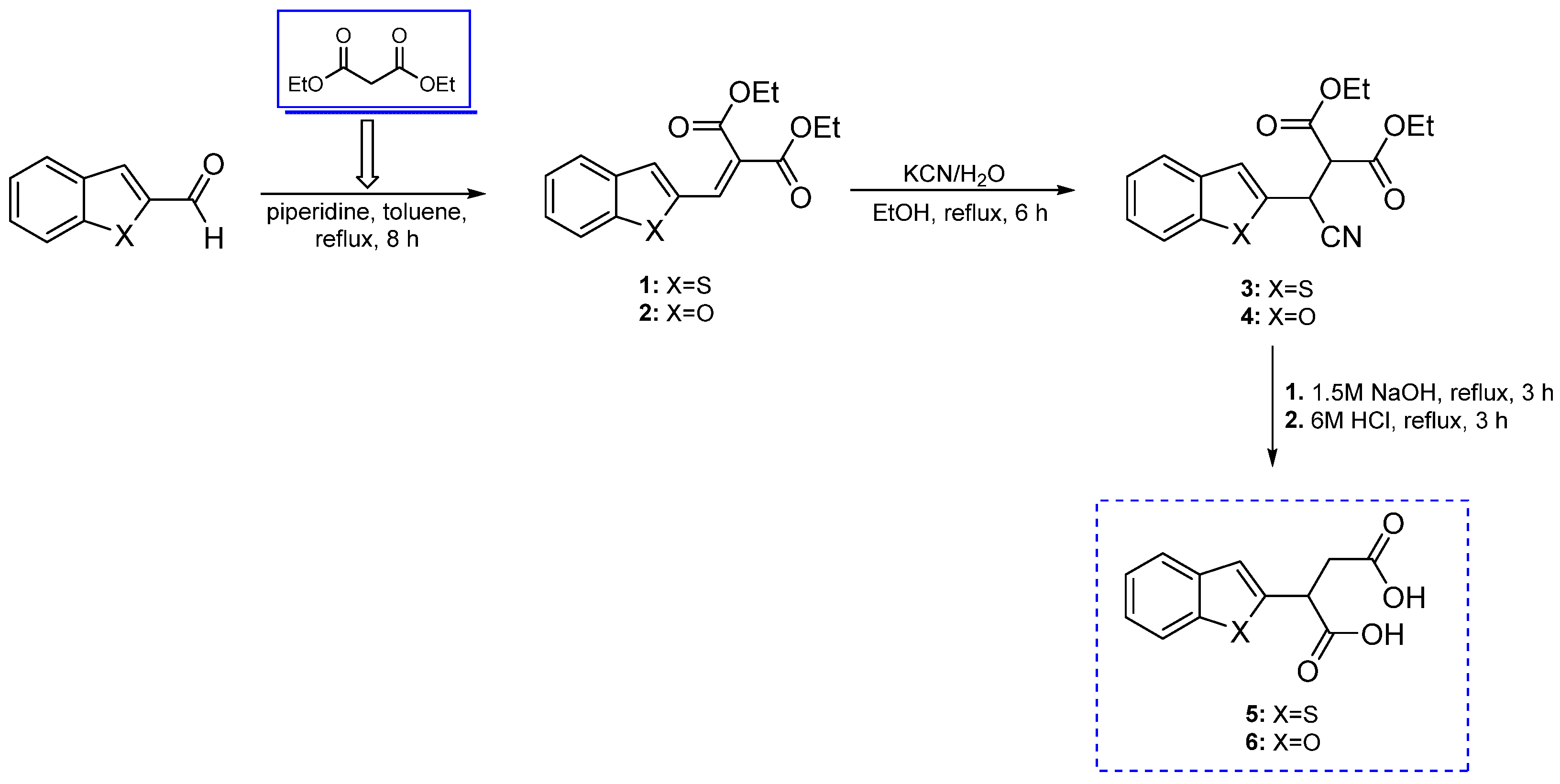
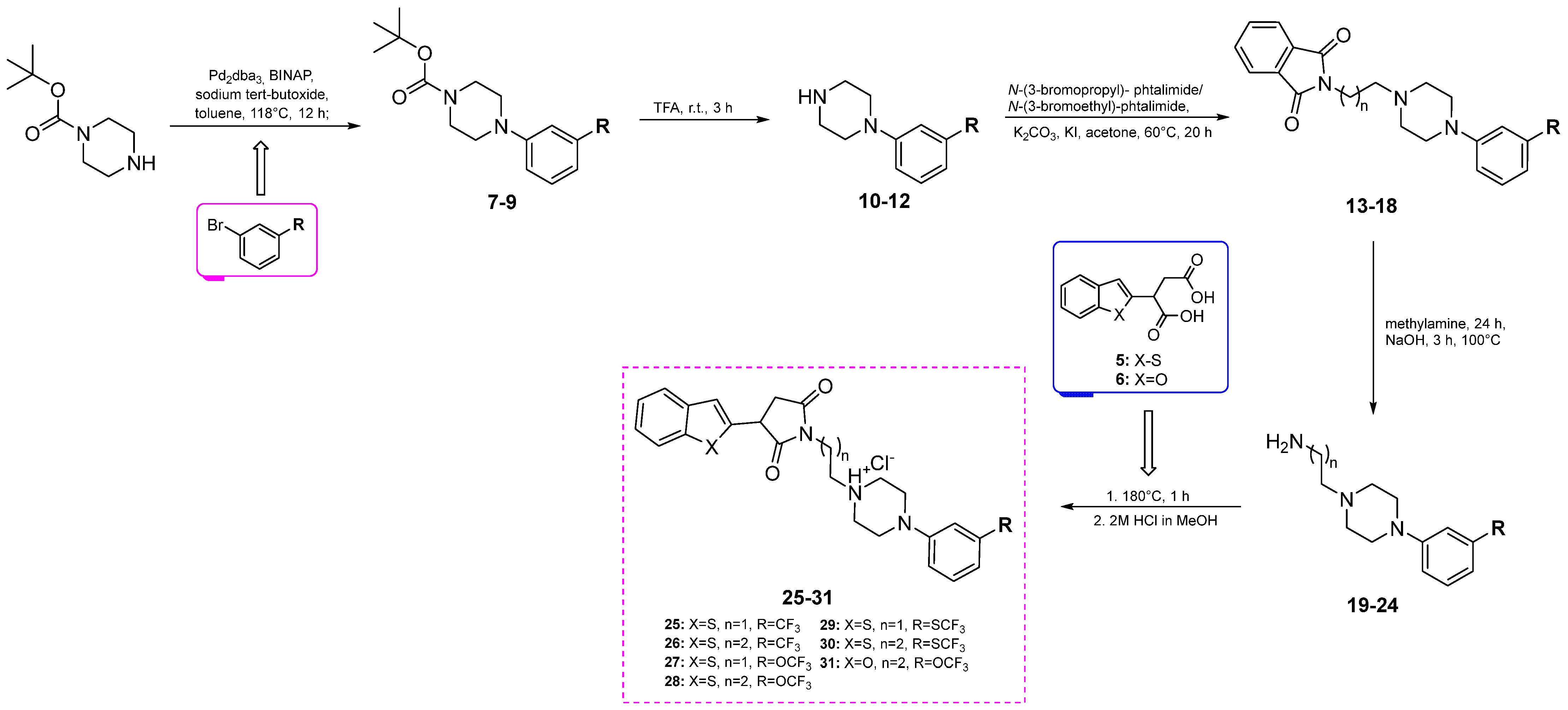
3.1. Synthesis
3.1.1. Synthetic Procedure for Diethyl Malonate Derivatives 1 and 2
3.1.2. Synthetic Procedure for Acids 5 and 6
3.1.3. Synthetic Procedure for Boc-Protected Derivatives (7–9)
3.1.4. Synthetic Procedure for Amines (10–12)
3.1.5. Synthetic Procedure for Phtalimide Derivatives (13–18)
3.1.6. Synthetic Procedure of Aminolysis—Synthesis of Amines (19–24)
3.1.7. Synthetic Procedure for Fusion Reaction (25–31)
3.1.8. Synthetic Procedure for Final Hydrochloride Derivatives (32–33)
3.2. In Vivo Studies
3.2.1. Animals
3.2.2. Drug Administration
3.2.3. Maximal Electroshock Seizure (MES) Test
3.2.4. The Six Herz (6 Hz) Electrical Stimulation Seizure Test
3.2.5. Subcutaneous Pentylenetetrazole Seizure (scPTZ) Test
3.2.6. Neurotoxicity Screening (NT)—Rotarod Test
3.2.7. Median Effective Dose (ED50), Median Toxic Dose (TD50), and Protective Index (PI)
3.2.8. Formalin Test
3.2.9. Oxaliplatin-Induced Neuropathic Pain Model
3.2.10. Data Analysis
3.3. In Vitro Studies
3.3.1. Hepatocytotoxicity and Neurocytotoxicity Assesment
3.3.2. Analysis of Mutagenicity by Ames Microplate Test
3.3.3. Data Analysis
3.3.4. In Vitro Sodium and Calcium Channel Binding Studies
3.4. In Silico Studies
4. Conclusions and Perspectives
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Thijs, R.D.; Surges, R.; O’Brien, T.J.; Sander, J.W. Epilepsy in Adults. Lancet 2019, 393, 689–701. [Google Scholar] [CrossRef] [PubMed]
- Klein, P.; Kaminski, R.M.; Koepp, M.; Löscher, W. New Epilepsy Therapies in Development. Nat. Rev. Drug Discov. 2024, 23, 682–708. [Google Scholar] [CrossRef]
- Tang, F.; Hartz, A.M.S.; Bauer, B. Drug-Resistant Epilepsy: Multiple Hypotheses, Few Answers. Front. Neurol. 2017, 8, 301. [Google Scholar] [CrossRef] [PubMed]
- Hange, N.; Poudel, S.; Ozair, S.; Paul, T.; Nambakkam, M.; Shrestha, R.; Greye, F.; Shah, S.; Raj Adhikari, Y.; Thapa, S.; et al. Managing Chronic Neuropathic Pain: Recent Advances and New Challenges. Neurol. Res. Int. 2022, 2022, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Colloca, L.; Ludman, T.; Bouhassira, D.; Baron, R.; Dickenson, A.H.; Yarnitsky, D.; Freeman, R.; Truini, A.; Attal, N.; Finnerup, N.B.; et al. Neuropathic Pain. Nat. Rev. Dis. Prim. 2017, 3, 17002. [Google Scholar] [CrossRef]
- Kremer, M.; Salvat, E.; Muller, A.; Yalcin, I.; Barrot, M. Antidepressants and Gabapentinoids in Neuropathic Pain: Mechanistic Insights. Neuroscience 2016, 338, 183–206. [Google Scholar] [CrossRef]
- Garcez, D.; Marques, J.; Fernandes, M.; Foreid, J.P. Paroxysmal Pain as the Only Presentation of Focal Epilepsy. Clin. Case Rep. 2020, 8, 1971–1973. [Google Scholar] [CrossRef]
- Siegel, A.M.; Williamson, P.D.; Roberts, D.W.; Thadani, V.M.; Darcey, T.M. Localized Pain Associated with Seizures Originating in the Parietal Lobe. Epilepsia 1999, 40, 845–855. [Google Scholar] [CrossRef]
- Landmark, C.J. Antiepileptic Drugs in Non-Epilepsy Disorders. CNS Drugs 2008, 22, 27–47. [Google Scholar] [CrossRef]
- Löscher, W. Single-Target Versus Multi-Target Drugs Versus Combinations of Drugs with Multiple Targets: Preclinical and Clinical Evidence for the Treatment or Prevention of Epilepsy. Front. Pharmacol. 2021, 12, 730257. [Google Scholar] [CrossRef]
- Yuan, Y.; Pei, J.; Lai, L. LigBuilder V3: A Multi-Target de Novo Drug Design Approach. Front. Chem. 2020, 8, 142. [Google Scholar] [CrossRef] [PubMed]
- Góra, M.; Czopek, A.; Rapacz, A.; Dziubina, A.; Głuch-Lutwin, M.; Mordyl, B.; Obniska, J. Synthesis, Anticonvulsant and Antinociceptive Activity of New Hybrid Compounds: Derivatives of 3-(3-Methylthiophen-2-Yl)-Pyrrolidine-2,5-Dione. Int. J. Mol. Sci. 2020, 21, 5750. [Google Scholar] [CrossRef] [PubMed]
- Kamiński, K.; Zagaja, M.; Łuszczki, J.J.; Rapacz, A.; Andres-Mach, M.; Latacz, G.; Kieć-Kononowicz, K. Design, Synthesis, and Anticonvulsant Activity of New Hybrid Compounds Derived from 2-(2,5-Dioxopyrrolidin-1-Yl)Propanamides and 2-(2,5-Dioxopyrrolidin-1-Yl)Butanamides. J. Med. Chem. 2015, 58, 5274–5286. [Google Scholar] [CrossRef] [PubMed]
- Abram, M.; Zagaja, M.; Mogilski, S.; Andres-Mach, M.; Latacz, G.; Baś, S.; Łuszczki, J.J.; Kieć-Kononowicz, K.; Kamiński, K. Multifunctional Hybrid Compounds Derived from 2-(2,5-Dioxopyrrolidin-1-Yl)-3-Methoxypropanamides with Anticonvulsant and Antinociceptive Properties. J. Med. Chem. 2017, 60, 8565–8579. [Google Scholar] [CrossRef]
- Rapacz, A.; Obniska, J.; Wiklik-Poudel, B.; Rybka, S.; Sałat, K.; Filipek, B. Anticonvulsant and Antinociceptive Activity of New Amides Derived from 3-Phenyl-2,5-Dioxo-Pyrrolidine-1-Yl-Acetic Acid in Mice. Eur. J. Pharmacol. 2016, 781, 239–249. [Google Scholar] [CrossRef]
- Abram, M.; Jakubiec, M.; Rapacz, A.; Mogilski, S.; Latacz, G.; Kamiński, R.M.; Kamiński, K. The Search for New Anticonvulsants in a Group of (2,5-Dioxopyrrolidin-1-Yl)(Phenyl)Acetamides with Hybrid Structure—Synthesis and In Vivo/In Vitro Studies. Int. J. Mol. Sci. 2020, 21, 8780. [Google Scholar] [CrossRef]
- Obniska, J.; Kaminski, K.; Skrzynska, D.; Pichor, J. Synthesis and Anticonvulsant Activity of New N-[(4-Arylpiperazin-1-Yl)-Alkyl] Derivatives of 3-Phenyl-Pyrrolidine-2,5-Dione. Eur. J. Med. Chem. 2009, 44, 2224–2233. [Google Scholar] [CrossRef]
- Obniska, J.; Kopytko, M.; Zagórska, A.; Chlebek, I.; Kamiński, K. Synthesis and Anticonvulsant Properties of New Mannich Bases Derived from 3-Aryl-Pyrrolidine-2,5-Diones. Part 1. Arch. Pharm. 2010, 343, 333–341. [Google Scholar] [CrossRef]
- Wolfe, J.P.; Wagaw, S.; Buchwald, S.L. An Improved Catalyst System for Aromatic Carbon−Nitrogen Bond Formation: The Possible Involvement of Bis(Phosphine) Palladium Complexes as Key Intermediates. J. Am. Chem. Soc. 1996, 118, 7215–7216. [Google Scholar] [CrossRef]
- Bickerton, G.R.; Paolini, G.V.; Besnard, J.; Muresan, S.; Hopkins, A.L. Quantifying the Chemical Beauty of Drugs. Nat. Chem. 2012, 4, 90–98. [Google Scholar] [CrossRef]
- Daina, A.; Michielin, O.; Zoete, V. SwissADME: A Free Web Tool to Evaluate Pharmacokinetics, Drug-Likeness and Medicinal Chemistry Friendliness of Small Molecules. Sci. Rep. 2017, 7, 42717. [Google Scholar] [CrossRef] [PubMed]
- Wager, T.T.; Hou, X.; Verhoest, P.R.; Villalobos, A. Central Nervous System Multiparameter Optimization Desirability: Application in Drug Discovery. ACS Chem. Neurosci. 2016, 7, 767–775. [Google Scholar] [CrossRef] [PubMed]
- Litchfield, J.T.; Wilcoxon, F.A. Simplified Method of Evaluating Dose-Effect Experiments. J. Pharmacol. Exp. Ther. 1949, 96, 99–113. [Google Scholar] [PubMed]
- Macdonald, R.L.; Barker, J.L. Specific Antagonism of GABA-Mediated Postsynaptic Inhibition in Cultured Mammalian Spinal Cord Neurons. Neurology 1978, 28, 325. [Google Scholar] [CrossRef]
- Dantas, L.L.S.F.R.; Fonseca, A.G.; Pereira, J.R.; Furtado, A.A.; Gomes, P.a.T.M.; Fernandes-Pedrosa, M.F.; Leite, A.C.L.; Rêgo, M.J.B.M.; Pitta, M.G.R.; Lemos, T.M.a.M. Anti-Inflammatory and Antinociceptive Effects of the Isatin Derivative (Z)-2-(5-Chloro-2-Oxoindolin-3-Ylidene)-N-Phenyl-Hydrazinecarbothioamide in Mice. Braz. J. Med. Biol. Res. 2020, 53, e10204. [Google Scholar] [CrossRef]
- Lopes, D.M.; Cater, H.L.; Thakur, M.; Wells, S.; McMahon, S.B. A Refinement to the Formalin Test in Mice. F1000Res 2019, 8, 891. [Google Scholar] [CrossRef]
- Laughlin, T.M.; Tram, K.V.; Wilcox, G.L.; Birnbaum, A.K. Comparison of Antiepileptic Drugs Tiagabine, Lamotrigine, and Gabapentin in Mouse Models of Acute, Prolonged, and Chronic Nociception. J. Pharmacol. Exp. Ther. 2002, 302, 1168–1175. [Google Scholar] [CrossRef]
- Nguyen, T.; Chen, X.; Chai, J.; Li, R.; Han, X.; Chen, X.; Liu, S.; Chen, M.; Xu, X. Antipyretic, Anti-Inflammatory and Analgesic Activities of Periplaneta americana Extract and Underlying Mechanisms. Biomed. Pharmacother. 2020, 123, 109753. [Google Scholar] [CrossRef]
- Salinas-Abarca, A.B.; Avila-Rojas, S.H.; Barragán-Iglesias, P.; Pineda-Farias, J.B.; Granados-Soto, V. Formalin Injection Produces Long-Lasting Hypersensitivity with Characteristics of Neuropathic Pain. Eur. J. Pharmacol. 2017, 797, 83–93. [Google Scholar] [CrossRef]
- Beyreuther, B.; Callizot, N.; Stöhr, T. Antinociceptive Efficacy of Lacosamide in a Rat Model for Painful Diabetic Neuropathy. Eur. J. Pharmacol. 2006, 539, 64–70. [Google Scholar] [CrossRef]
- Attal, N.; Bouhassira, D. Advances in the Treatment of Neuropathic Pain. Curr. Opin. Neurol. 2021, 34, 631. [Google Scholar] [CrossRef] [PubMed]
- Rapacz, A.; Głuch-Lutwin, M.; Mordyl, B.; Filipek, B.; Abram, M.; Kamiński, K. Evaluation of Anticonvulsant and Analgesic Activity of New Hybrid Compounds Derived from N-Phenyl-2-(2,5-Dioxopyrrolidin-1-Yl)-Propanamides and -Butanamides. Epilepsy Res. 2018, 143, 11–19. [Google Scholar] [CrossRef] [PubMed]
- Rapacz, A.; Obniska, J.; Koczurkiewicz, P.; Wójcik-Pszczoła, K.; Siwek, A.; Gryboś, A.; Rybka, S.; Karcz, A.; Pękala, E.; Filipek, B. Antiallodynic and Antihyperalgesic Activity of New 3,3-Diphenyl-Propionamides with Anticonvulsant Activity in Models of Pain in Mice. Eur. J. Pharmacol. 2018, 821, 39–48. [Google Scholar] [CrossRef] [PubMed]
- Kaplan, D.I.; Isom, L.L.; Petrou, S. Role of Sodium Channels in Epilepsy. Cold Spring Harb. Perspect. Med. 2016, 6, a022814. [Google Scholar] [CrossRef]
- Nicita, F.; Spalice, A.; Raucci, U.; Iannetti, P.; Parisi, P. The Possible Use of the L-Type Calcium Channel Antagonist Verapamil in Drug-Resistant Epilepsy. Expert Rev. Neurother. 2016, 16, 9–15. [Google Scholar] [CrossRef]
- Russo, E.; Constanti, A.; Ferreri, G.; Citraro, R.; De Sarro, G. Nifedipine Affects the Anticonvulsant Activity of Topiramate in Various Animal Models of Epilepsy. Neuropharmacology 2004, 46, 865–878. [Google Scholar] [CrossRef]
- Mantegazza, M.; Curia, G.; Biagini, G.; Ragsdale, D.S.; Avoli, M. Voltage-Gated Sodium Channels as Therapeutic Targets in Epilepsy and Other Neurological Disorders. Lancet Neurol. 2010, 9, 413–424. [Google Scholar] [CrossRef]
- Barbieri, R.; Nizzari, M.; Zanardi, I.; Pusch, M.; Gavazzo, P. Voltage-Gated Sodium Channel Dysfunctions in Neurological Disorders. Life 2023, 13, 1191. [Google Scholar] [CrossRef]
- Kushnarev, M.; Pirvulescu, I.P.; Candido, K.D.; Knezevic, N.N. Neuropathic Pain: Preclinical and Early Clinical Progress with Voltage-Gated Sodium Channel Blockers. Expert Opin. Investig. Drugs 2020, 29, 259–271. [Google Scholar] [CrossRef]
- Sałat, K.; Cios, A.; Wyska, E.; Sałat, R.; Mogilski, S.; Filipek, B.; Więckowski, K.; Malawska, B. Antiallodynic and Antihyperalgesic Activity of 3-[4-(3-Trifluoromethyl-Phenyl)-Piperazin-1-Yl]-Dihydrofuran-2-One Compared to Pregabalin in Chemotherapy-Induced Neuropathic Pain in Mice. Pharmacol. Biochem. Behav. 2014, 122, 173–181. [Google Scholar] [CrossRef]
- Löscher, W.; Fassbender, C.P.; Nolting, B. The Role of Technical, Biological and Pharmacological Factors in the Laboratory Evaluation of Anticonvulsant Drugs. II. Maximal Electroshock Seizure Models. Epilepsy Res. 1991, 8, 79–94. [Google Scholar] [CrossRef] [PubMed]
- Brown, W.C.; Schiffman, D.O.; Swinyard, E.A.; Goodman, L.S. Comparative Assay of Antiepileptic Drugs by “Psychomotor” Seizure Test and Minimal Electroshock Threshold Test. J. Pharmacol. Exp. Ther. 1953, 107, 273–283. [Google Scholar] [PubMed]
- Barton, M.E.; Klein, B.D.; Wolf, H.H.; Steve White, H. Pharmacological Characterization of the 6 Hz Psychomotor Seizure Model of Partial Epilepsy. Epilepsy Res. 2001, 47, 217–227. [Google Scholar] [CrossRef] [PubMed]
- Leclercq, K.; Kaminski, R.M. Genetic Background of Mice Strongly Influences Treatment Resistance in the 6 Hz Seizure Model. Epilepsia 2015, 56, 310–318. [Google Scholar] [CrossRef]
- Florek-Luszczki, M.; Wlaz, A.; Kondrat-Wrobel, M.W.; Tutka, P.; Luszczki, J.J. Effects of WIN 55,212-2 (a Non-Selective Cannabinoid CB1 and CB2 Receptor Agonist) on the Protective Action of Various Classical Antiepileptic Drugs in the Mouse 6 Hz Psychomotor Seizure Model. J. Neural Transm. 2014, 121, 707–715. [Google Scholar] [CrossRef]
- Ferreri, G.; Chimirri, A.; Russo, E.; Gitto, R.; Gareri, P.; De Sarro, A.; De Sarro, G. Comparative Anticonvulsant Activity of N-Acetyl-1-Aryl-6,7-Dimethoxy-1,2,3,4-Tetrahydroisoquinoline Derivatives in Rodents. Pharmacol. Biochem. Behav. 2004, 77, 85–94. [Google Scholar] [CrossRef]
- Świąder, M.J.; Świąder, K.; Zakrocka, I.; Krzyżanowski, M.; Wróbel, A.; Łuszczki, J.J.; Czuczwar, S.J. Long-Term Vigabatrin Treatment Modifies Pentylenetetrazole-Induced Seizures in Mice: Focused on GABA Brain Concentration. Pharmacol. Rep. 2020, 72, 322–330. [Google Scholar] [CrossRef]
- Tchekalarova, J.; da Conceição Machado, K.; Gomes Júnior, A.L.; de Carvalho Melo Cavalcante, A.A.; Momchilova, A.; Tzoneva, R. Pharmacological Characterization of the Cannabinoid Receptor 2 Agonist, β-Caryophyllene on Seizure Models in Mice. Seizure 2018, 57, 22–26. [Google Scholar] [CrossRef]
- Löscher, W.; Nolting, B. The Role of Technical, Biological and Pharmacological Factors in the Laboratory Evaluation of Anticonvulsant Drugs. IV. Protective Indices. Epilepsy Res. 1991, 9, 1–10. [Google Scholar] [CrossRef]
- Reifferscheid, G.; Maes, H.m.; Allner, B.; Badurova, J.; Belkin, S.; Bluhm, K.; Brauer, F.; Bressling, J.; Domeneghetti, S.; Elad, T.; et al. International Round-Robin Study on the Ames Fluctuation Test. Environ. Mol. Mutagen. 2012, 53, 185–197. [Google Scholar] [CrossRef]
- Koczurkiewicz-Adamczyk, P.; Klaś, K.; Gunia-Krzyżak, A.; Piska, K.; Andrysiak, K.; Stępniewski, J.; Lasota, S.; Wójcik-Pszczoła, K.; Dulak, J.; Madeja, Z.; et al. Cinnamic Acid Derivatives as Cardioprotective Agents against Oxidative and Structural Damage Induced by Doxorubicin. Int. J. Mol. Sci. 2021, 22, 6217. [Google Scholar] [CrossRef] [PubMed]
- Brown, G.B. 3H-Batrachotoxinin-A Benzoate Binding to Voltage-Sensitive Sodium Channels: Inhibition by the Channel Blockers Tetrodotoxin and Saxitoxin. J. Neurosci. 1986, 6, 2064–2070. [Google Scholar] [CrossRef] [PubMed]
- Gould, R.J.; Murphy, K.M.; Snyder, S.H. [3H]Nitrendipine-Labeled Calcium Channels Discriminate Inorganic Calcium Agonists and Antagonists. Proc. Natl. Acad. Sci. USA 1982, 79, 3656–3660. [Google Scholar] [CrossRef] [PubMed]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compd | Lipinski Rule | Veber Rule | CNS MPO | ||||
|---|---|---|---|---|---|---|---|
| MW a ≤ 500 | Log P b ≤ 5 | HBD c ≤ 5 | HBA d ≤ 10 | NBR e ≤ 10 | TPSA f ≤ 140 Å2 | ||
| 25 | 487.54 | 4.42 | 0 | 6 | 6 | 72.10 | 3.35 |
| 26 | 501.56 | 4.74 | 0 | 6 | 7 | 72.10 | 3.21 |
| 27 | 503.54 | 4.25 | 0 | 7 | 7 | 81.33 | 2.76 |
| 28 | 517.56 | 4.54 | 0 | 7 | 8 | 81.33 | 2.83 |
| 29 | 519.60 | 4.82 | 0 | 6 | 7 | 97.40 | 2.75 |
| 30 | 533.63 | 5.12 | 0 | 6 | 8 | 97.40 | 2.83 |
| 31 | 501.50 | 3.96 | 0 | 8 | 8 | 66.23 | 3.59 |
| 32 | 344.43 | 2.25 | 0 | 4 | 4 | 78.09 | 6.00 |
| 33 | 358.46 | 2.55 | 0 | 4 | 5 | 78.09 | 6.00 |
| Cmpd | TPE (h) a | ED50 MES (mg/kg) b | ED50 6 Hz (mg/kg) c | TD50 (mg/kg) d | PI (TD50/ED50) e |
|---|---|---|---|---|---|
| 28 | 0.5 | 33.5 (23.3–48.3) | 41.1 (27.2–62.0) | >300 | >8.9 (MES) >7.3 (6 Hz) |
| 30 | 0.5 | 41.7 (25.1–69.4) | 57.7 (34.0–97.9) | >200 | >4.8 (MES) >3.5 (6 Hz) |
| 31 | 0.5 | 27.3 (22.8–32.7) | 36.3 (24.9–53.1) | <200 | |
| 32 | 0.5 | 62.9 (45.7–86.7) | 37.7 (17.1–81.5) | >200 | >3.2 (MES) >5.3 (6 Hz) |
| 33 | 0.5 | 24.1 (19.7–29.4) | 30.8 (18.2–52.1) | >200 | >8.3 (MES) >6.5 (6 Hz) |
| ETX | 0.25 | - | >200 | 722.1 (647.0−805.8) | |
| LEV f | 1 | - | 14.8 (11.2−18.4) | >500 | >33.8 (6 Hz) |
| VPA f | 0.5 | 216.9 (207.5−226.3) | 130.1 (116.3−143.9) | 372.9 (356.0−389.8) | 1.7 (MES) 2.9 (6 Hz) |
| Cmpd | Concentration [µM] | Na+ Channel (Site 2) | L-Type Ca2+ Channel |
|---|---|---|---|
| % Inhibition of Control Specific Binding | |||
| 32 | 100 | 15.8 | 28.2 |
| 33 | 100 | 62.2 | 12.2 |
| Phenytoin a | 10 | −1.6 | 6.6 |
| 100 | 53.9 | 57.8 | |
| Topiramate a | 100 | - | 7.9 |
| Cmpd | Concentration (µM) | FIB | |||
|---|---|---|---|---|---|
| S. typhimurium—TA98 | S. typhimurium—TA100 | ||||
| −S9 | +S9 | −S9 | +S9 | ||
| 32 | 10 | 0.33 | 0.66 | 0.44 | 0.83 |
| 50 | 0.33 | 0.16 | 1.77 | 0.77 | |
| 100 | 0.66 | 1 | 1.33 | 0.55 | |
| 33 | 10 | 1 | 0.66 | 0.55 | 1.11 |
| 50 | 0 | 0.5 | 0.77 | 0.88 | |
| 100 | 0.66 | 0.66 | 0.88 | 1 | |
| Positive control * | - | 23.8 | 23.16 | 10.6 | 3.6 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rapacz, A.; Jakubiec, M.; Abram, M.; Jasiński, J.; Chrzan, K.; Góra, M.; Dziubina, A.; Wójcik-Pszczoła, K.; Koczurkiewicz-Adamczyk, P.; Ciepiela, K.; et al. Discovery of New 3-(Benzo[b]Thiophen-2-yl)Pyrrolidine-2,5-Dione Derivatives as Potent Antiseizure and Antinociceptive Agents—In Vitro and In Vivo Evaluation. Pharmaceuticals 2024, 17, 1532. https://doi.org/10.3390/ph17111532
Rapacz A, Jakubiec M, Abram M, Jasiński J, Chrzan K, Góra M, Dziubina A, Wójcik-Pszczoła K, Koczurkiewicz-Adamczyk P, Ciepiela K, et al. Discovery of New 3-(Benzo[b]Thiophen-2-yl)Pyrrolidine-2,5-Dione Derivatives as Potent Antiseizure and Antinociceptive Agents—In Vitro and In Vivo Evaluation. Pharmaceuticals. 2024; 17(11):1532. https://doi.org/10.3390/ph17111532
Chicago/Turabian StyleRapacz, Anna, Marcin Jakubiec, Michał Abram, Jakub Jasiński, Karolina Chrzan, Małgorzata Góra, Anna Dziubina, Katarzyna Wójcik-Pszczoła, Paulina Koczurkiewicz-Adamczyk, Katarzyna Ciepiela, and et al. 2024. "Discovery of New 3-(Benzo[b]Thiophen-2-yl)Pyrrolidine-2,5-Dione Derivatives as Potent Antiseizure and Antinociceptive Agents—In Vitro and In Vivo Evaluation" Pharmaceuticals 17, no. 11: 1532. https://doi.org/10.3390/ph17111532
APA StyleRapacz, A., Jakubiec, M., Abram, M., Jasiński, J., Chrzan, K., Góra, M., Dziubina, A., Wójcik-Pszczoła, K., Koczurkiewicz-Adamczyk, P., Ciepiela, K., Pękala, E., Obniska, J., & Kamiński, K. (2024). Discovery of New 3-(Benzo[b]Thiophen-2-yl)Pyrrolidine-2,5-Dione Derivatives as Potent Antiseizure and Antinociceptive Agents—In Vitro and In Vivo Evaluation. Pharmaceuticals, 17(11), 1532. https://doi.org/10.3390/ph17111532