
Discovery of Potent Indolyl-Hydrazones as Kinase Inhibitors for Breast Cancer: Synthesis, X-ray Single-Crystal Analysis, and In Vitro and In Vivo Anti-Cancer Activity Evaluation

 ,
,  , ,
, ,  ,
,  and
and
Abstract
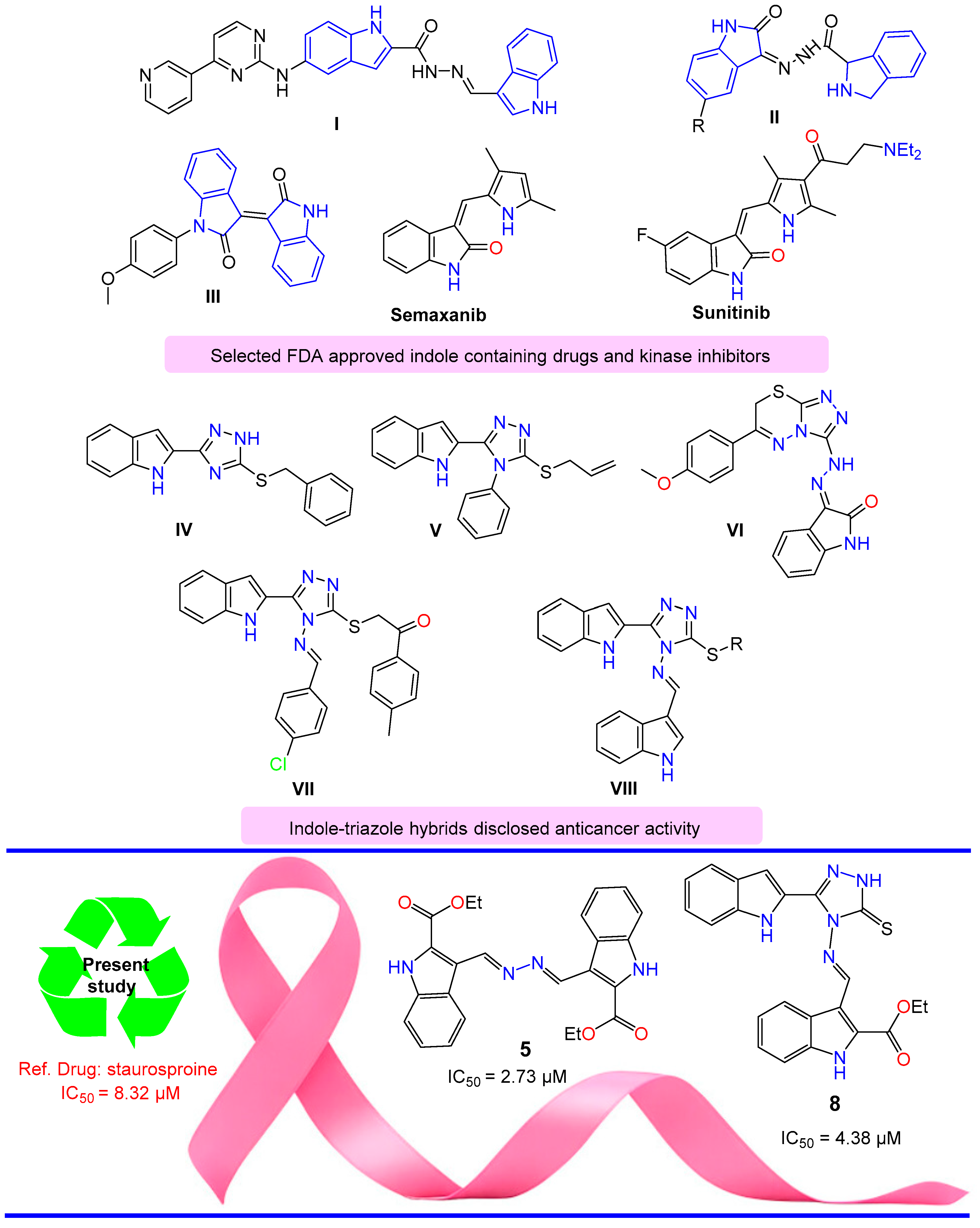
1. Introduction
2. Results and Discussion
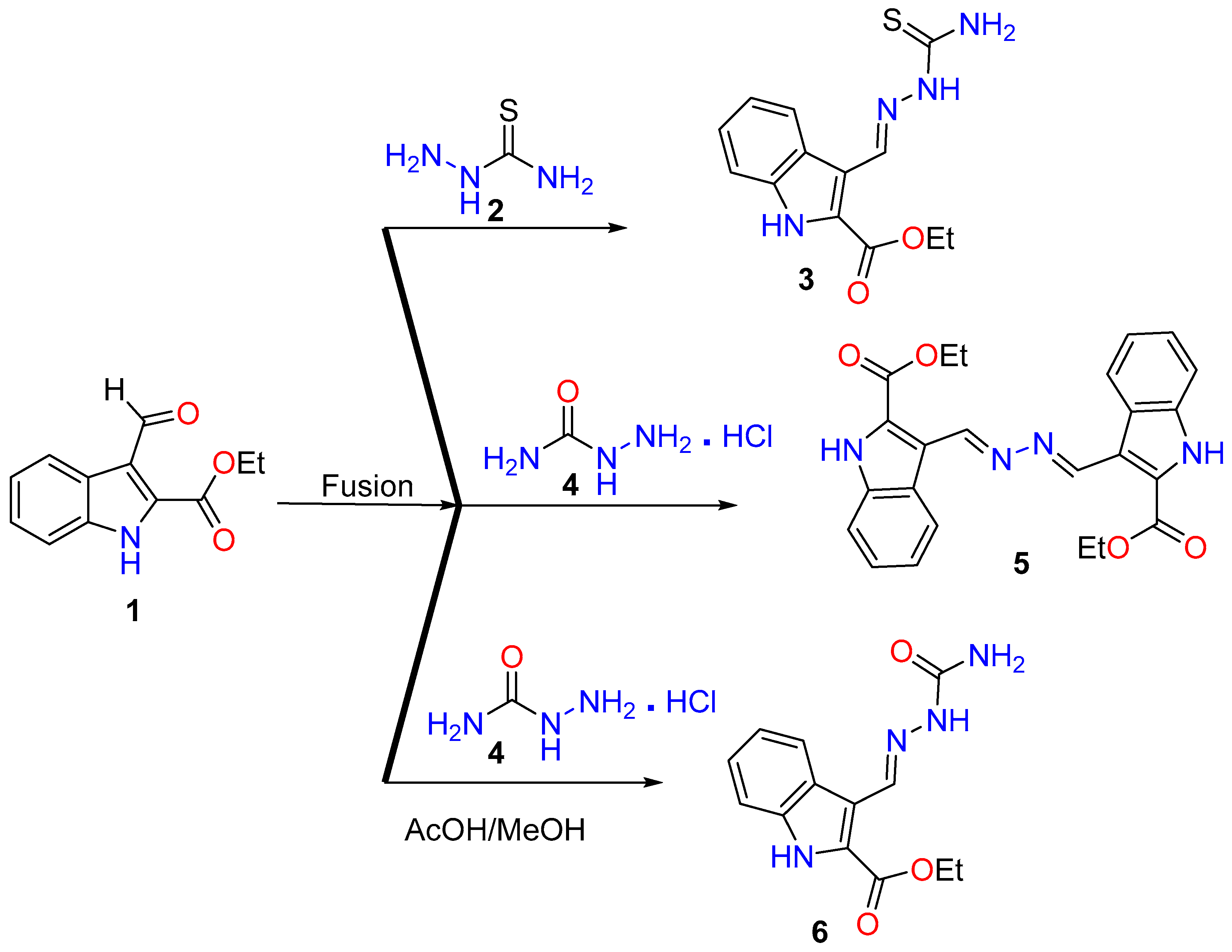
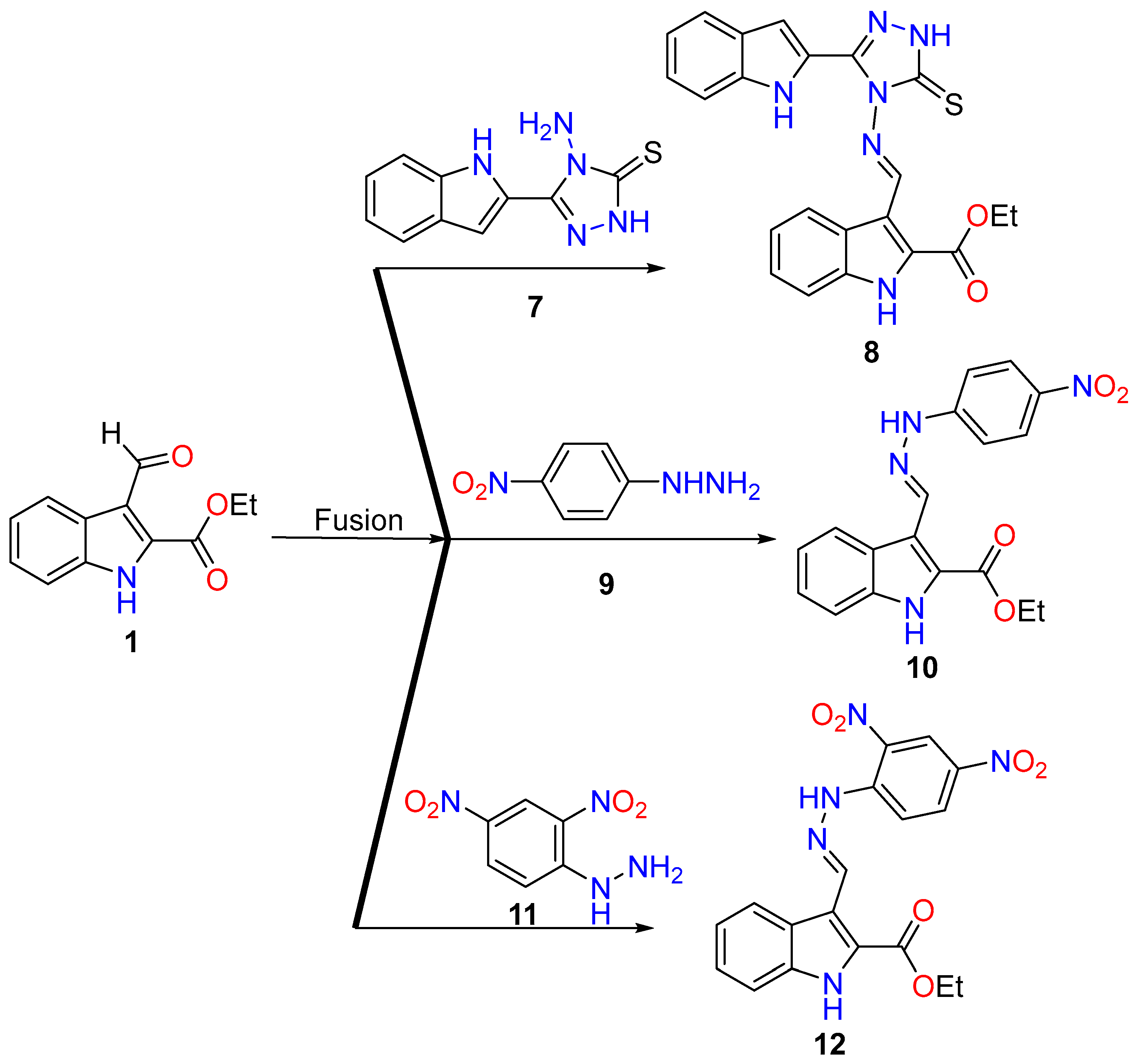
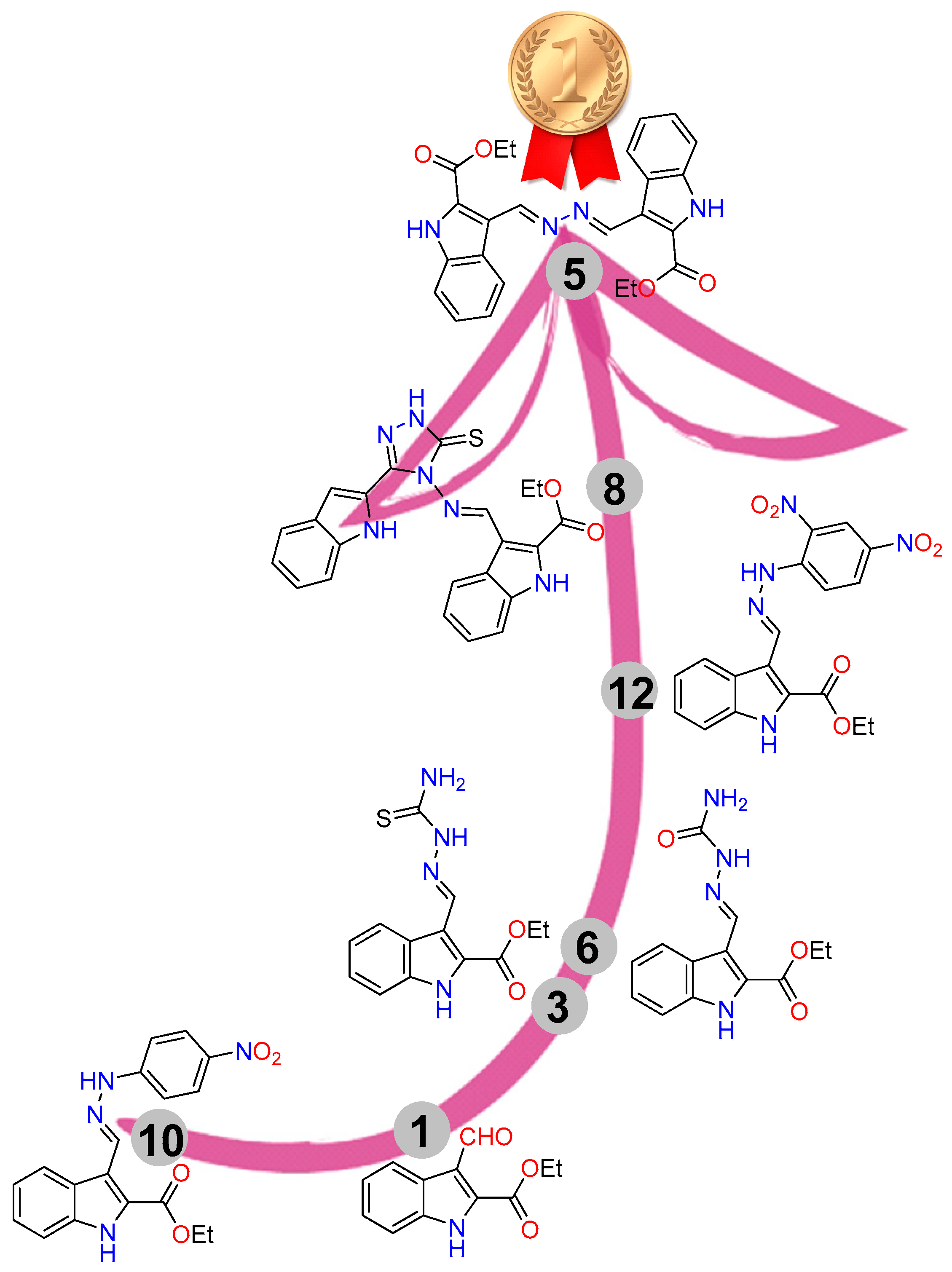
2.1. Synthesis
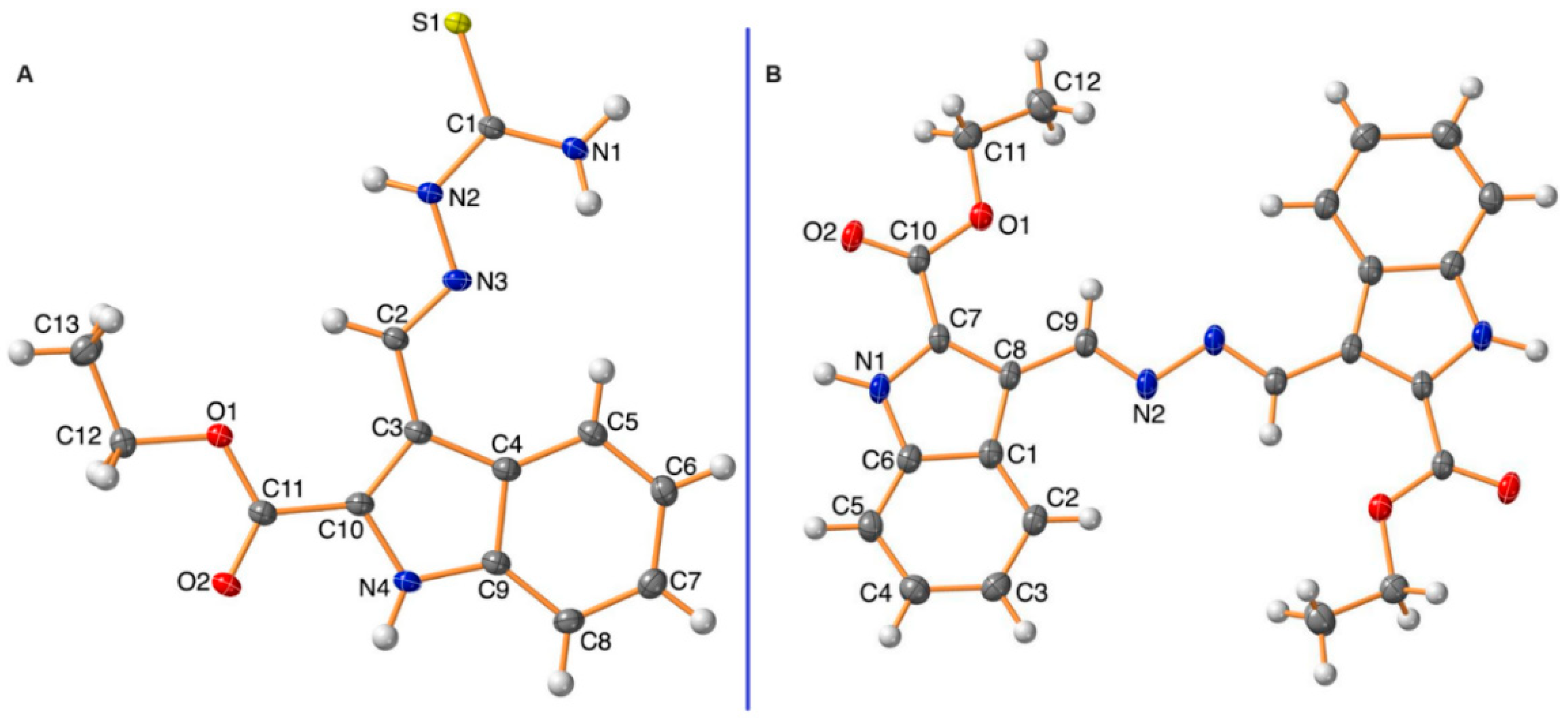
2.2. X-ray Single-Crystal Analysis for Compounds 3 and 5
2.3. Biology
2.3.1. MTT Assay for the Synthesized Compounds
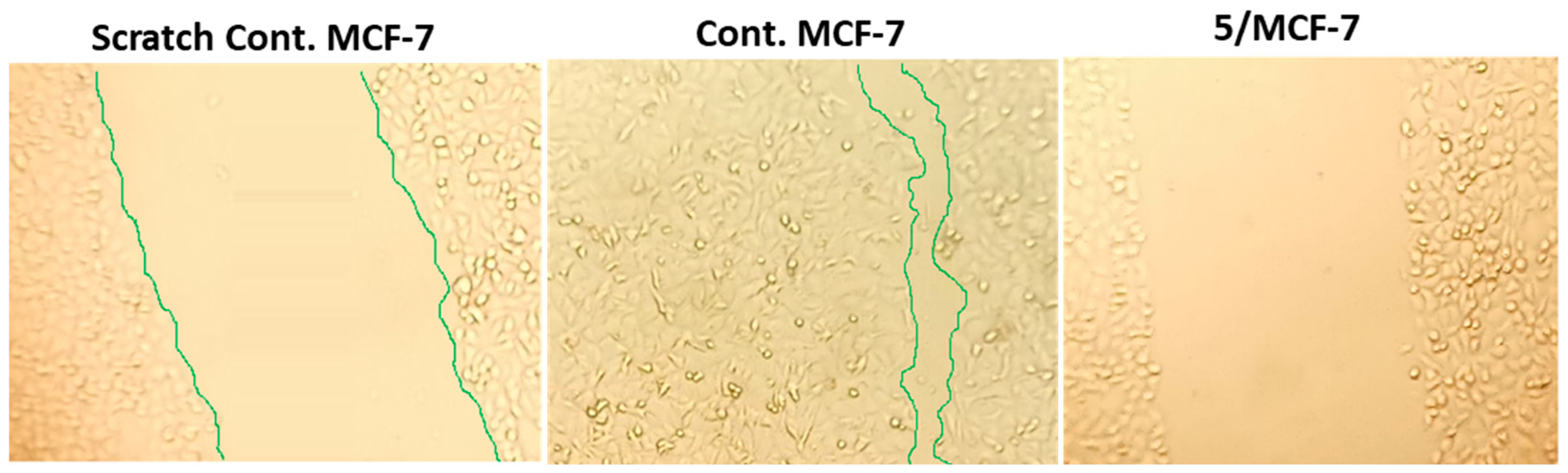
2.3.2. Wound-Healing Activity
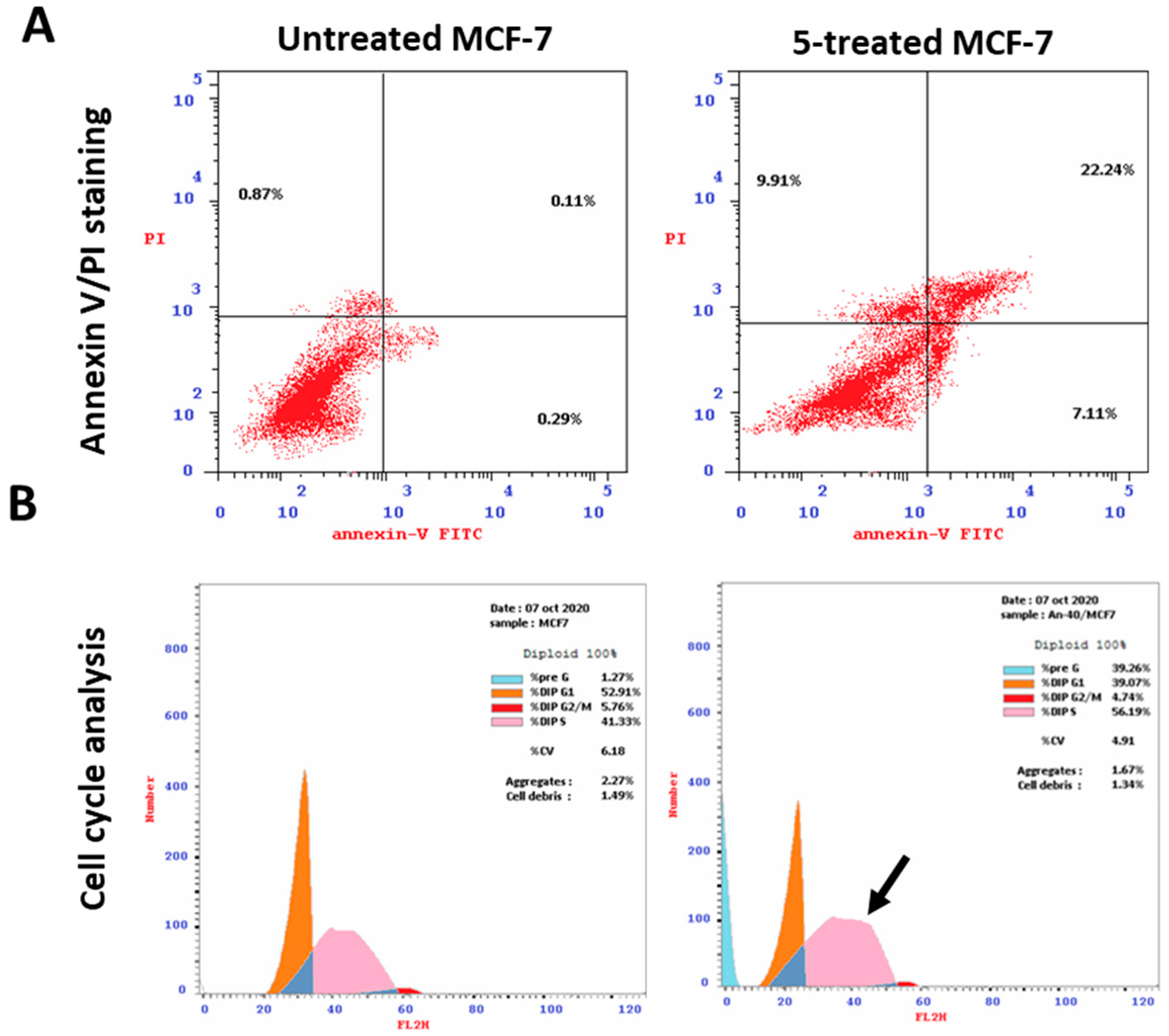
2.3.3. Apoptotic Induction Activity
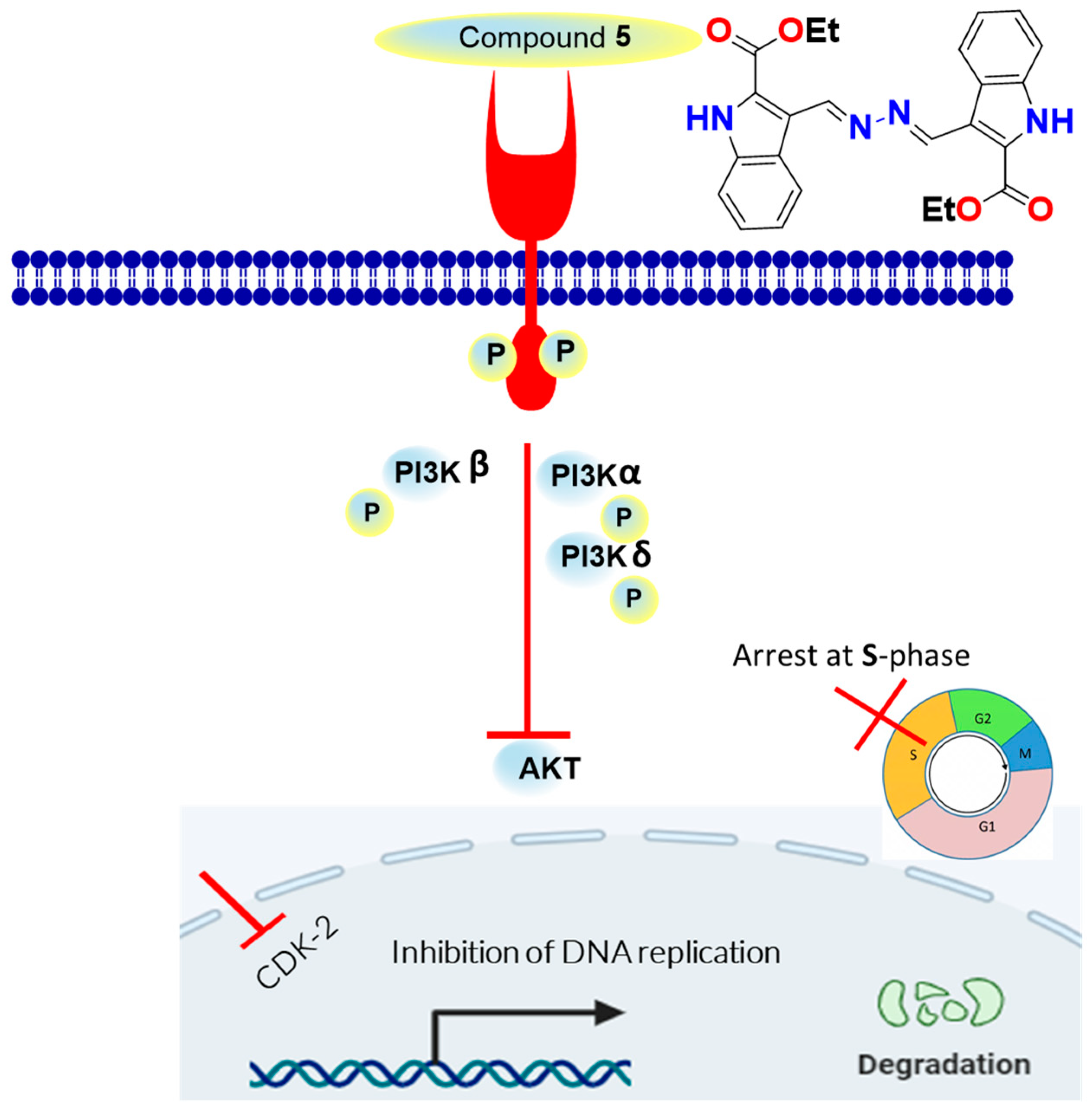
2.3.4. Kinase-Inhibition Activity
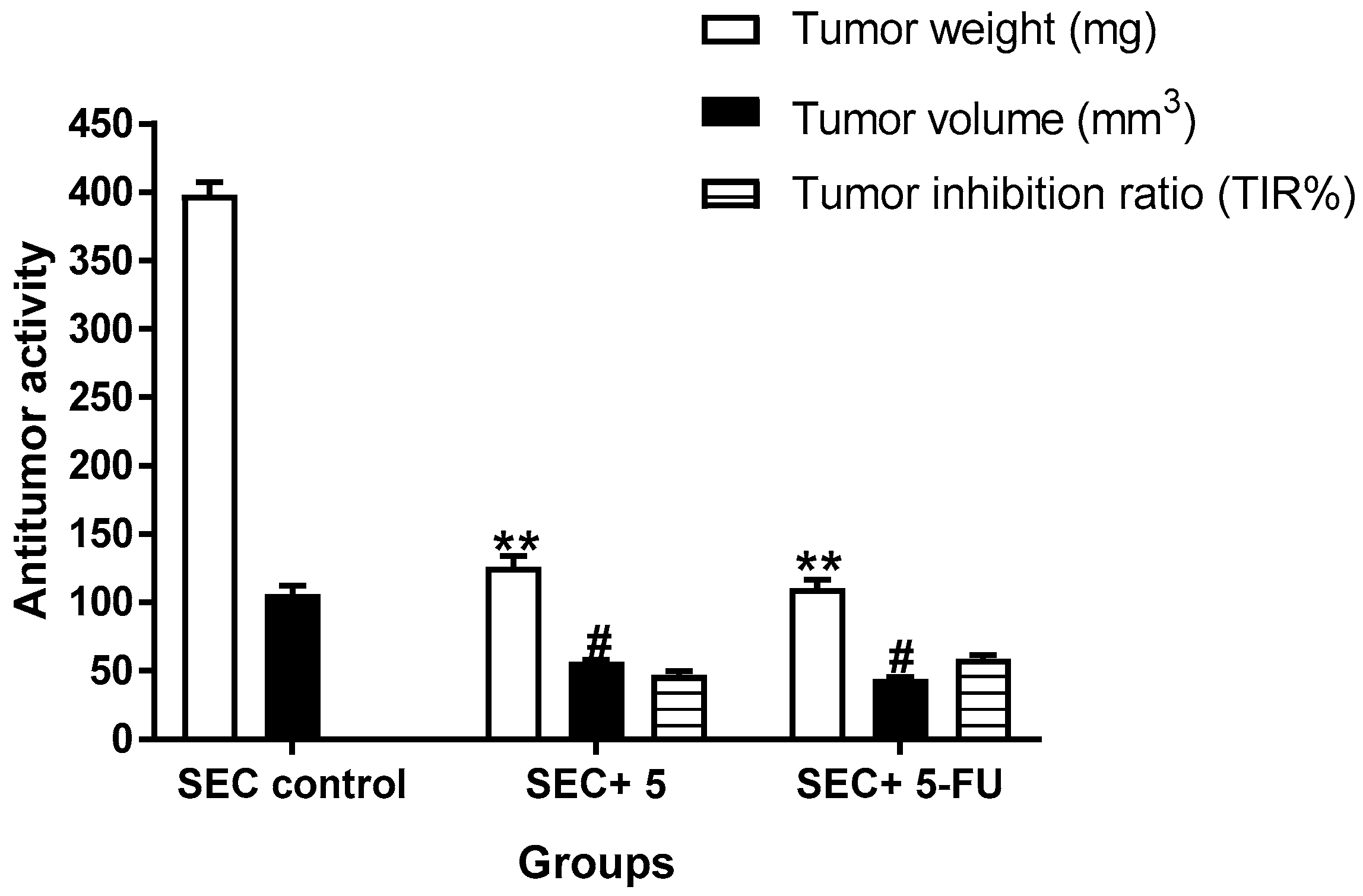
2.3.5. In Vivo (SEC-Bearing Mice)
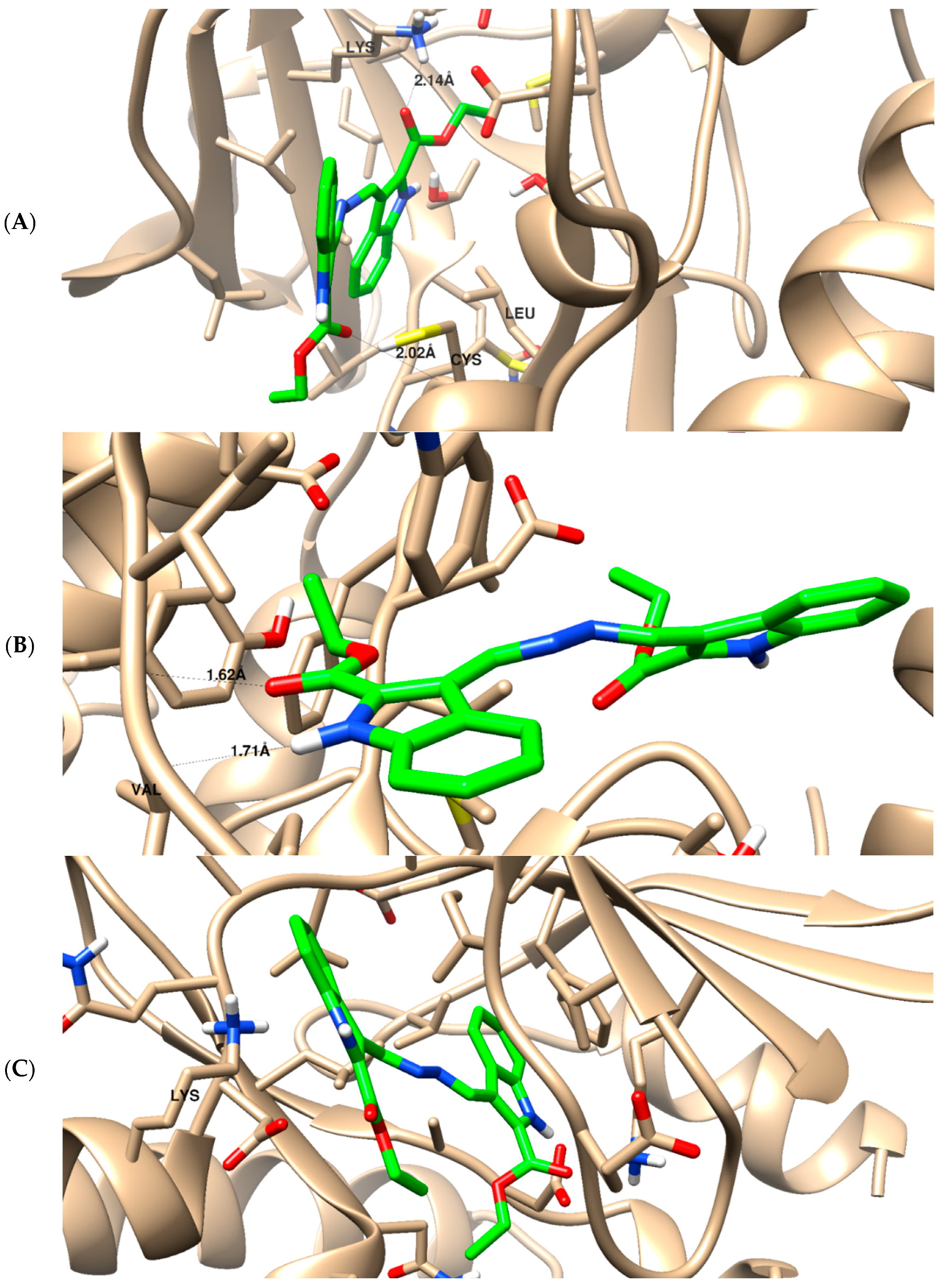
2.3.6. Molecular Docking
2.3.7. SAR
3. Materials and Methods
3.1. Chemistry
3.1.1. General
3.1.2. Synthesis
3.1.3. X-ray Structure Determination
4. Cytotoxicity
4.1. Investigation of Apoptosis
4.2. Wound-Healing Assay (Scratch Assay)
4.3. Kinase Inhibitory Assays
4.4. In Vivo (SEC-Bearing Model)
4.5. Molecular Docking
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Correction Statement
References
- Ye, F.; Dewanjee, S.; Li, Y.; Jha, N.K.; Chen, Z.-S.; Kumar, A.; Vishakha; Behl, T.; Jha, S.K.; Tang, H. Advancements in clinical aspects of targeted therapy and immunotherapy in breast cancer. Mol. Cancer 2023, 22, 105. [Google Scholar] [CrossRef] [PubMed]
- Senapati, S.; Mahanta, A.K.; Kumar, S.; Maiti, P. Controlled drug delivery vehicles for cancer treatment and their performance. Signal Transduct. Target. Ther. 2018, 3, 7. [Google Scholar] [CrossRef] [PubMed]
- Ward, R.A.; Goldberg, F.W. Introduction to Kinase Drug Discovery: Modern Approaches; Royal Society of Chemistry: London, UK, 2018; Volume 67. [Google Scholar]
- Zhao, Z.; Bourne, P.E. Progress with covalent small-molecule kinase inhibitors. Drug Discov. Today 2018, 23, 727–735. [Google Scholar] [CrossRef]
- Bhanumathy, K.K.; Balagopal, A.; Vizeacoumar, F.S.; Vizeacoumar, F.J.; Freywald, A.; Giambra, V. Protein Tyrosine Kinases: Their roles and their targeting in leukemia. Cancers 2021, 13, 184. [Google Scholar] [CrossRef]
- Patterson, H.; Nibbs, R.; McInnes, I.; Siebert, S. Protein kinase inhibitors in the treatment of inflammatory and autoimmune diseases. Clin. Exp. Immunol. 2014, 176, 1–10. [Google Scholar] [CrossRef]
- Bhullar, K.S.; Lagarón, N.O.; McGowan, E.M.; Parmar, I.; Jha, A.; Hubbard, B.P.; Rupasinghe, H.P.V. Kinase-targeted cancer therapies: Progress, challenges and future directions. Mol. Cancer 2018, 17, 48. [Google Scholar] [CrossRef]
- Bononi, A.; Agnoletto, C.; De Marchi, E.; Marchi, S.; Patergnani, S.; Bonora, M.; Giorgi, C.; Missiroli, S.; Poletti, F.; Rimessi, A.; et al. Protein kinases and phosphatases in the control of cell fate. Enzym. Res. 2011, 2011, 329098. [Google Scholar] [CrossRef]
- Valdespino-Gómez, V.M.; Valdespino-Castillo, P.M.; Valdespino-Castillo, V.E. Cell signaling pathways interaction in cellular proliferation: Potential target for therapeutic interventionism. Cirugía Cir. 2015, 83, 165–174. [Google Scholar] [CrossRef] [PubMed]
- Zhong, L.; Li, Y.; Xiong, L.; Wang, W.; Wu, M.; Yuan, T.; Yang, W.; Tian, C.; Miao, Z.; Wang, T.; et al. Small molecules in targeted cancer therapy: Advances, challenges, and future perspectives. Signal Transduct. Target. Ther. 2021, 6, 201. [Google Scholar] [CrossRef] [PubMed]
- You, K.S.; Yi, Y.W.; Cho, J.; Park, J.S.; Seong, Y.S. Potentiating therapeutic effects of epidermal growth factor receptor inhibition in triple-negative breast cancer. Pharmaceuticals 2021, 14, 589. [Google Scholar] [CrossRef] [PubMed]
- Roskoski, R., Jr. Properties of FDA-approved small molecule protein kinase inhibitors: A 2021 update. Pharmacol. Res. 2021, 165, 105463. [Google Scholar] [CrossRef]
- Spanò, V.; Barreca, M.; Rocca, R.; Bortolozzi, R.; Bai, R.; Carbone, A.; Raimondi, M.V.; Piccionello, A.P.; Montalbano, A.; Alcaro, S.; et al. Insight on [1,3]thiazolo[4,5-e]isoindoles as tubulin polymerization inhibitors. Eur. J. Med. Chem. 2021, 212, 113122. [Google Scholar] [CrossRef] [PubMed]
- Ostacolo, C.; Di Sarno, V.; Lauro, G.; Pepe, G.; Musella, S.; Ciaglia, T.; Vestuto, V.; Autore, G.; Bifulco, G.; Marzocco, S.; et al. Identification of an indol-based multi-target kinase inhibitor through phenotype screening and tar-get fishing using inverse virtual screening approach. Eur. J. Med. Chem. 2019, 167, 61–75. [Google Scholar] [CrossRef] [PubMed]
- Kryshchyshyn-Dylevych, A.; Radko, L.; Finiuk, N.; Garazd, M.; Kashchak, N.; Posyniak, A.; Niemczuk, K.; Stoika, R.; Lesyk, R. Synthesis of novel indole-thiazolidinone hybrid structures as promising scaffold with anticancer potential. Bioorganic Med. Chem. 2021, 50, 116453. [Google Scholar] [CrossRef] [PubMed]
- Wang, G.; He, M.; Liu, W.; Fan, M.; Li, Y.; Peng, Z. Design, synthesis and biological evaluation of novel 2-phenyl-4,5,6,7-tetrahydro-1H- indole derivatives as potential anticancer agents and tubulin polymerization inhibitors. Arab. J. Chem. 2022, 15, 103504. [Google Scholar] [CrossRef]
- Ponnam, D.; Arigari, N.K.; Kalvagunta Venkata Naga, S.S.; Jonnala, K.K.; Singh, S.; Meena, A.; Misra, P.; Luqman, S. Synthesis of non-toxic anticancer active forskolin-indole-triazole conjugates along with their in silico succinate dehydrogenase inhibition studies. J. Heterocycl. Chem. 2021, 58, 2090–2101. [Google Scholar] [CrossRef]
- Saruengkhanphasit, R.; Butkinaree, C.; Ornnork, N.; Lirdprapamongkol, K.; Niwetmarin, W.; Svasti, J.; Ruchirawat, S.; Eurtivong, C. Identification of new 3-phenyl-1H-indole-2-carbohydrazide derivatives and their structure-activity relationships as potent tubulin inhibitors and anticancer agents: A combined in silico, in vitro and synthetic study. Bioorganic Chem. 2021, 110, 104795. [Google Scholar] [CrossRef]
- Hu, H.; Wu, J.; Ao, M.; Zhou, X.; Li, B.; Cui, Z.; Wu, T.; Wang, L.; Xue, Y.; Wu, Z.; et al. Design, synthesis and biological evaluation of methylenehydrazine-1-carboxamide derivatives with (5-((4-(pyridin-3-yl)pyrimidin-2-yl)amino)-1H-indole scaffold: Novel potential CDK9 inhibitors. Bioorganic Chem. 2020, 102, 104064. [Google Scholar] [CrossRef]
- Al-Warhi, T.; El Kerdawy, A.M.; Aljaeed, N.; Ismael, O.E.; Ayyad, R.R.; Eldehna, W.M.; Abdel-Aziz, H.A.; Al-Ansary, G.H. Synthesis, biological evaluation and in silico studies of certain oxindole–indole conjugates as anticancer CDK inhibitors. Molecules 2020, 25, 2031. [Google Scholar] [CrossRef]
- Zhao, P.; Li, Y.; Gao, G.; Wang, S.; Yan, Y.; Zhan, X.; Liu, Z.; Mao, Z.; Chen, S.; Wang, L. Design, synthesis and biological evaluation of N-alkyl or aryl substituted isoindigo derivatives as potential dual Cyclin-Dependent Kinase 2 (CDK2)/Glycogen Synthase Kinase 3β (GSK-3β) phosphorylation inhibitors. Eur. J. Med. Chem. 2014, 86, 165–174. [Google Scholar] [CrossRef]
- Oudard, S.; Beuselinck, B.; Decoene, J.; Albers, P. Sunitinib for the treatment of metastatic renal cell carcinoma. Cancer Treat. Rev. 2011, 37, 178–184. [Google Scholar] [CrossRef]
- Xu, D.; Wang, T.-L.; Sun, L.-P.; You, Q.-D. Recent progress of small molecular VEGFR inhibitors as anticancer agents. Mini-Rev. Med. Chem. 2011, 11, 18–31. [Google Scholar] [CrossRef]
- Gridelli, C.; Maione, P.; Del Gaizo, F.; Colantuoni, G.; Guerriero, C.; Ferrara, C.; Nicolella, D.; Comunale, D.; De Vita, A.; Rossi, A. Sorafenib and Sunitinib in the Treatment of Advanced Non-Small Cell Lung Cancer. Oncologist 2007, 12, 191–200. [Google Scholar] [CrossRef] [PubMed]
- Imming, P.; Sinning, C.; Meyer, A. Drugs, their targets and the nature and number of drug targets. Nat. Rev. Drug Discov. 2006, 5, 821–834. [Google Scholar] [CrossRef] [PubMed]
- Lockhart, A.C.; Cropp, G.F.; Berlin, J.D.; Donnelly, E.; Schumaker, R.D.; Schaaf, L.J.; Hande, K.R.; Fleischer, A.C.; Hannah, A.L.; Rothenberg, M.L. Phase I/pilot study of SU5416 (semaxinib) in combination with irinotecan/bolus 5-FU/LV (IFL) in patients with metastatic colorectal cancer. Am. J. Clin. Oncol. 2006, 29, 109–115. [Google Scholar] [CrossRef] [PubMed]
- Boraei, A.T.A.; Gomaa, M.S.; El Ashry, E.S.H.; Duerkop, A. Design, selective alkylation and X-ray crystal structure determination of dihydro-indolyl-1,2,4-triazole-3-thione and its 3-benzylsulfanyl analogue as potent anticancer agents. Eur. J. Med. Chem. 2017, 125, 360–371. [Google Scholar] [CrossRef] [PubMed]
- Boraei, A.T.A.; Singh, P.K.; Sechi, M.; Satta, S. Discovery of novel functionalized 1,2,4-triazoles as PARP-1 inhibitors in breast cancer: Design, synthesis and antitumor activity evaluation. Eur. J. Med. Chem. 2019, 182, 111621. [Google Scholar] [CrossRef] [PubMed]
- Mohamady, S.; Galal, M.; Eldehna, W.M.; Gutierrez, D.C.; Ibrahim, H.S.; Elmazar, M.M.; Ali, H.I. Dual Targeting of VEGFR2 and C-Met Kinases via the Design and Synthesis of Substituted 3-(Triazolo-thiadiazin-3-yl)indolin-2-one Derivatives as Angiogenesis Inhibitors. ACS Omega 2020, 5, 18872–18886. [Google Scholar] [CrossRef]
- Al-Hussain, S.A.; Farghaly, T.A.; Zaki, M.E.A.; Abdulwahab, H.G.; Al-Qurashi, N.T.; Muhammad, Z.A. Discovery of novel indolyl-1,2,4-triazole hybrids as potent vascular endothelial growth factor receptor-2 (VEGFR-2) inhibitors with potential anti-renal cancer activity. Bioorganic Chem. 2020, 105, 104330. [Google Scholar] [CrossRef]
- Nafie, M.S.; Boraei, A.T.A. Exploration of novel VEGFR2 tyrosine kinase inhibitors via design and synthesis of new alkylated indolyl-triazole Schiff bases for targeting breast cancer. Bioorg. Chem. 2022, 122, 105708. [Google Scholar] [CrossRef]
- Lee, D.H.; Szczepanski, M.J.; Lee, Y.J. Magnolol induces apoptosis via inhibiting the EGFR/PI3K/Akt signaling pathway in human prostate cancer cells. J. Cell. Biochem. 2009, 106, 1113–1122. [Google Scholar] [CrossRef] [PubMed]
- Sreenivasulu, R.; Reddy, K.T.; Sujitha, P.; Kumar, C.G.; Raju, R.R. Synthesis, antiproliferative and apoptosis induction potential activities of novel bis (indolyl) hydrazide-hydrazone derivatives. Bioorg. Med. Chem. 2019, 27, 1043–1055. [Google Scholar] [CrossRef] [PubMed]
- Das Mukherjee, D.; Kumar, N.M.; Tantak, M.P.; Das, A.; Ganguli, A.; Datta, S.; Kumar, D.; Chakrabarti, G. Development of novel bis(indolyl)-hydrazide–hydrazone derivatives as potent microtubule-targeting cytotoxic agents against A549 lung cancer cells. Biochemistry 2016, 55, 3020–3035. [Google Scholar] [CrossRef] [PubMed]
- Kilic-Kurt, Z.; Acar, C.; Ergul, M.; Bakar-Ates, F.; Altuntas, T.G. Novel indole hydrazide derivatives: Synthesis and their antiproliferative activities through inducing apoptosis and DNA damage. Arch. Pharm. 2020, 353, 2000059. [Google Scholar] [CrossRef]
- Rikagu Oxford Diffraction. CrysAlisPro; Rikagu Oxford Diffraction Inc.: Yarnton, UK, 2020. [Google Scholar]
- Sheldrick, G.M. SHELXT–Integrated space-group and crystal-structure determination. Acta Cryst. 2015, A71, 3–8. [Google Scholar] [CrossRef]
- Hübschle, C.B.; Sheldrick, G.M.; Dittrich, B. ShelXle: A Qt graphical user interface for SHELXL. J. Appl. Crystallogr. 2011, 44, 1281–1284. [Google Scholar] [CrossRef]
- Van Meerloo, J.; Kaspers, G.J.L.; Cloos, J. Cell Sensitivity Assays: The MTT Assay. Methods Mol. Biol. 2011, 731, 237–245. [Google Scholar]
- Mohamed, F.A.M.; Gomaa, H.A.M.; Hendawy, O.M.; Ali, A.T.; Farghaly, H.S.; Gouda, A.M.; Abdelazeem, A.H.; Abdelrahman, M.H.; Trembleau, L.; Youssif, B.G.M. Design, Synthesis, and Biological Evaluation of Novel EGFR Inhibitors Containing 5-Chloro-3-Hydroxymethyl-Indole-2-Carboxamide Scaffold with Apoptotic Antiproliferative Activity. Bioorganic Chem. 2021, 112, 104960. [Google Scholar] [CrossRef]
- Ali, G.M.E.; Ibrahim, D.A.; Elmetwali, A.M.; Ismail, N.S.M. Design, Synthesis and Biological Evaluation of Certain CDK2 Inhibitors Based on Pyrazole and Pyrazolo[1,5-a] Pyrimidine Scaffold with Apoptotic Activity. Bioorganic Chem. 2019, 86, 1–14. [Google Scholar] [CrossRef]
- Qin, X.; Han, X.; Hu, L.; Li, Z.; Geng, Z.; Wang, Z.; Zeng, C.; Xiao, X. Design, Synthesis and Biological Evaluation of Quinoxalin-2(1H)-One Derivatives as EGFR Tyrosine Kinase Inhibitors. Anticancer. Agents Med. Chem. 2015, 15, 267–273. [Google Scholar] [CrossRef]
- AkgÜl, Ö.; ErdoĞan, M.A.; Bİrİm, D.; KayabaŞi, Ç.; GÜndÜz, C.; ArmaĞan, G. Design, Synthesis, Cytotoxic Activity, and Apoptosis Inducing Effects of 4- and N-Substituted Benzoyltaurinamide Derivatives. Turk. J. Chem. 2020, 44, 1674–1693. [Google Scholar] [CrossRef]
- Turner, D.P.; Moussa, O.; Sauane, M.; Fisher, P.B.; Watson, D.K. Prostate-derived ETS factor is a mediator of metastatic potential through the inhibition of migration and invasion in breast cancer. Cancer Res. 2007, 67, 1618–1625. [Google Scholar] [CrossRef]
- Chen, Y.; Lu, B.; Yang, Q.; Fearns, C.; Yates, J.R.; Lee, J.-D. Combined Integrin Phosphoproteomic Analyses and siRNA-based Functional Screening Identified Key Regulators for Cancer Cell Adhesion and Migration. Cancer Res. 2009, 69, 3713–3720. [Google Scholar] [CrossRef] [PubMed]
- Barghash, R.F.; Eldehna, W.M.; Kovalová, M.; Vojáčková, V.; Kryštof, V.; Abdel-Aziz, H.A. One-Pot Three-Component Synthesis of Novel Pyrazolo[3,4-b]Pyridines as Potent Antileukemic Agents. Eur. J. Med. Chem. 2022, 227, 113952. [Google Scholar] [CrossRef]
- Noser, A.A.; Abdelmonsef, A.H.; Salem, M.M. Design, Synthesis and Molecular Docking of Novel Substituted Azepines as Inhibitors of PI3K/Akt/TSC2/mTOR Signaling Pathway in Colorectal Carcinoma. Bioorganic Chem. 2023, 131, 106299. [Google Scholar] [CrossRef]
- Lapillo, M.; Tuccinardi, T.; Martinelli, A.; Macchia, M.; Giordano, A.; Poli, G. Extensive Reliability Evaluation of Docking-Based Target-Fishing Strategies. Int. J. Mol. Sci. 2019, 20, 1023. [Google Scholar] [CrossRef] [PubMed]
- Dhanalakshmi, B.; Anil Kumar, B.M.; Srinivasa Murthy, V.; Srinivasa, S.M.; Vivek, H.K.; Sennappan, M.; Rangappa, S. Design, Synthesis and Docking Studies of Novel 4-Aminophenol-1,2,4-Oxadiazole Hybrids as Apoptosis Inducers against Triple Negative Breast Cancer Cells Targeting MAP Kinase. J. Biomol. Struct. Dyn. 2023, 1–17. [Google Scholar] [CrossRef] [PubMed]
- Dicato, M.; Plawny, L.; Diederich, M. Anemia in cancer. Ann. Oncol. 2010, 21, vii167–vii172. [Google Scholar] [CrossRef]
- HEl Zahabi, S.A.; Nafie, M.S.; Osman, D.; Elghazawy, N.H.; Soliman, D.H.; EL-Helby, A.A.H.; Arafa, R.K. Design, synthesis and evaluation of new quinazolin-4-one derivatives as apoptotic enhancers and autophagy inhibitors with potent antitumor activity. Eur. J. Med. Chem. 2021, 222, 113609. [Google Scholar]
- Boraei, A.T.A.; Eltamany, E.H.; Ali, I.A.I.; Gebriel, S.M.; Nafie, M.S. Synthesis of new substituted pyridine derivatives as potent anti-liver cancer agents through apoptosis induction: In vitro, in vivo, and in silico integrated approaches. Bioorganic Chem. 2021, 111, 104877. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| 3 | 5 | |
|---|---|---|
| CCDC | 2293777 | 2293778 |
| empirical formula | C13H14N4O2S | C24H22N4O4 |
| fw | 290.34 | 430.45 |
| temp (K) | 120(2) | 120(2) |
| Λ (Å) | 1.54184 | 0.71073 |
| cryst syst | Monoclinic | Triclinic |
| space group | P21/c | P 1 |
| a (Å) | 9.23490(10) | 5.4774(3) |
| b (Å) | 19.4168(2) | 9.2031(5) |
| c (Å) | 7.89280(10) | 10.6234(8) |
| A (deg) | 90 | 93.995(5) |
| β (deg) | 103.8110(10) | 94.008(6) |
| γ (deg) | 90 | 94.201(5) |
| V (Å3) | 1374.36(3) | 531.26(6) |
| Z | 4 | 1 |
| ρcalc (Mg/m3) | 1.403 | 1.345 |
| μ(Mo Kα) (mm−1) | 2.168 | 0.094 |
| No. reflns. | 16497 | 8485 |
| Unique reflns. | 2884 | 2637 |
| Completeness to θ = 67.684° | 99.8% | |
| Completeness to θ = 25.242° | 99.9% | |
| GOOF (F2) | 1.058 | 1.046 |
| Rint | 0.0187 | 0.0300 |
| R1 a (I ≥ 2σ) | 0.0279 | 0.0469 |
| wR2 b (I ≥ 2σ) | 0.0780 | 0.1123 |
| Compounds | IC50 ± SD [μM] |
|---|---|
| 1 | 19.7 ± 2.31 |
| 3 | 10.2 ± 0.53 |
| 5 | 2.73 ± 0.14 |
| 6 | 9.42 ± 0.57 |
| 8 | 4.38 ± 0.23 |
| 10 | 25.4 ± 1.54 |
| 12 | 7.03 ± 0.37 |
| Staurosporine | 8.32 ± 0.43 |
| Compound | %Closure *, MCF-7 |
|---|---|
| 5 | 48.88 # ± 2.7 |
| 8 | 60.74 # ± 3.43 |
| 12 | 51.85 # ± 2.92 |
| Untreated control | 94.07 ± 5.5 |
| Compound | Annexin V/PI Staining | DNA Content | ||||||
|---|---|---|---|---|---|---|---|---|
| Total | Early | Late | Necrosis | %G0-G1 | %S | %G2/M | %Pre-G1 | |
| Cont.MCF7 | 1.27 | 0.29 | 0.11 | 0.87 | 52.91 | 41.33 | 5.76 | 1.27 |
| 5 | 39.26 | 7.11 | 22.24 | 9.91 | 39.07 | 56.19 | 4.74 | 39.26 |
| 8 | 24.38 | 2.27 | 13.45 | 8.66 | 47.10 | 46.31 | 6.57 | 24.38 |
| 12 | 37.05 | 4.59 | 17.44 | 15.02 | 59.33 | 38.10 | 2.55 | 37.05 |
| Compound | IC50 [μM] ± SD * | |||||
|---|---|---|---|---|---|---|
| PI3K-α | PI3K-β | PI3K-δ | CDK2 | AKT-1 | EGFR | |
| 5 | 1.73 ± 0.1 | 2.27 ± 0.11 | 2.68 ± 0.15 | 0.156 ± 0.01 | 0.602 ± 0.03 | 0.058 ± 0.029 |
| LY294002 | 8.52 ± 0.48 | 0.44 ± 0.02 | 0.85 ± 0.05 | NT | NT | NT |
| erlotinib | NT | NT | NT | 0.173 ± 0.01 | NT | 0.038 ± 0.019 |
| A-674563 | NT | NT | NT | NT | 0.26 ± 0.01 | NT |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Salama, E.E.; Youssef, M.F.; Aboelmagd, A.; Boraei, A.T.A.; Nafie, M.S.; Haukka, M.; Barakat, A.; Sarhan, A.A.M. Discovery of Potent Indolyl-Hydrazones as Kinase Inhibitors for Breast Cancer: Synthesis, X-ray Single-Crystal Analysis, and In Vitro and In Vivo Anti-Cancer Activity Evaluation. Pharmaceuticals 2023, 16, 1724. https://doi.org/10.3390/ph16121724
Salama EE, Youssef MF, Aboelmagd A, Boraei ATA, Nafie MS, Haukka M, Barakat A, Sarhan AAM. Discovery of Potent Indolyl-Hydrazones as Kinase Inhibitors for Breast Cancer: Synthesis, X-ray Single-Crystal Analysis, and In Vitro and In Vivo Anti-Cancer Activity Evaluation. Pharmaceuticals. 2023; 16(12):1724. https://doi.org/10.3390/ph16121724
Chicago/Turabian StyleSalama, Eid E., Mohamed F. Youssef, Ahmed Aboelmagd, Ahmed T. A. Boraei, Mohamed S. Nafie, Matti Haukka, Assem Barakat, and Ahmed A. M. Sarhan. 2023. "Discovery of Potent Indolyl-Hydrazones as Kinase Inhibitors for Breast Cancer: Synthesis, X-ray Single-Crystal Analysis, and In Vitro and In Vivo Anti-Cancer Activity Evaluation" Pharmaceuticals 16, no. 12: 1724. https://doi.org/10.3390/ph16121724
APA StyleSalama, E. E., Youssef, M. F., Aboelmagd, A., Boraei, A. T. A., Nafie, M. S., Haukka, M., Barakat, A., & Sarhan, A. A. M. (2023). Discovery of Potent Indolyl-Hydrazones as Kinase Inhibitors for Breast Cancer: Synthesis, X-ray Single-Crystal Analysis, and In Vitro and In Vivo Anti-Cancer Activity Evaluation. Pharmaceuticals, 16(12), 1724. https://doi.org/10.3390/ph16121724