

Novel Coumarin Derivatives as Potential Urease Inhibitors for Kidney Stone Prevention and Antiulcer Therapy: From Synthesis to In Vivo Evaluation

 , , ,
, , ,
Abstract
:1. Introduction
2. Results and Discussion
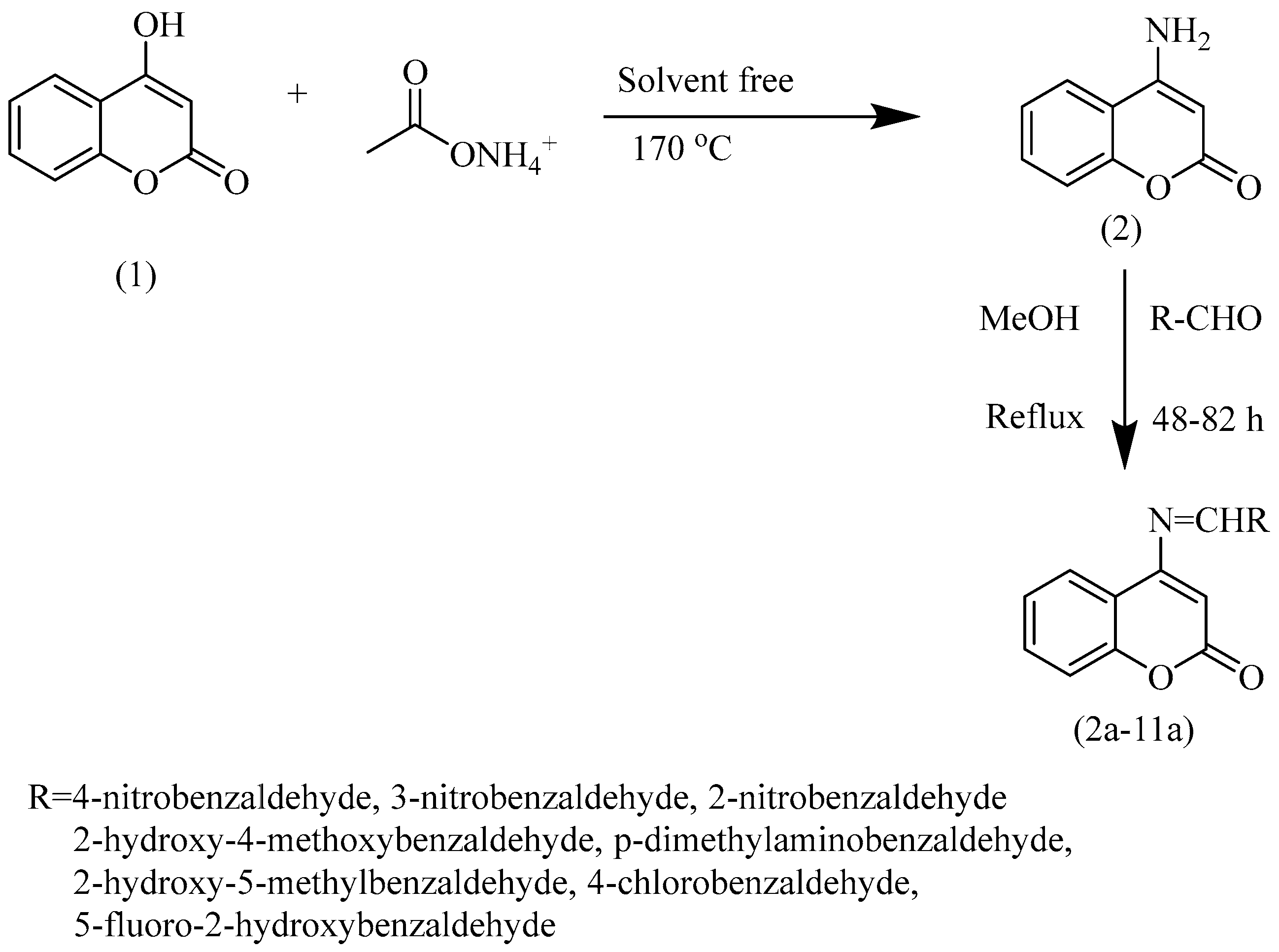
- 4-aminocoumarin
- Schiff bases (2a–11a)
- (E)-4-(5-fluoro-2-hydroxybenzylideneamino)-2H-chromen-2-one (2a):
- (E)-4-(3-nitrobenzylideneamino)-2H-chromen-2-one (3a):
- (E)-4-(2-hydroxy-4-methoxybenzylideneamino)-2H-chromen-2-one (4a):
- (E)-4-(4-nitrobenzylideneamino)-2H-chromen-2-one (5a):
- (E)-4-(4-hydroxybenzylideneamino)-2H-chromen-2-one (6a):
- (E)-4-(2-hydroxy-5-methylbenzylideneamino)-2H-chromen-2-one (7a):
- (E)-4-(2-nitrobenzylideneamino)-2H-chromen-2-one (8a):
- (E)-4-(4-chlorobenzylideneamino)-2H-chromen-2-one (9a):
- (E)-4-(5-bromo-2-hydroxybenzylideneamino)-2H-chromen-2-one (10a):
- (E)-4-(4-ethoxybenzylideneamino)-2H-chromen-2-one (11a):
2.1. UV-Vis Analysis
2.2. FT-IR Analysis
2.3. NMR Spectral Analysis
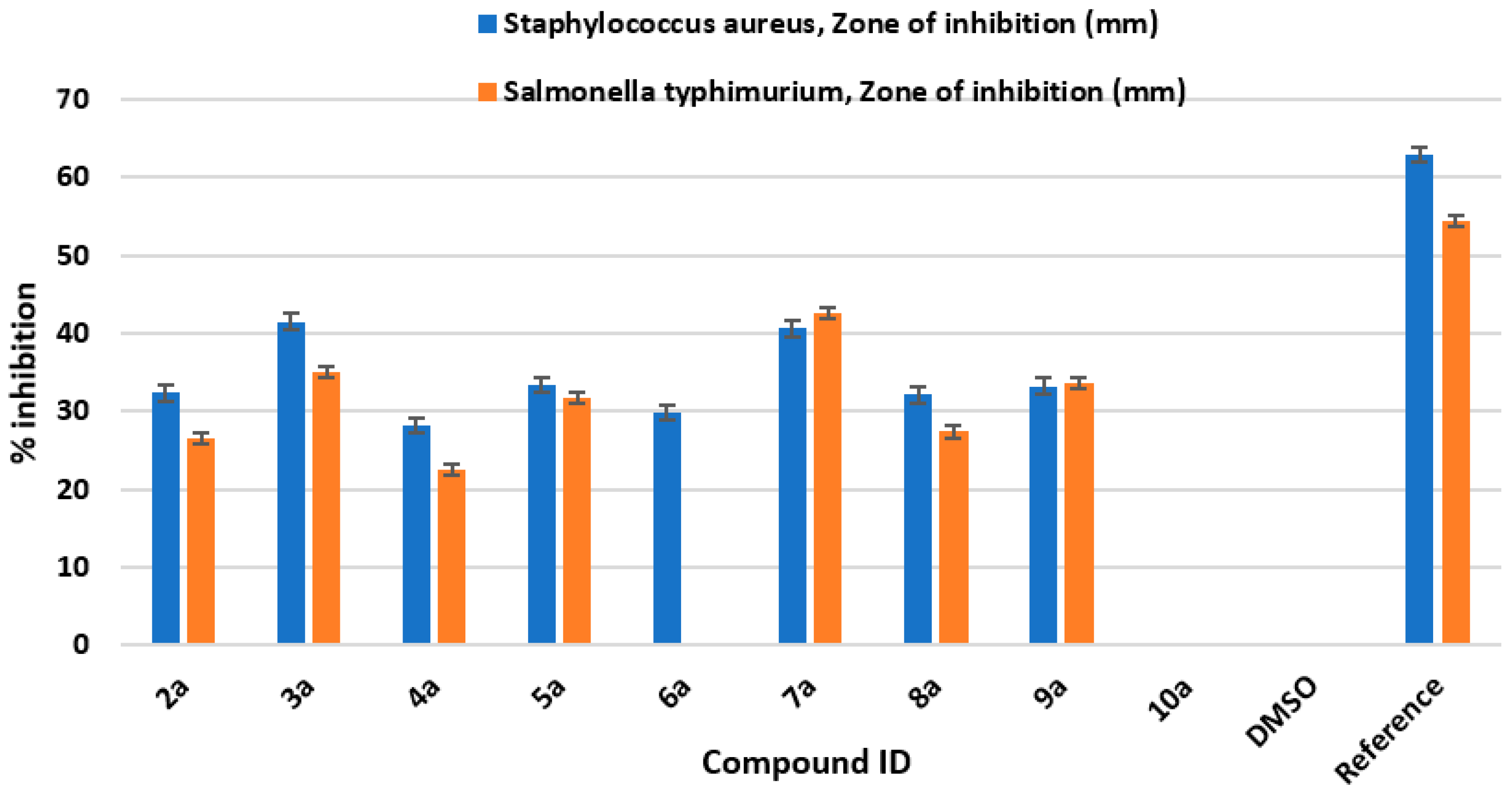
2.4. Antibacterial Activity
2.5. In Vitro Urease Inhibition
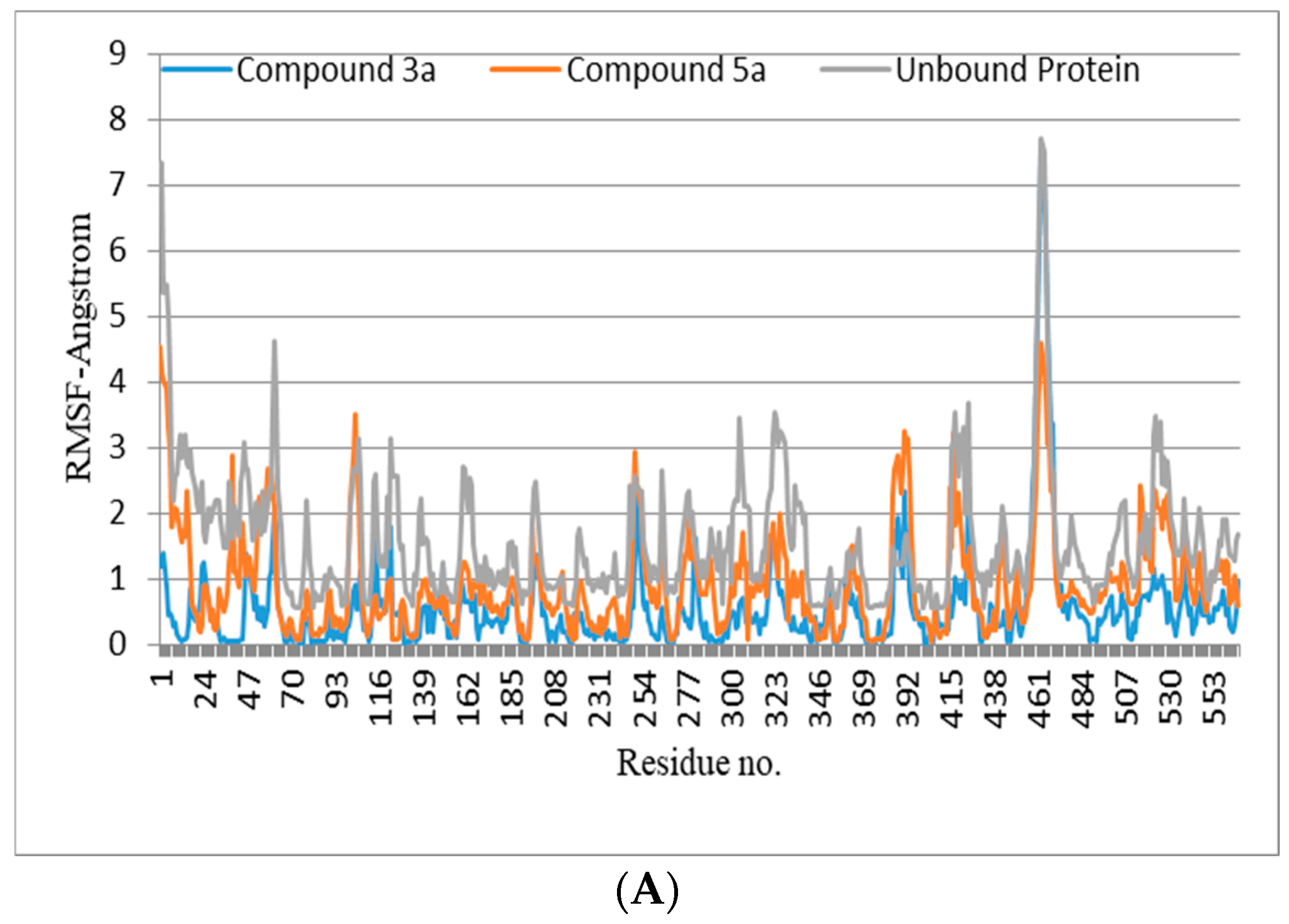
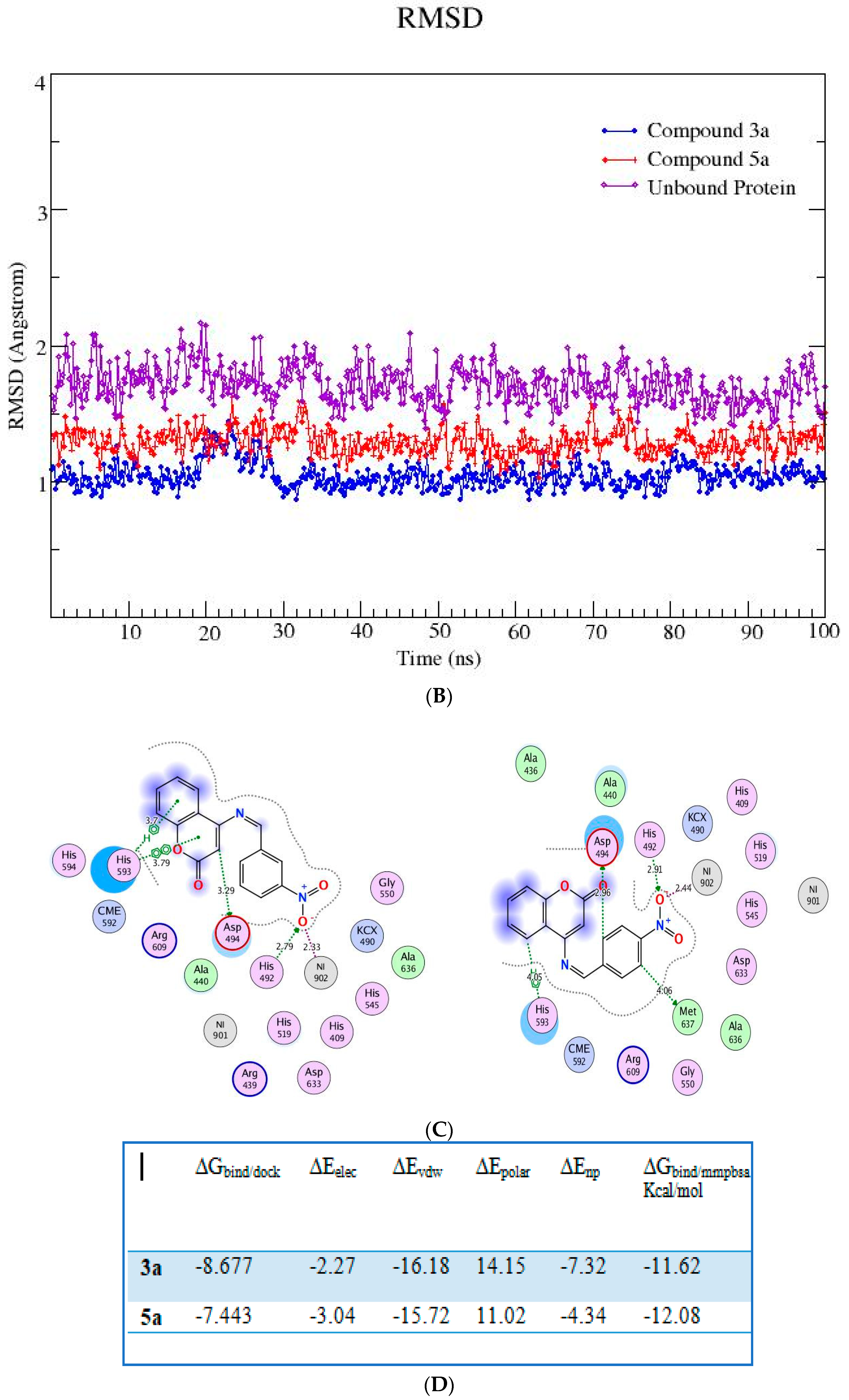
2.6. In Silico Studies
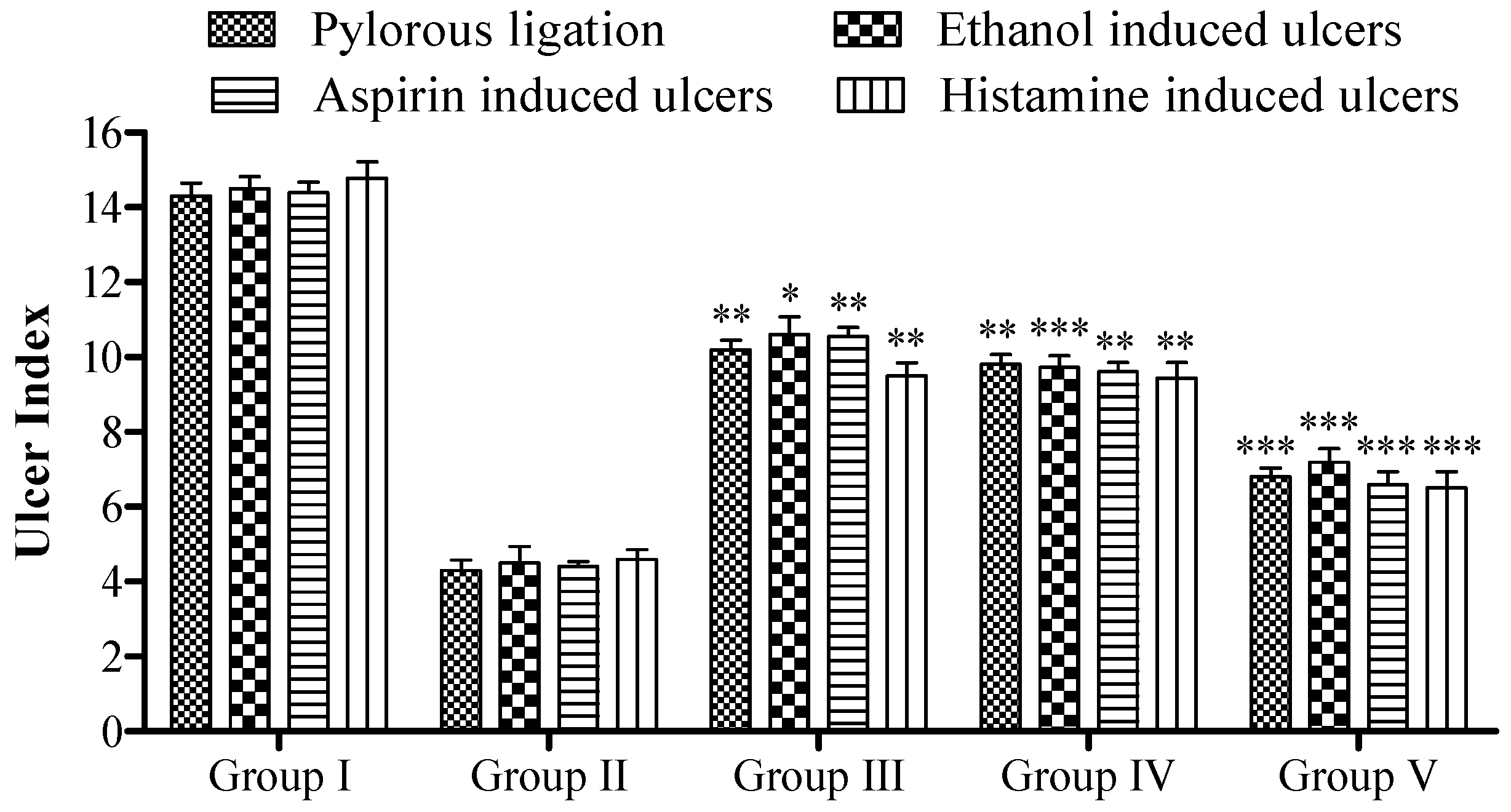
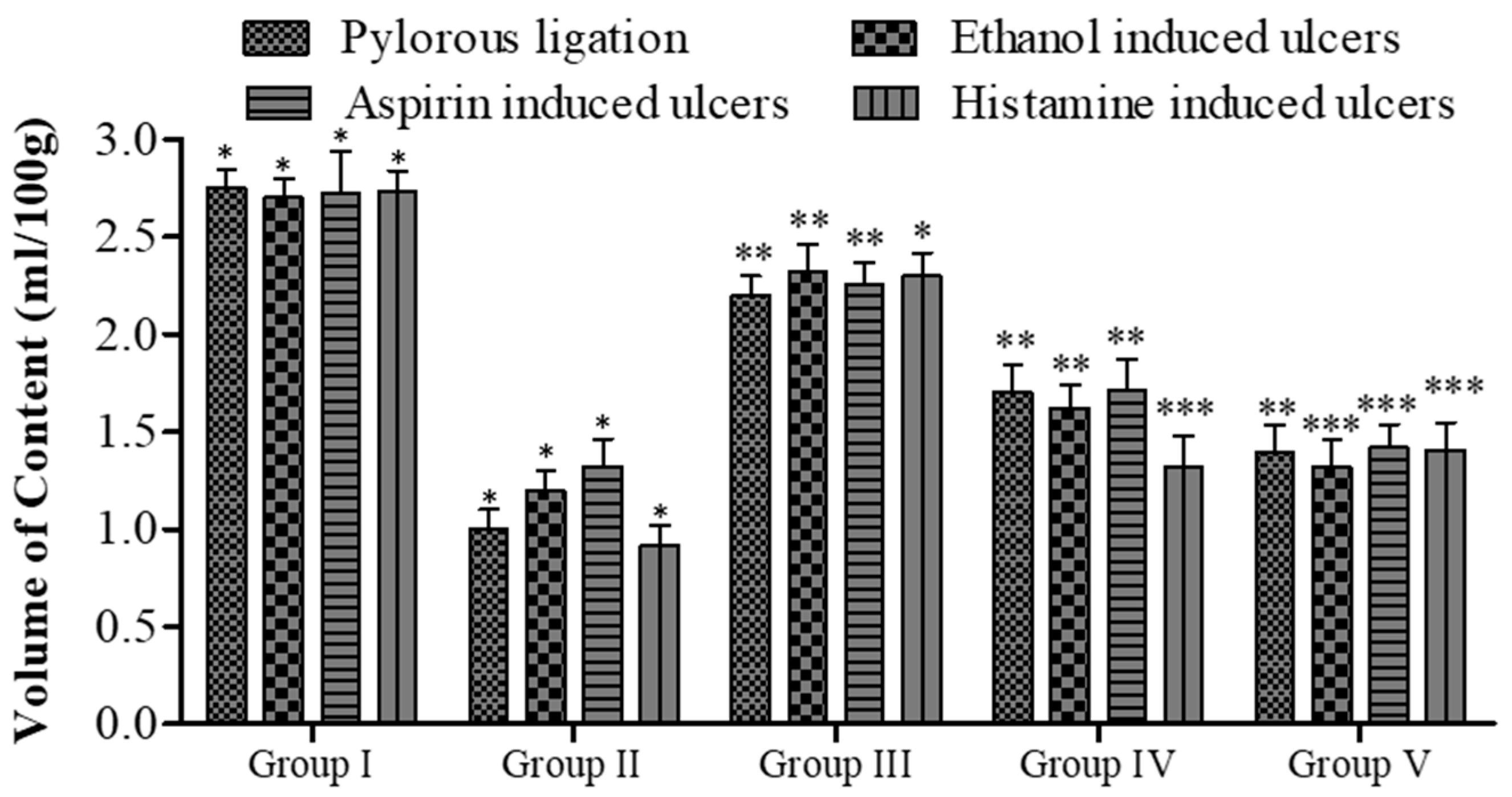
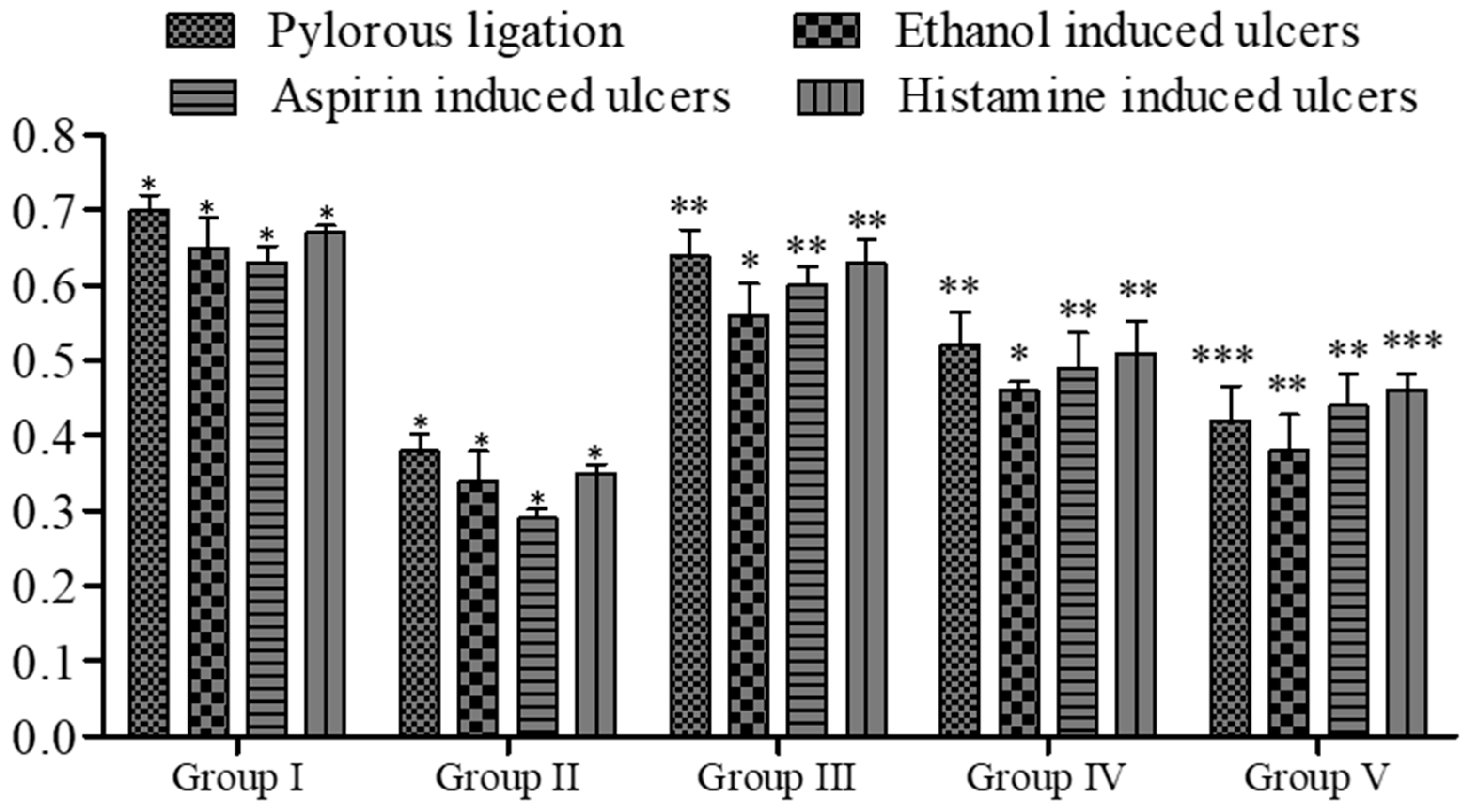
2.7. In Vivo Pharmacology
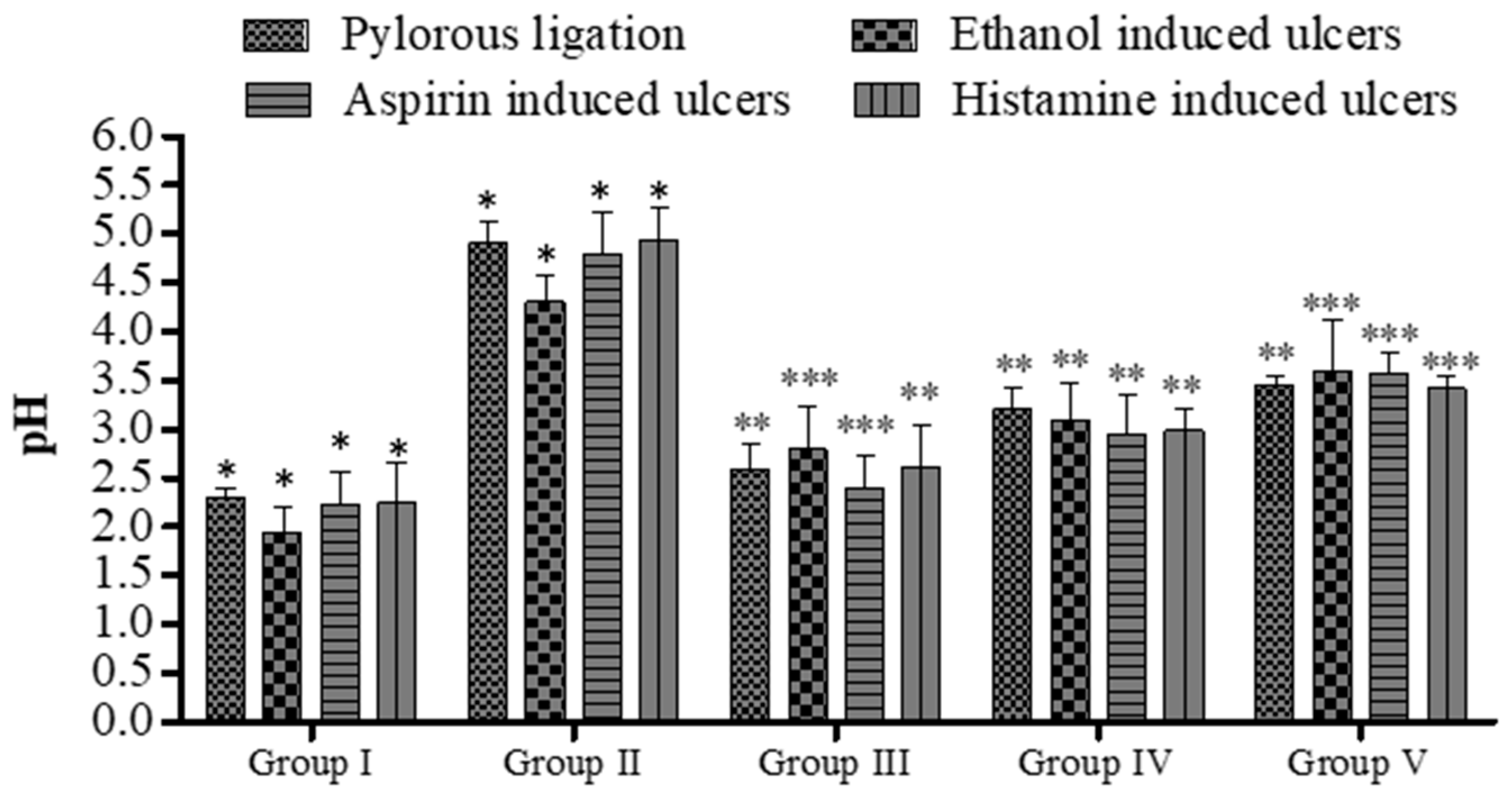
2.7.1. Pylorus Ligation Activity
2.7.2. Ethanol-Induced Ulcer
2.7.3. Aspirin-Induced Ulcer
2.7.4. Histamine-Induced Ulcer
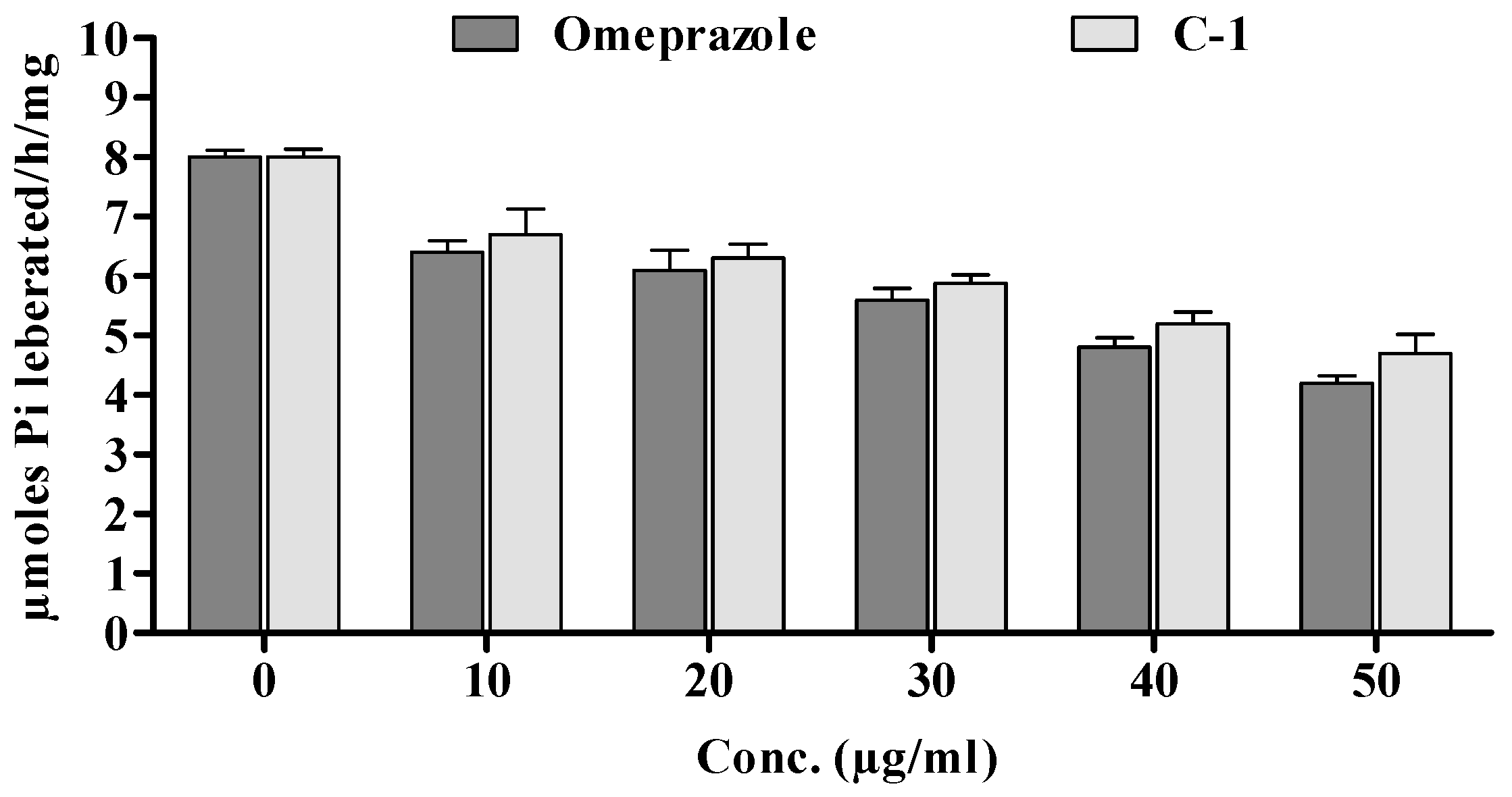
2.7.5. H+–K+ ATPase Assay
- Purity of the synthesized compounds
3. Materials and Methods
3.1. Chemicals and Reagents
3.2. Experimental Synthesis of 4-Aminocoumarin and Its Schiff Base Derivatives
3.3. Antibacterial Activity
3.4. Urease Inhibition Assay
3.5. In Silico Studies
Selection and Refinement of Protein Structure
3.6. Validation of Model, Active Site Determination, and Molecular Docking Studies
3.7. MD Simulation Studies of Protein–Ligand Complexes as Well as Rescoring of Binding Energies
3.8. In Vivo Pharmacology
Experimental Animals
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Konieczna, I.; Żarnowiec, P.; Kwinkowski, M.; Kolesińska, B.; Frączyk, J.; Kamiński, Z.; Kaca, W. Bacterial urease and its role in long-lasting human diseases. Curr. Protein Pept. Sci. 2012, 13, 789–806. [Google Scholar] [CrossRef]
- Vekariya, R.H.; Patel, H.D. Recent advances in the synthesis of coumarin derivatives via knoevenagel condensation: A review. Synth. Commun. 2014, 44, 2756–2788. [Google Scholar] [CrossRef]
- Stefanachi, A.; Leonetti, F.; Pisani, L.; Catto, M.; Carotti, A. Coumarin: A natural, privileged and versatile scaffold for bioactive compounds. Molecules 2018, 23, 250. [Google Scholar] [CrossRef] [PubMed]
- Valizadeh, H.; Shockravi, A. An efficient procedure for the synthesis of coumarin derivatives using TiCl4 as catalyst under solvent-free conditions. Tetrahedron Lett. 2005, 46, 3501–3503. [Google Scholar] [CrossRef]
- Abdou, M.M. 3-acetyl-4-hydroxycoumarin: Synthesis, reactions and applications. Arab. J. Chem. 2017, 10, S3664–S3675. [Google Scholar] [CrossRef]
- Kostova, I.; Bhatia, S.; Grigorov, P.; Balkansky, S.; Parmar, S.V.; Prasad, K.A.; Saso, l. Coumarins as antioxidants. Curr. Med. Chem. 2011, 18, 3929–3951. [Google Scholar] [CrossRef]
- Chohan, Z.H.; Shaikh, A.U.; Rauf, A.; Supuran, C.T. Antibacterial, antifungal and cytotoxic properties of novel N-substituted sulfonamides from 4-hydroxycoumarin. J. Enzyme Inhib. Med. Chem. 2006, 21, 741–748. [Google Scholar] [CrossRef]
- Mitra, A.K.; Misra, S.K.; Patra, A. New synthesis of 3-alkyl coumarins. Synth. Commun. 1980, 10, 915–919. [Google Scholar] [CrossRef]
- Bose, D.S.; Rudradas, A.; Babu, M.H. The indium (III) chloride-catalyzed von Pechmann reaction: A simple and effective procedure for the synthesis of 4-substituted coumarins. Tetrahedron Lett. 2002, 43, 9195–9197. [Google Scholar] [CrossRef]
- Ghosh, R.; Singha, P.S.; Das, L.K.; Ghosh, D.; Firdaus, S.B. Antiinflammatory activirty of natural coumarin compounds from plants of the Indo-Gangetic plain. AIMS Mol. Sci. 2010, 10, 79–98. [Google Scholar] [CrossRef]
- Satyanarayana, V.; Sreevani, P.; Sivakumar, A.; Vijayakumar, V. Synthesis and antimicrobial activity of new Schiff bases containing coumarin moiety and their spectral characterization. Arkovic 2008, 17, 221–233. [Google Scholar] [CrossRef]
- Paul, M.K.; Singh, Y.D.; Dey, A.; Saha, S.K.; Anwar, S.; Chattopadhyay, A.P. Coumarin based emissive rod shaped new schiff base mesogens and their zinc (II) complexes: Synthesis, photophysical, mesomorphism, gelation and DFT studies. Liq. Cryst. 2016, 43, 343–360. [Google Scholar] [CrossRef]
- Creaven, B.S.; Devereux, M.; Karcz, D.; Kellett, A.; McCann, M.; Noble, A.; Walsh, M. Copper (II) complexes of coumarin-derived Schiff bases and their anti-candida activity. J. Inorg. Biochem. 2009, 103, 1196–1203. [Google Scholar] [CrossRef] [PubMed]
- Kulkarni, A.; Avaji, P.G.; Bagihalli, G.B.; Patil, S.A.; Badami, P.S. Synthesis, spectral, electrochemical and biological studies of Co (II), Ni (II) and Cu (II) complexes with Schiff bases of 8-formyl-7-hydroxy-4-methyl coumarin. J. Coord. Chem. 2009, 62, 481–492. [Google Scholar] [CrossRef]
- Al-Majedy, Y.K.; Kadhum, A.A.H.; Al-Amiery, A.A.; Mohamad, A.B. Coumarins: The antimicrobial agents. Syst. Rev. Pharm. 2017, 8, 62–70. [Google Scholar] [CrossRef]
- Baboukani, A.R.; Sharifi, E.; Akhavan, S.; Saatchi, A. Co complexes as a corrosion inhibitor for 316 l stainless steel in H2SO4 solution. J. Mater. Sci. Chem. Eng. 2016, 4, 28–35. [Google Scholar]
- Kadhum, A.A.H.; Al-Amiery, A.A.; Musa, A.Y.; Mohamad, A.B. The antioxidant activity of new coumarin derivatives. Int. J. Mol. Sci. 2011, 12, 5747–5761. [Google Scholar] [CrossRef] [PubMed]
- Hassan, S.T.; Žemlička, M. Plant-derived urease inhibitors as alternative chemotherapeutic agents. Arch. Pharm. 2016, 349, 507–522. [Google Scholar] [CrossRef]
- Stamboliyska, B.; Janevska, V.; Shivachev, B.; Nikolova, R.P.; Stojkovic, G.; Mikhova, B.; Popovski, E. Experimental and theoretical investigation of the structure and nucleophilic properties of 4-aminocoumarin. Arkivoc 2010, 10, 62–76. [Google Scholar]
- Arshad, N.; Perveen, F.; Saeed, A.; Channar, P.A.; Farooqi, S.I.; Larik, F.A.; Ismail, H.; Mirza, B. Spectroscopic, molecular docking and structural activity studies of (E)-N′-(substituted benzylidene/methylene) isonicotinohydrazide derivatives for DNA binding and their biological screening. J. Mol. Struct. 2017, 1139, 371–380. [Google Scholar] [CrossRef]
- Ali, M.; Bukhari, S.M.; Zaidi, A.; Khan, F.A.; Rashid, U.; Tahir, N.; Rabbani, B.; Farooq, U. Inhibition profiling of urease and carbonic anhydrase II by high throughput screening and molecular docking studies of structurally diverse organic compounds. Lett. Drug Des. Discov. 2021, 18, 299–312. [Google Scholar] [CrossRef]
- Vilar, S.; Cozza, G.; Moro, S. Medicinal chemistry and the molecular operating environment (MOE): Application of QSAR and molecular docking to drug discovery. Curr. Top. Med. Chem. 2008, 8, 1555–1572. [Google Scholar] [CrossRef] [PubMed]
- Case, D.A.; Aktulga, H.M.; Belfon, K.; Ben-Shalom, I.; Brozell, S.R.; Cerutti, D.; Cheatham, T., III; Cisneros, G.; Cruzeiro, V.; Darden, T. Amber 2021; University of California Press: San Francisco, CA, USA, 2021. [Google Scholar]
- Zaman, Z.; Khan, S.; Nouroz, F.; Farooq, U.; Urooj, A. Targeting protein tyrosine phosphatase to unravel possible inhibitors for Streptococcus pneumoniae using molecular docking, molecular dynamics simulations coupled with free energy calculations. Life Sci. 2021, 264, 118621. [Google Scholar] [CrossRef] [PubMed]
- Mahmood, F.; Khan, J.A.; Mahnashi, M.H.; Jan, M.S.; Javed, M.A.; Rashid, U.; Sadiq, A.; Hussan, S.S.; Bungau, S. Anti-Inflammatory, Analgesic and Antioxidant Potential of New (2S,3S)-2-(4-isopropylbenzyl)-2-methyl-4-nitro-3-phenylbutanals and Their Corresponding Carboxylic Acids through In Vitro, In Silico and In Vivo Studies. Molecules 2022, 27, 4068. [Google Scholar] [CrossRef]
- Shah, S.M.M.; Ullah, F.; Shah, S.M.H.; Zahoor, M.; Sadiq, A. Analysis of chemical constituents and ntinociceptive potential of essential oil of Teucrium Stocksianum bioss collected from the North West of Pakistan. BMC Complement. Med. 2012, 12, 244. [Google Scholar]
- Wang, X.-Y.; Yin, J.-Y.; Zhao, M.-M.; Liu, S.-Y.; Nie, S.-P.; Xie, M.-Y. Gastroprotective activity of polysaccharide from Hericium erinaceus against ethanol-induced gastric mucosal lesion and pylorus ligation-induced gastric ulcer, and its antioxidant activities. Carbohydr. Polym. 2018, 186, 100–109. [Google Scholar] [CrossRef]
- Al-Qarawi, A.A.; Abdel-Rahman, H.; Ali, B.H. The ameliorative effect of dates (Phoenix dactylifera L.) on ethanol-induced gastric ulcer in rats. J. Ethnopharmacol. 2005, 98, 313–317. [Google Scholar] [CrossRef]
- Choi, J.-I.; Raghavendran, H.R.B.; Sung, N.-Y.; Kim, J.-H.; Chun, B.S.; Ahn, D.H.; Choi, H.-S.; Kang, K.-W.; Lee, J.-W. Effect of fucoidan on asprin-induced stomachulceration in rats. Chem. Biol. Interact. 2010, 183, 249–254. [Google Scholar] [CrossRef]
- Bodhankar, S.L.; Jain, B.B.; Ahire, B.P.; Daude, R.B.; Shitole, P.P. The effect of rabeprazole and its isomers on asprin and histamine-induced ulcers in rats. Indian J. Pharmacol. 2006, 38, 357–358. [Google Scholar] [CrossRef]
- Razzaq, S.; Minhas, A.M.; Qazi, N.G.; Nadeem, H.; Khan, A.U.; Ali, F.; ul Hassan, S.S.; Bangau, S. Novel Isoxazole Derivative Attenuates Ethanol-Induced Gastric Mucosal Injury through Inhibition of H+/K+-ATPase Pump, Oxidative Stress and Inflammatory Pathways. Molecules 2022, 27, 5065. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| 1st Reactant (Equiv.) | 2nd Reactant (Equiv.) | Solvents | Temp. (°C) | Time (h) | Results |
|---|---|---|---|---|---|
| 1 | 1 | Ethoxyethanol | 170 | 48 | Too many side products are present |
| 1 | 2 | Glacial acetic acid | 180 | 72 | Too many side products are present |
| 1 | 2 | Solvent-free | 130 | 05 | No reaction |
| 1 | 5 | Solvent-free | 170 | 14–15 | 70% yield |
| 1st Reactant (Equiv.) | 2nd Reactant (Equiv.) | Solvents | Temp. (°C) | Time (h) | Results |
|---|---|---|---|---|---|
| 1 | 1 | Ethanol | 170 | 48 | 10% yield |
| 1 | 1.5 | Ethanol + glacial acetic acid (3–4 drops) | 170 | 72 | 10% yield |
| 1 | 1.5 | Methanol + glacial acetic acid (3–4 drops) | 170 | 72 | 15% yield |
| 1 | 1 | Analytical grade methanol | 170 | 36–82 Continuous reflux | 65–75% yield |
| Compound No. | % Inhibition ± STD | IC50 ± SEM (µM) |
|---|---|---|
| 3a | 64.0 ± 0.42 | 0.412 ± 0.10 |
| 5a | 77.7 ± 0.64 | 0.322 ± 0.13 |
| 7a | 60.2 ± 0.32 | 0.112 ± 0.26 |
| Thiourea | 82.0 ± 0.15 | 0.140 ± 0.22 |
| Compounds ID | Binding Kcal/mol | Ni Binding |
|---|---|---|
| 2a | −3.547 | Absent |
| 3a | −8.677 | Present (Ni-O 2.3 Å) |
| 4a | −5.180 | Present (Ni-O 2.5 Å) |
| 5a | −7.443 | Present (Ni-O 2.4 Å) |
| 6a | −5.892 | Present (Ni-O 2.7 Å) |
| 7a | −6.834 | Present (Ni-O 2.3 Å) |
| 8a | −4.131 | Absent |
| 9a | −6.967 | Present (Ni-O 2.6 Å) |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Shahzadi, K.; Bukhari, S.M.; Zaidi, A.; Wani, T.A.; Jan, M.S.; Zargar, S.; Rashid, U.; Farooq, U.; Khushal, A.; Khan, S. Novel Coumarin Derivatives as Potential Urease Inhibitors for Kidney Stone Prevention and Antiulcer Therapy: From Synthesis to In Vivo Evaluation. Pharmaceuticals 2023, 16, 1552. https://doi.org/10.3390/ph16111552
Shahzadi K, Bukhari SM, Zaidi A, Wani TA, Jan MS, Zargar S, Rashid U, Farooq U, Khushal A, Khan S. Novel Coumarin Derivatives as Potential Urease Inhibitors for Kidney Stone Prevention and Antiulcer Therapy: From Synthesis to In Vivo Evaluation. Pharmaceuticals. 2023; 16(11):1552. https://doi.org/10.3390/ph16111552
Chicago/Turabian StyleShahzadi, Kiran, Syed Majid Bukhari, Asma Zaidi, Tanveer A. Wani, Muhammad Saeed Jan, Seema Zargar, Umer Rashid, Umar Farooq, Aneela Khushal, and Sara Khan. 2023. "Novel Coumarin Derivatives as Potential Urease Inhibitors for Kidney Stone Prevention and Antiulcer Therapy: From Synthesis to In Vivo Evaluation" Pharmaceuticals 16, no. 11: 1552. https://doi.org/10.3390/ph16111552
APA StyleShahzadi, K., Bukhari, S. M., Zaidi, A., Wani, T. A., Jan, M. S., Zargar, S., Rashid, U., Farooq, U., Khushal, A., & Khan, S. (2023). Novel Coumarin Derivatives as Potential Urease Inhibitors for Kidney Stone Prevention and Antiulcer Therapy: From Synthesis to In Vivo Evaluation. Pharmaceuticals, 16(11), 1552. https://doi.org/10.3390/ph16111552