
Virtual Prospection of Marine Cyclopeptides as Therapeutics by Means of Conceptual DFT and Computational ADMET




Abstract
:
1. Introduction
2. Results and Discussion
3. Materials and Methods
3.1. Conceptual DFT Studies
3.2. Computational ADMET
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Kim, S.K. Encyclopedia of Marine Biotechnology; Wiley-Blackwell: Hoboken, NJ, USA, 2020. [Google Scholar]
- Bertrand, B.; Munoz-Garay, C. Marine Antimicrobial Peptides: A Promising Source of New Generation Antibiotics and Other Bio-active Molecules. Int. J. Pept. Res. Ther. 2018, 25, 1441–1450. [Google Scholar] [CrossRef]
- Negi, B.; Kumar, D.; Rawat, D.S. Marine Peptides as Anticancer Agents: A Remedy to Mankind by Nature. Curr. Protein Pept. Sci. 2017, 18, 885–904. [Google Scholar] [CrossRef] [PubMed]
- Bottegoni, G.; Cavalli, A. Computational Methods in Multitarget Drug Discovery. In Design of Hybrid Molecules for Drug Development; Elsevier: Oxford, UK, 2017; pp. 239–258. [Google Scholar]
- Khan, F.; Yadav, D.K.; Maurya, A.; Srivastava, S.K. Modern Methods & Web Resources in Drug Design & Discovery. Lett. Drug Des. Discov. 2011, 8, 469–490. [Google Scholar] [CrossRef]
- Parr, R.; Yang, W. Density-Functional Theory of Atoms and Molecules; Oxford University Press: New York, NY, USA, 1989. [Google Scholar]
- Chermette, H. Chemical Reactivity Indexes in Density Functional Theory. J. Comput. Chem. 1999, 20, 129–154. [Google Scholar] [CrossRef]
- Geerlings, P.; De Proft, F.; Langenaeker, W. Conceptual Density Functional Theory. Chem. Rev. 2003, 103, 1793–1874. [Google Scholar] [CrossRef]
- Geerlings, P.; Chamorro, E.; Chattaraj, P.K.; De Proft, F.; Gázquez, J.L.; Liu, S.; Morell, C.; Toro-Labbé, A.; Vela, A.; Ayers, P. Conceptual Density Functional Theory: Status, Prospects, Issues. Theor. Chem. Acc. 2020, 139, 36. [Google Scholar] [CrossRef]
- Toro-Labbé, A. (Ed.) Theoretical Aspects of Chemical Reactivity; Elsevier Science: Amsterdam, The Netherlands, 2007. [Google Scholar]
- Chattaraj, P.K. (Ed.) Chemical Reactivity Theory—A Density Functional View; CRC Press. Taylor & Francis Group: Boca Raton, FL, USA, 2009. [Google Scholar]
- Chakraborty, D.; Chattaraj, P.K. Conceptual Density Functional Theory Based Electronic Structure Principles. Chem. Sci. 2021, 12, 6264–6279. [Google Scholar] [CrossRef]
- Heidar-Zadeh, F.; Ayers, P.W.; Carbó-Dorca, R. A Statistical Perspective on Molecular Similarity. In Conceptual Density Functional Theory and Its Application in the Chemical Domain; Islam, N., Kaya, S., Eds.; Apple Academic Press: Toronto, ON, Canada, 2018; Chapter 10; pp. 263–273. [Google Scholar]
- Carbó, R.; Leyda, L.; Arnau, M. How Similar is a Molecule to Another? An Electron Density Measure of Similarity between Two Molecular Structures. Int. J. Quantum Chem. 1980, 17, 1185–1189. [Google Scholar] [CrossRef]
- Ayers, P.; Parr, R. The Variational Principles for Describing Chemical Reactions: The Fukui Function and Chemical Hardness Revisited. J. Am. Chem. Soc. 2000, 122, 2010–2018. [Google Scholar] [CrossRef]
- Poater, A.; Saliner, A.G.; Carbó-Dorca, R.; Poater, J.; Solà, M.; Cavallo, L.; Worth, A.P. Modeling the Structure-Property Relationships of Nanoneedles: A Journey Toward Nanomedicine. J. Comput. Chem. 2009, 30, 275–284. [Google Scholar] [CrossRef]
- Poater, A.; Saliner, A.G.; Solá, M.; Cavallo, L.; Worth, A.P. Computational Methods to Predict the Reactivity of Nanoparticles Through Structure-Property Relationships. Expert Opin. Drug Deliv. 2010, 7, 295–305. [Google Scholar] [CrossRef] [PubMed]
- Tan, L.T. Marine Cyanobacteria. In Bioactive Natural Products; Elsevier: Amsterdam, The Netherlands, 2012; pp. 67–110. [Google Scholar]
- Gutiérrez, M.; Suyama, T.L.; Engene, N.; Wingerd, J.S.; Matainaho, T.; Gerwick, W.H. Apratoxin D, a Potent Cytotoxic Cyclodepsipeptide from Papua New Guinea Collections of the Marine Cyanobacteria Lyngbya majuscula and Lyngbya sordida. J. Nat. Prod. 2008, 71, 1099–1103. [Google Scholar] [CrossRef] [PubMed]
- Liu, L.; Rein, K.S. New Peptides Isolated from Lyngbya Species: A Review. Mar. Drugs 2010, 8, 1817–1837. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ahmad, I.Z.; Parvez, S.; Tabassum, H. Cyanobacterial Peptides with Respect to Anticancer Activity: Structural and Functional Perspective. In Bioactive Natural Products; Elsevier: Amsterdam, The Netherlands, 2020; pp. 345–388. [Google Scholar]
- Tidgewell, K.; Engene, N.; Byrum, T.; Media, J.; Doi, T.; Valeriote, F.A.; Gerwick, W.H. Evolved Diversification of a Modular Natural Product Pathway: Apratoxins F and G, Two Cytotoxic Cyclic Depsipeptides from a Palmyra Collection of Lyngbya bouillonii. ChemBioChem 2010, 11, 1458–1466. [Google Scholar] [CrossRef]
- Flores-Holguín, N.; Frau, J.; Glossman-Mitnik, D. A Fast and Simple Evaluation of the Chemical Reactivity Properties of the Pristinamycin Family of Antimicrobial Peptides. Chem. Phys. Lett. 2020, 739, 137021. [Google Scholar] [CrossRef]
- Flores-Holguín, N.; Frau, J.; Glossman-Mitnik, D. Conceptual DFT-Based Computational Peptidology of Marine Natural Compounds: Discodermins A–H. Molecules 2020, 25, 4158. [Google Scholar] [CrossRef]
- Flores-Holguín, N.; Frau, J.; Glossman-Mitnik, D. Virtual Screening of Marine Natural Compounds by Means of Chemoinformatics and CDFT-Based Computational Peptidology. Mar. Drugs 2020, 18, 478. [Google Scholar] [CrossRef]
- Flores-Holguín, N.; Frau, J.; Glossman-Mitnik, D. Conceptual DFT as a Helpful Chemoinformatics Tool for the Study of the Clavanin Family of Antimicrobial Marine Peptides. In Density Functional Theory; De Lazaro, S.R., Da Silveira Lacerda, L.H., Pontes Ribeiro, R.A., Eds.; IntechOpen: London, UK, 2021; Chapter 3; pp. 57–67. [Google Scholar]
- Flores-Holguín, N.; Ortega-Castro, J.; Frau, J.; Glossman-Mitnik, D. Conceptual DFT-Based Computational Peptidology, Pharmacokinetics Study and ADMET Report of the Veraguamides A–G Family of Marine Natural Drugs. Mar. Drugs 2022, 20, 97. [Google Scholar] [CrossRef]
- Lewars, E. Computational Chemistry—Introduction to the Theory and Applications of Molecular and Quantum Mechanics; Kluwer Academic Publishers: Dordrecht, The Netherlands, 2003. [Google Scholar]
- Young, D. Computational Chemistry—A Practical Guide for Applying Techniques to Real-World Problems; John Wiley & Sons: New York, NY, USA, 2001. [Google Scholar]
- Jensen, F. Introduction to Computational Chemistry, 2nd ed.; John Wiley & Sons: Chichester, UK, 2007. [Google Scholar]
- Cramer, C.J. Essentials of Computational Chemistry—Theories and Models, 2nd ed.; John Wiley & Sons: Chichester, UK, 2004. [Google Scholar]
- Janak, J. Proof that ∂E/∂ni=ϵ in Density Functional Theory. Phys. Rev. B 1978, 18, 7165–7168. [Google Scholar] [CrossRef]
- Kar, R.; Song, J.W.; Hirao, K. Long-Range Corrected Functionals Satisfy Koopmans’ Theorem: Calculation of Correlation and Relaxation Energies. J. Comput. Chem. 2013, 34, 958–964. [Google Scholar] [CrossRef]
- Tsuneda, T.; Song, J.W.; Suzuki, S.; Hirao, K. On Koopmans’ Theorem in Density Functional Theory. J. Chem. Phys. 2010, 133, 174101. [Google Scholar] [CrossRef] [PubMed]
- Tsuneda, T.; Hirao, K. Long-Range Correction for Density Functional Theory. Wiley Interdiscip. Rev. Comput. Mol. Sci. 2014, 4, 375–390. [Google Scholar] [CrossRef]
- Kanchanakungwankul, S.; Truhlar, D.G. Examination of How Well Long-Range-Corrected Density Functionals Satisfy the Ionization Energy Theorem. J. Chem. Theory Comput. 2021, 17, 4823–4830. [Google Scholar] [CrossRef] [PubMed]
- Gázquez, J.; Cedillo, A.; Vela, A. Electrodonating and Electroaccepting Powers. J. Phys. Chem. A 2007, 111, 1966–1970. [Google Scholar] [CrossRef] [PubMed]
- Chattaraj, P.; Chakraborty, A.; Giri, S. Net Electrophilicity. J. Phys. Chem. A 2009, 113, 10068–10074. [Google Scholar] [CrossRef]
- Domingo, L.R.; Chamorro, E.; Perez, P. Understanding the Reactivity of Captodative Ethylenes in Polar Cycloaddition Reactions. A Theoretical Study. J. Org. Chem. 2008, 73, 4615–4624. [Google Scholar] [CrossRef]
- Jaramillo, P.; Domingo, L.R.; Chamorro, E.; Pérez, P. A Further Exploration of a Nucleophilicity Index Based on the Gas-Phase Ionization Potentials. J. Mol. Struct. THEOCHEM 2008, 865, 68–72. [Google Scholar] [CrossRef]
- Domingo, L.R.; Sáez, J.A. Understanding the Mechanism of Polar Diels-Alder Reactions. Org. Biomol. Chem. 2009, 7, 3576–3583. [Google Scholar] [CrossRef]
- Domingo, L.R.; Perez, P. The Nucleophilicity N Index in Organic Chemistry. Org. Biomol. Chem. 2011, 9, 7168–7175. [Google Scholar] [CrossRef]
- Domingo, L.R.; Ríos-Gutiérrez, M.; Pérez, P. Applications of the Conceptual Density Functional Theory Indices to Organic Chemistry Reactivity. Molecules 2016, 21, 748. [Google Scholar] [CrossRef] [Green Version]
- Domingo, L.R.; Aurell, M.; Pérez, P.; Contreras, R. Quantitative Characterization of the Global Electrophilicity Power of Common diene/Dienophile Pairs in Diels-Alder Reactions. Tetrahedron 2002, 58, 4417–4423. [Google Scholar] [CrossRef]
- Pérez, P.; Domingo, L.R.; Aurell, M.J.; Contreras, R. Quantitative Characterization of the Global Electrophilicity Pattern of Some Reagents Involved in 1,3-Dipolar Cycloaddition Reactions. Tetrahedron 2003, 59, 3117–3125. [Google Scholar] [CrossRef]
- Frau, J.; Hernández-Haro, N.; Glossman-Mitnik, D. Computational Prediction of the pKas of Small Peptides through Conceptual DFT Descriptors. Chem. Phys. Lett. 2017, 671, 138–141. [Google Scholar] [CrossRef]
- Morell, C.; Grand, A.; Toro-Labbé, A. New Dual Descriptor for Chemical Reactivity. J. Phys. Chem. A 2005, 109, 205–212. [Google Scholar] [CrossRef]
- Morell, C.; Grand, A.; Toro-Labbé, A. Theoretical Support for Using the Δf(r) Descriptor. Chem. Phys. Lett. 2006, 425, 342–346. [Google Scholar] [CrossRef]
- Martínez-Araya, J.I. Revisiting Caffeate’s Capabilities as a Complexation Agent to Silver Cation in Mining Processes by means of the Dual Descriptor – A Conceptual DFT Approach. J. Mol. Model. 2012, 18, 4299–4307. [Google Scholar] [CrossRef]
- Martínez-Araya, J.I. Explaining Reaction Mechanisms Using the Dual Descriptor: A Complementary Tool to the Molecular Electrostatic Potential. J. Mol. Model. 2012, 19, 2715–2722. [Google Scholar] [CrossRef]
- Martínez-Araya, J.I. Why is the Dual Descriptor a More Accurate Local Reactivity Descriptor than Fukui Functions? J. Math. Chem. 2015, 53, 451–465. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Petersson, G.A.; Nakatsuji, H.; et al. Gaussian 16 Revision C.01; Gaussian Inc.: Wallingford, CT, USA, 2016. [Google Scholar]
- Peverati, R.; Truhlar, D.G. Screened-Exchange Density Functionals with Broad Accuracy for Chemistry and Solid-State Physics. Phys. Chem. Chem. Phys. 2012, 14, 16187. [Google Scholar] [CrossRef]
- Weigend, F.; Ahlrichs, R. Balanced Basis Sets of Split Valence, Triple Zeta Valence and Quadruple Zeta Valence Quality for H to Rn: Design and Assessment of Accuracy. Phys. Chem. Chem. Phys. 2005, 7, 3297. [Google Scholar] [CrossRef]
- Weigend, F. Accurate Coulomb-fitting Basis Sets for H to Rn. Phys. Chem. Chem. Phys. 2006, 8, 1057. [Google Scholar] [CrossRef] [PubMed]
- Marenich, A.V.; Cramer, C.J.; Truhlar, D.G. Universal Solvation Model Based on Solute Electron Density and on a Continuum Model of the Solvent Defined by the Bulk Dielectric Constant and Atomic Surface Tensions. J. Phys. Chem. B 2009, 113, 6378–6396. [Google Scholar] [CrossRef] [PubMed]
- Yang, H.; Lou, C.; Sun, L.; Li, J.; Cai, Y.; Wang, Z.; Li, W.; Liu, G.; Tang, Y. admetSAR 2.0: Web-Service for Prediction and Optimization of Chemical ADMET Properties. Bioinformatics 2018, 35, 1067–1069. [Google Scholar] [CrossRef] [PubMed]
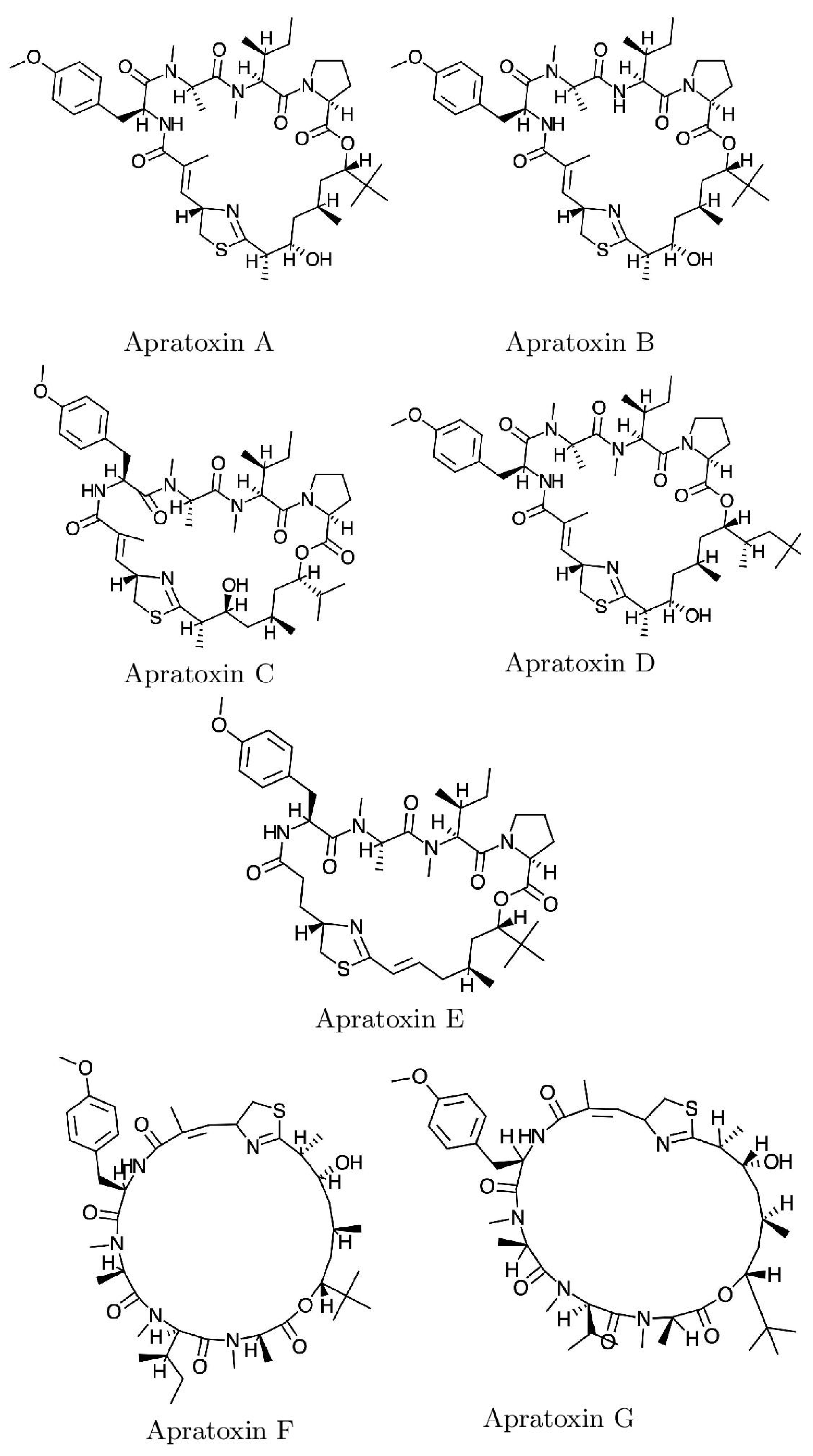
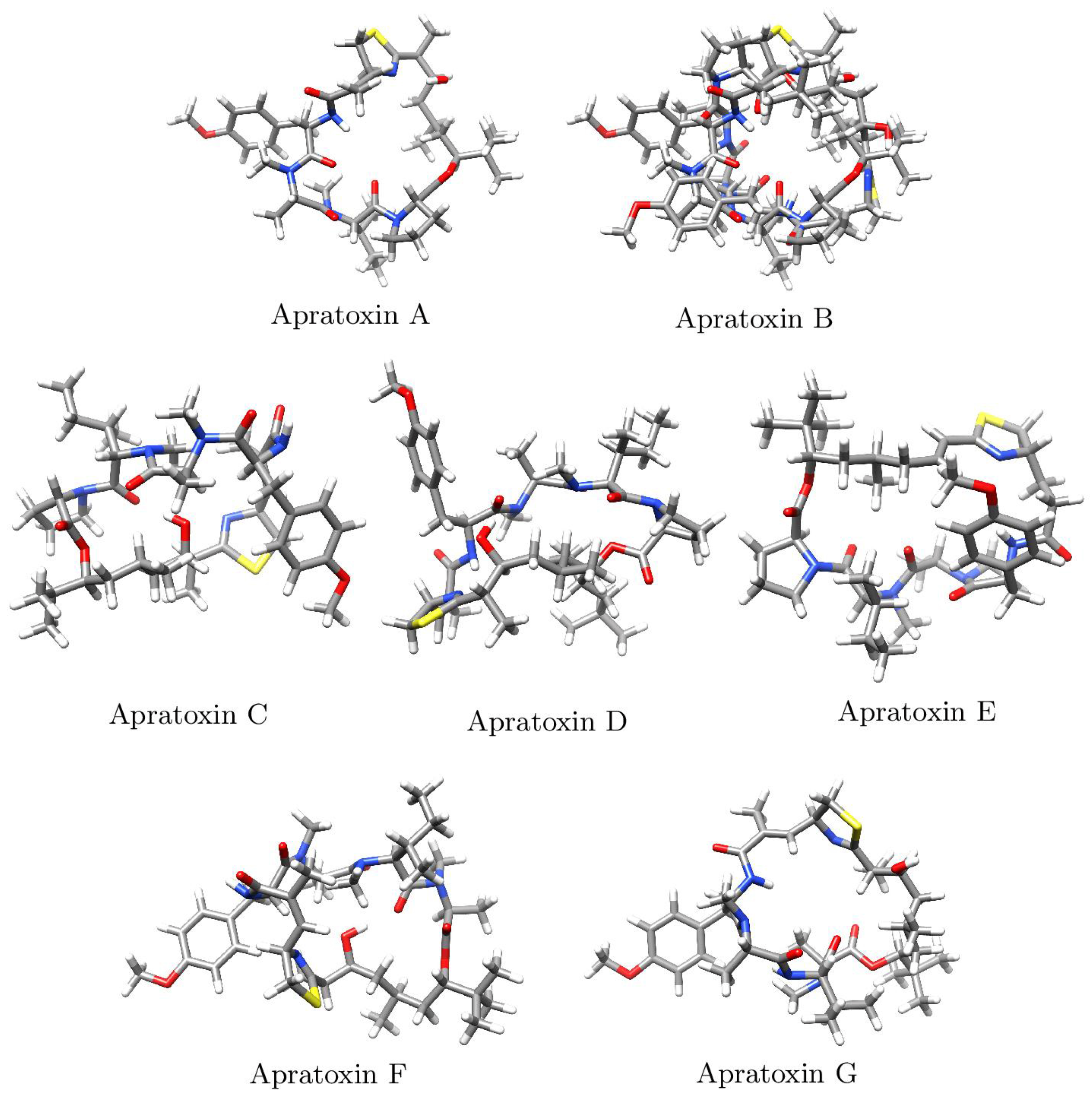
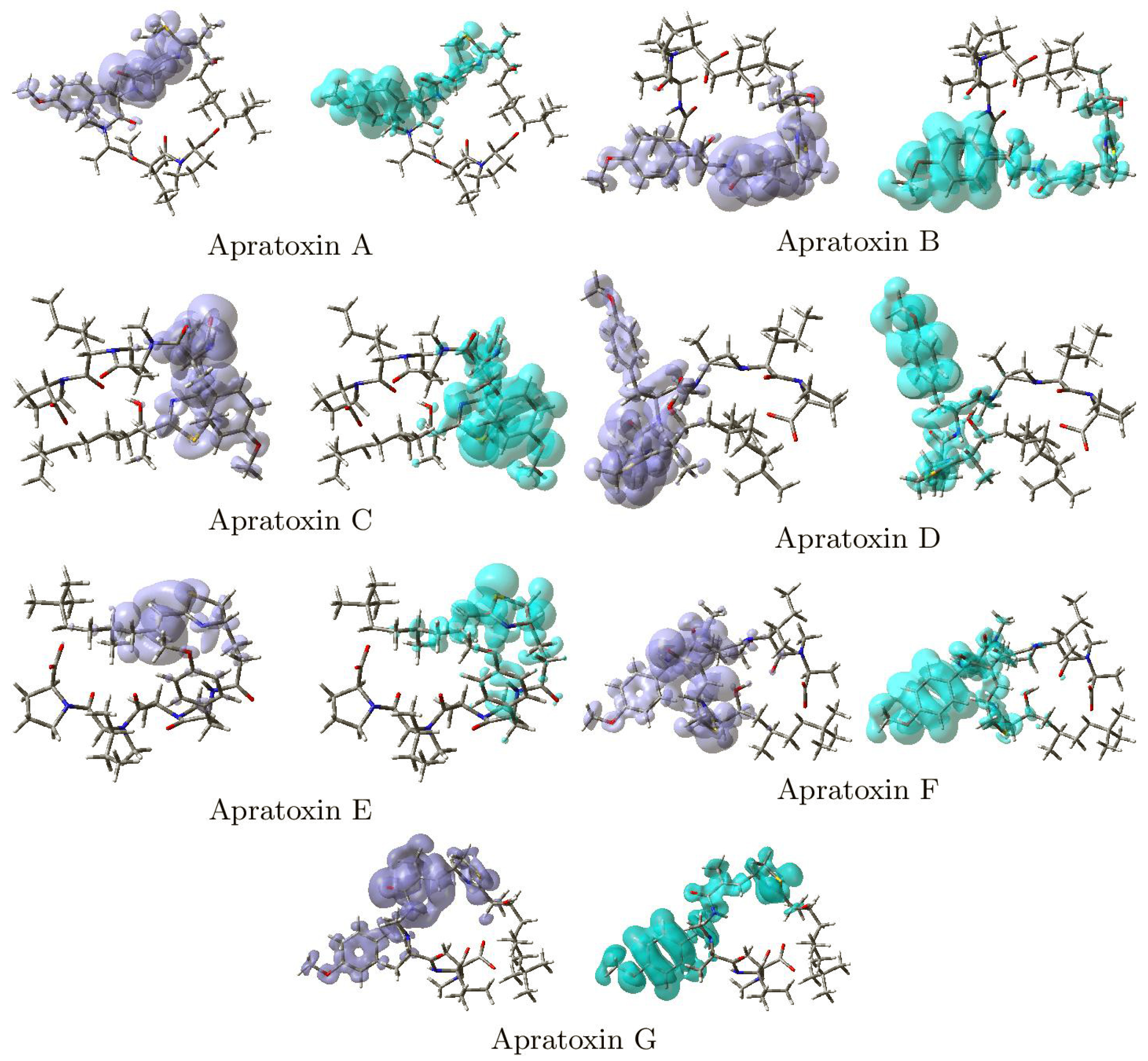

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Apratoxin | HOMO | LUMO | SOMO | H-L Gap | J | J | J | ΔSL |
|---|---|---|---|---|---|---|---|---|
| A | −6.24 | −1.22 | −1.18 | 5.02 | 0.025 | 0.019 | 0.032 | 0.038 |
| B | −6.20 | −1.74 | −1.70 | 4.47 | 0.023 | 0.017 | 0.028 | 0.037 |
| C | −6.04 | −1.76 | −1.73 | 4.28 | 0.091 | 0.016 | 0.092 | 0.029 |
| D | −6.22 | −1.38 | −1.38 | 4.84 | 0.013 | 0.005 | 0.014 | 0.005 |
| E | −6.08 | −1.69 | −1.67 | 4.39 | 0.117 | 0.007 | 0.117 | 0.015 |
| F | −6.19 | −1.40 | −1.37 | 4.79 | 0.015 | 0.019 | 0.024 | 0.037 |
| G | −6.12 | −1.79 | −1.77 | 4.33 | 0.100 | 0.010 | 0.100 | 0.022 |
| Apratoxin | S | N | ||||||
|---|---|---|---|---|---|---|---|---|
| A | 3.73 | 5.02 | 1.39 | 0.20 | 2.56 | 4.95 | 1.22 | 6.17 |
| B | 3.97 | 4.47 | 1.76 | 0.22 | 2.59 | 5.79 | 1.82 | 7.61 |
| C | 3.90 | 4.28 | 1.78 | 0.23 | 2.75 | 5.78 | 1.87 | 7.65 |
| D | 3.80 | 4.84 | 1.50 | 0.21 | 2.57 | 5.19 | 1.39 | 6.58 |
| E | 3.88 | 4.39 | 1.72 | 0.23 | 2.72 | 5.65 | 1.76 | 7.41 |
| F | 3.80 | 4.79 | 1.51 | 0.21 | 2.60 | 5.21 | 1.41 | 6.62 |
| G | 3.96 | 4.33 | 1.81 | 0.23 | 2.67 | 5.86 | 1.91 | 7.77 |
| Apratoxin | ΔG of Solvation | pKa | logP | TPSA | Molecular Volume |
|---|---|---|---|---|---|
| (kcal/mol) | (Å) | (Å) | |||
| A | −25.86 | 12.90 | 5.49 | 158.16 | 809.91 |
| B | −31.99 | 12.97 | 5.26 | 166.94 | 792.97 |
| C | −27.41 | 12.90 | 5.11 | 158.16 | 793.67 |
| D | −23.32 | 12.90 | 7.41 | 158.16 | 860.10 |
| E | −26.30 | 12.47 | 6.21 | 137.93 | 768.72 |
| F | −25.54 | 12.73 | 6.07 | 158.16 | 803.47 |
| G | −31.00 | 12.76 | 5.54 | 166.94 | 769.72 |
| Apratoxin | GPCR | Ion Channel | Nuclear Receptor | Kinase | Protease | Enzyme |
|---|---|---|---|---|---|---|
| Ligand | Modulator | Ligand | Inhibitor | Inhibitor | Inhibitor | |
| A | −1.95 | −3.12 | −3.14 | −3.02 | −1.11 | −2.42 |
| B | −1.73 | −3.00 | −2.97 | −2.86 | −0.92 | −2.19 |
| C | −1.75 | −2.96 | −2.92 | −2.90 | −0.96 | −2.24 |
| D | −2.69 | −3.51 | −3.59 | −3.53 | −1.85 | −2.98 |
| E | −1.46 | −2.74 | −2.69 | −2.48 | −0.79 | −1.89 |
| F | −3.77 | −3.88 | −3.88 | −3.89 | −3.67 | −3.81 |
| G | −3.76 | −3.87 | −3.87 | −3.88 | −3.65 | −3.80 |
| Apratoxins | |||||||
|---|---|---|---|---|---|---|---|
| Property | A | B | C | D | E | F | G |
| HI Absorption | + | + | + | + | + | + | + |
| BBB Permeability | − | − | − | − | − | − | − |
| Caco-2 | + | + | + | + | + | + | + |
| P-gp Substrate | + | + | + | + | + | + | + |
| P-gp I Inhibitor | + | + | + | + | + | + | + |
| P-gp II Inhibitor | + | + | + | + | − | + | + |
| CYP2D6 Substrate | − | + | − | − | − | − | − |
| CYP3A4 Substrate | + | − | + | + | + | + | + |
| CYP1A2 Inhibitor | − | + | − | − | − | − | − |
| CYP2C19 Inhibitor | − | − | − | − | − | − | − |
| CYP2C9 Inhibitor | − | − | − | − | − | − | − |
| CYP2D6 Inhibitor | − | − | − | − | − | − | − |
| CYP3A4 Inhibitor | + | − | + | + | + | + | − |
| OCT2 Substrate | − | − | − | − | − | − | − |
| AMES Toxicity | − | − | − | − | − | − | − |
| hERG I Inhibitor | − | − | − | − | − | − | − |
| hERG II Inhibitor | − | − | − | − | − | + | − |
| Hepatoxicity | + | + | + | + | + | + | + |
| Skin Sensitization | − | − | − | − | − | − | − |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Flores-Holguín, N.; Frau, J.; Glossman-Mitnik, D. Virtual Prospection of Marine Cyclopeptides as Therapeutics by Means of Conceptual DFT and Computational ADMET. Pharmaceuticals 2022, 15, 509. https://doi.org/10.3390/ph15050509
Flores-Holguín N, Frau J, Glossman-Mitnik D. Virtual Prospection of Marine Cyclopeptides as Therapeutics by Means of Conceptual DFT and Computational ADMET. Pharmaceuticals. 2022; 15(5):509. https://doi.org/10.3390/ph15050509
Chicago/Turabian StyleFlores-Holguín, Norma, Juan Frau, and Daniel Glossman-Mitnik. 2022. "Virtual Prospection of Marine Cyclopeptides as Therapeutics by Means of Conceptual DFT and Computational ADMET" Pharmaceuticals 15, no. 5: 509. https://doi.org/10.3390/ph15050509
APA StyleFlores-Holguín, N., Frau, J., & Glossman-Mitnik, D. (2022). Virtual Prospection of Marine Cyclopeptides as Therapeutics by Means of Conceptual DFT and Computational ADMET. Pharmaceuticals, 15(5), 509. https://doi.org/10.3390/ph15050509