
A Quantum Chemical Deep-Dive into the π-π Interactions of 3-Methylindole and Its Halogenated Derivatives—Towards an Improved Ligand Design and Tryptophan Stacking

 , ,
, , 
Abstract
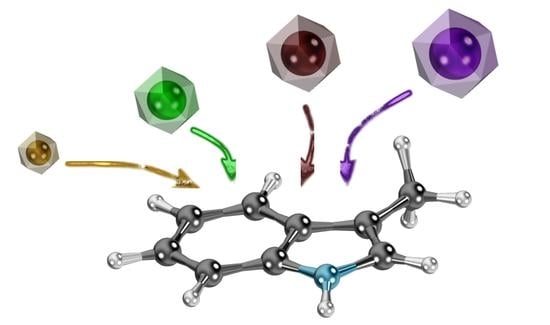
:
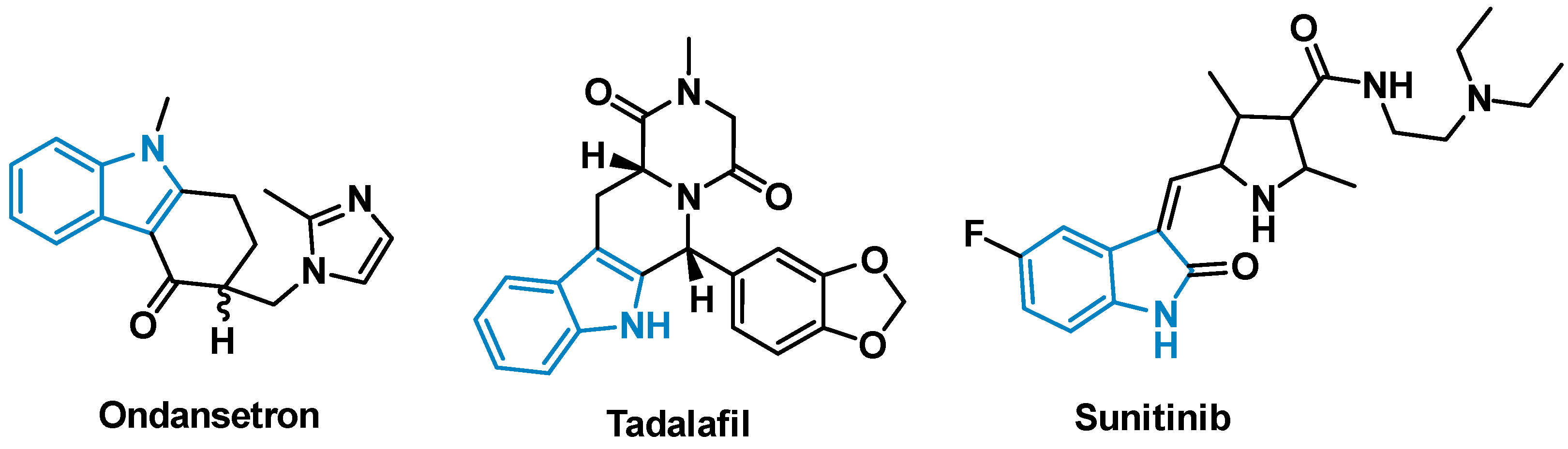
1. Introduction
2. Results and Discussion
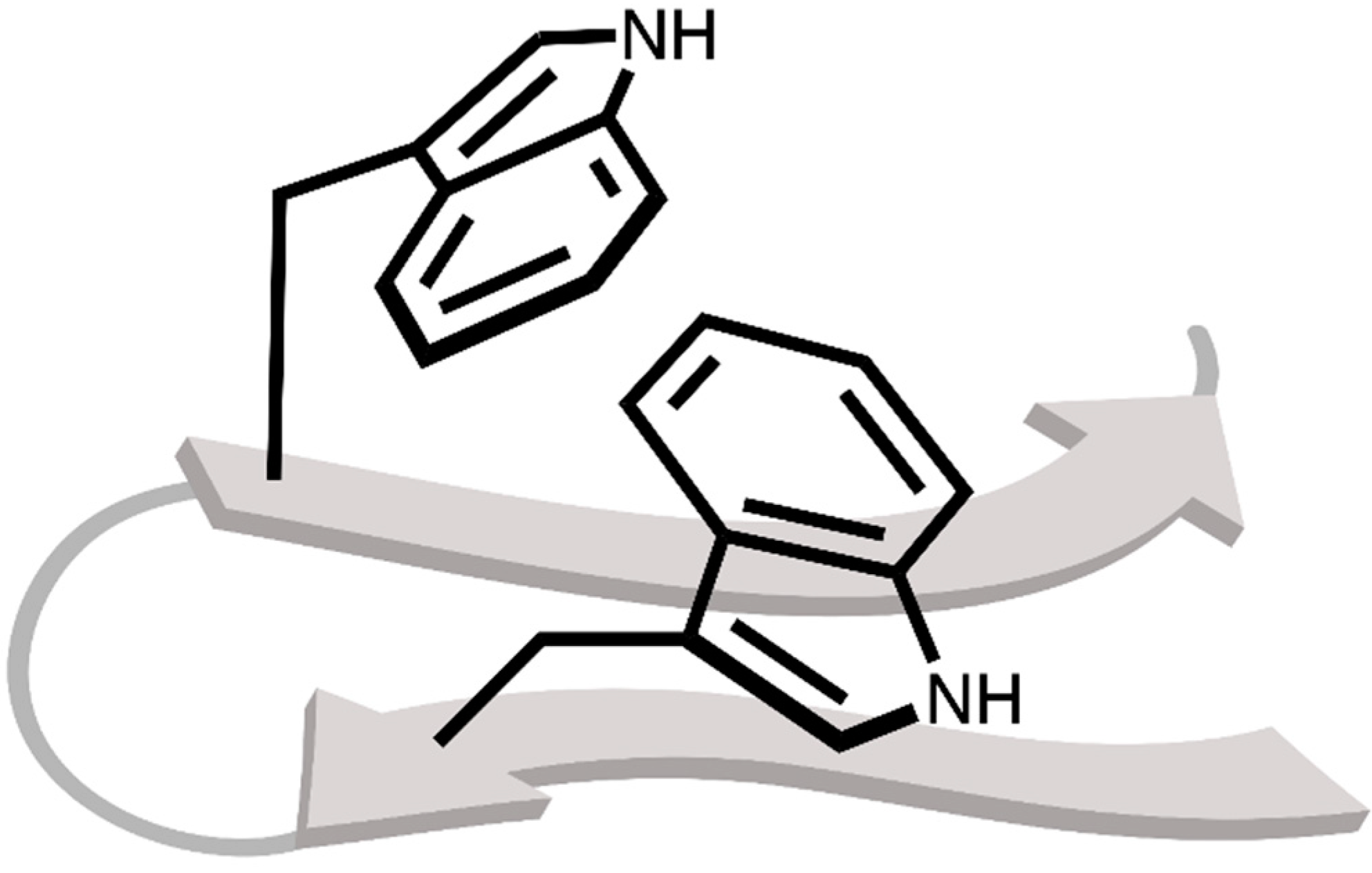
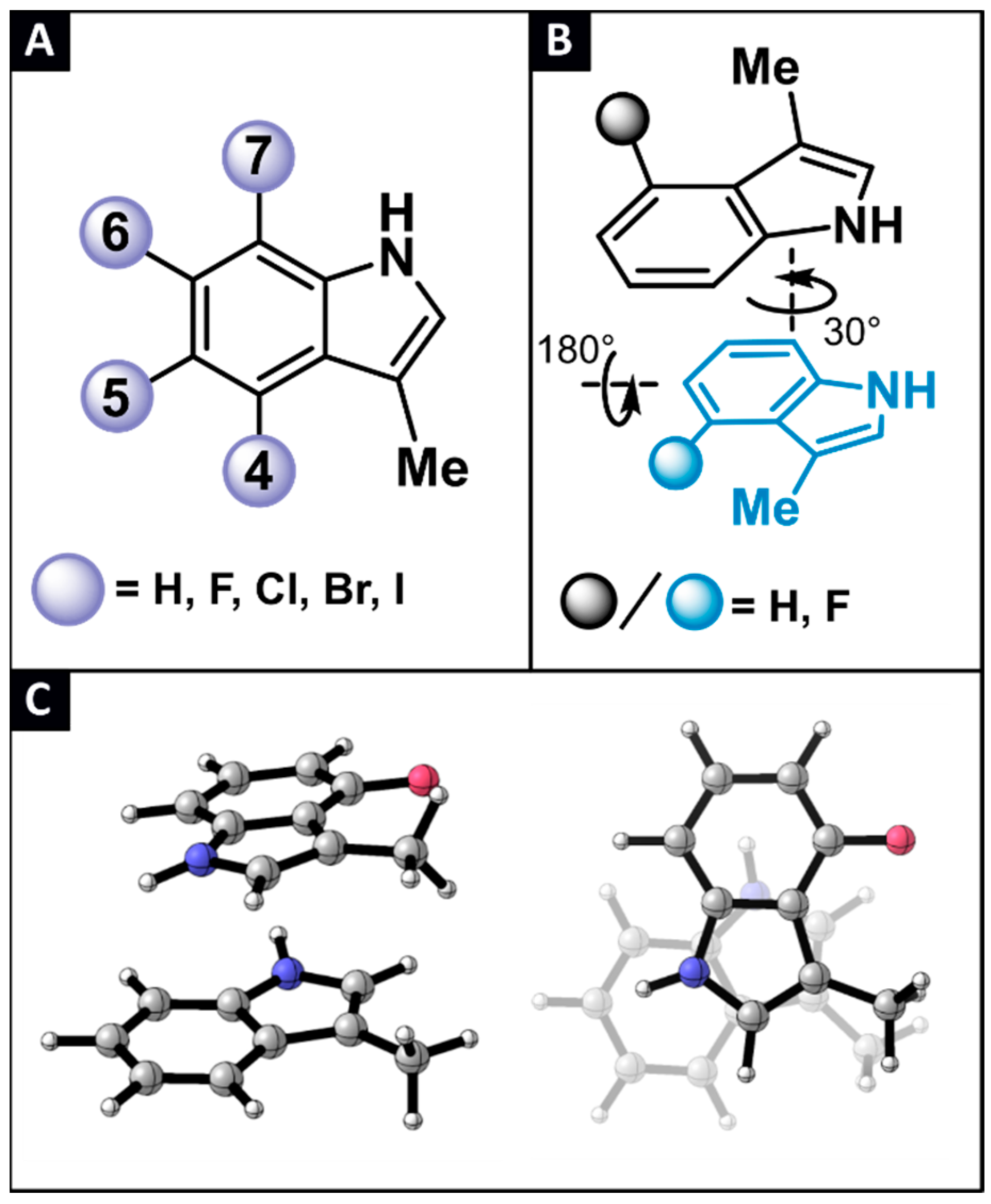
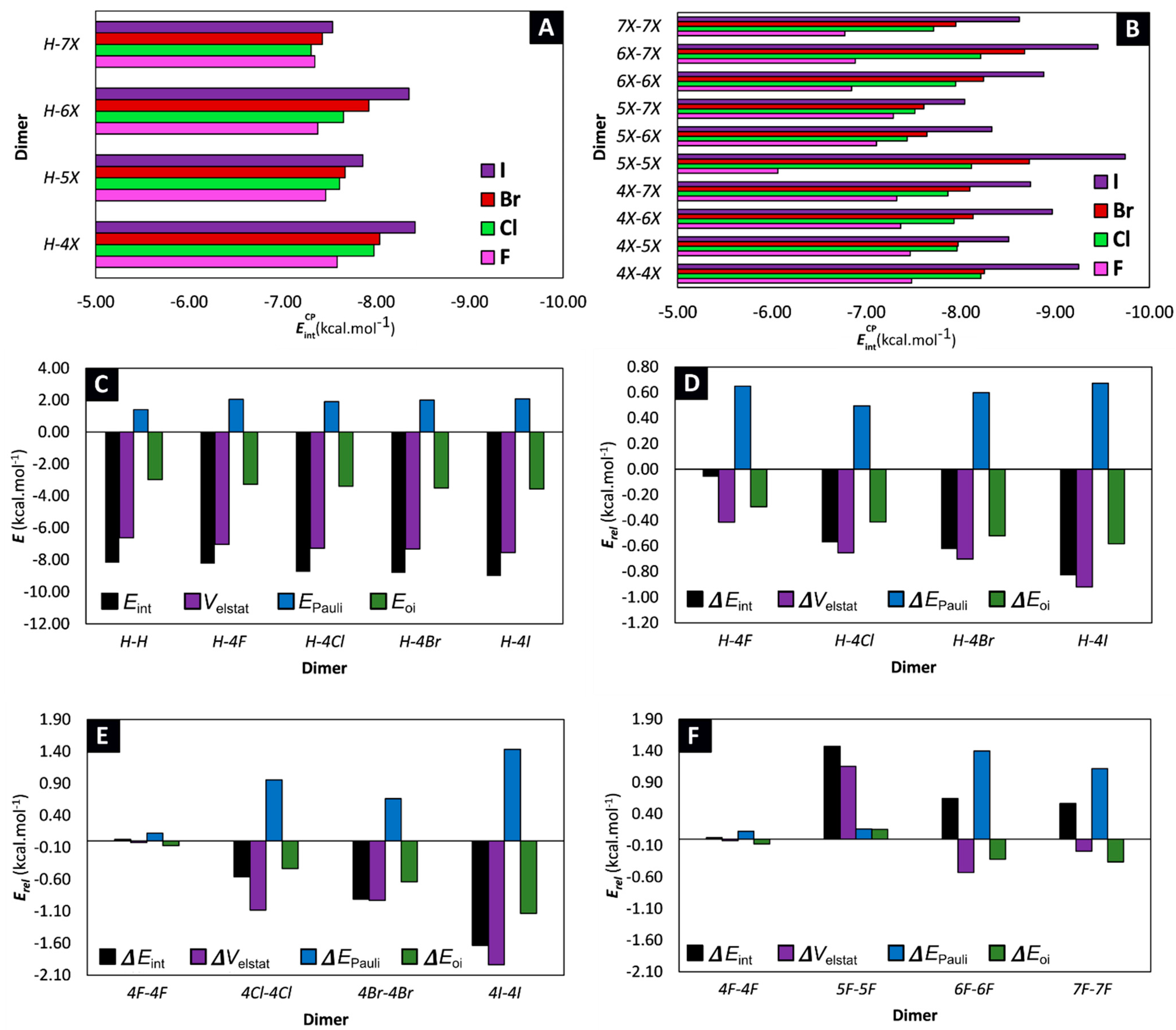
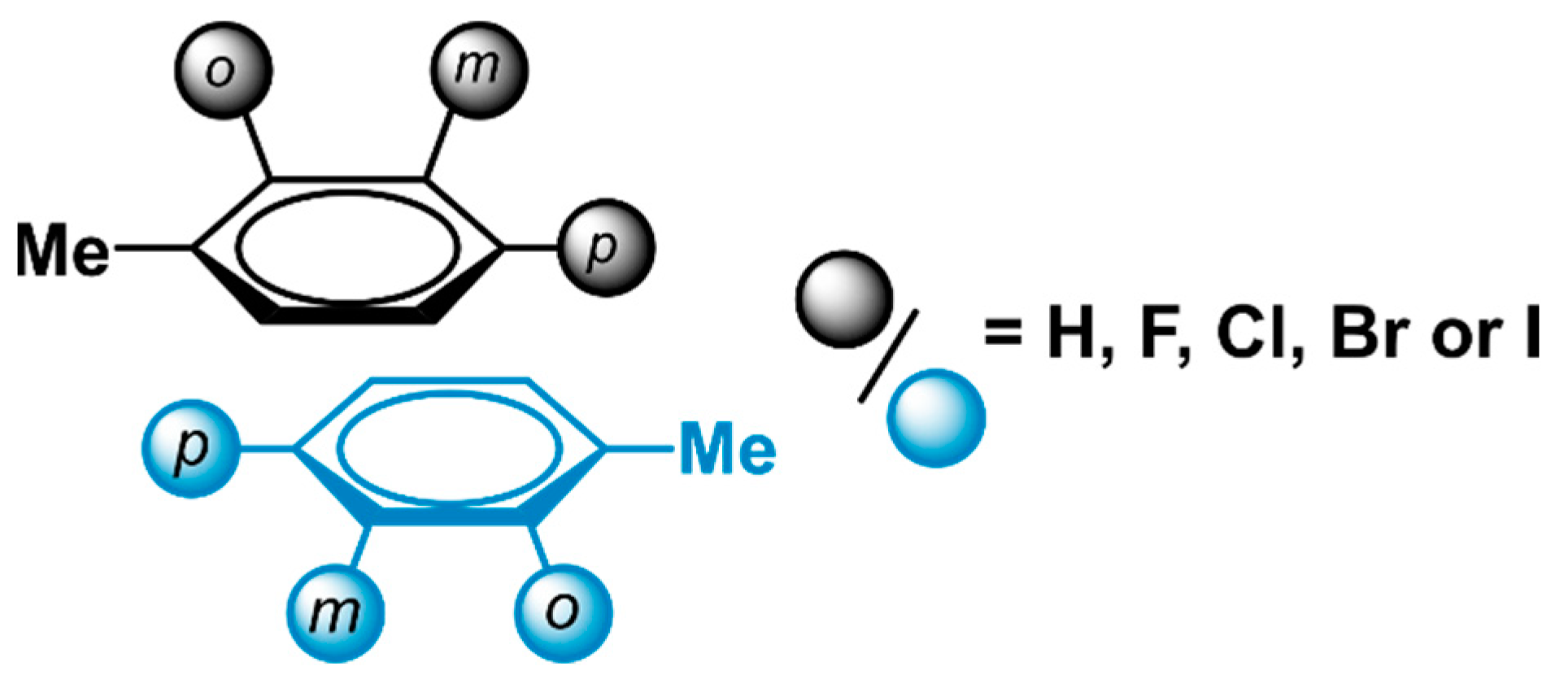
2.1. Locating Stacked Dimers and Revealing the Roots of Their Stability through Energy Decomposition Analyses
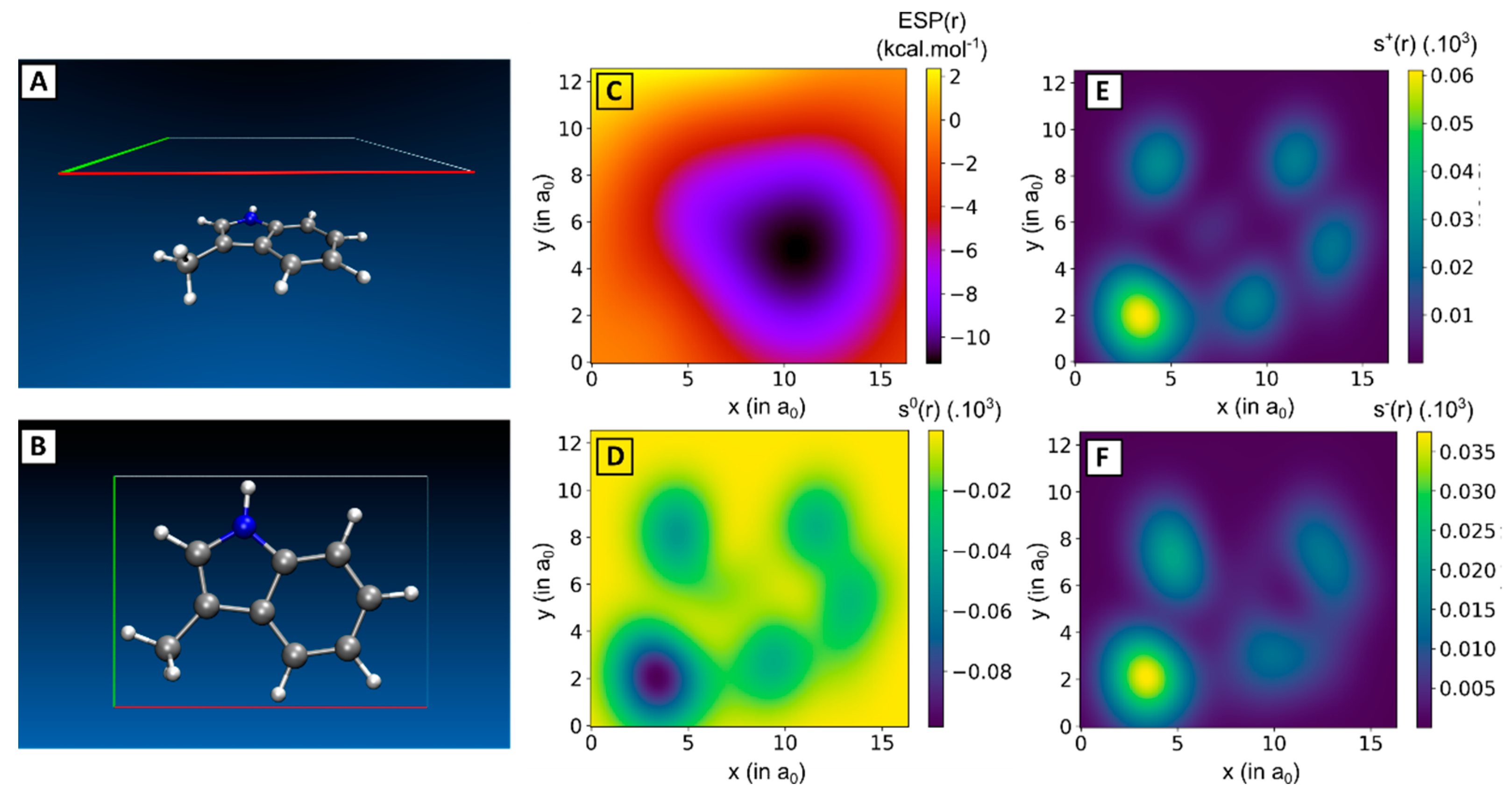
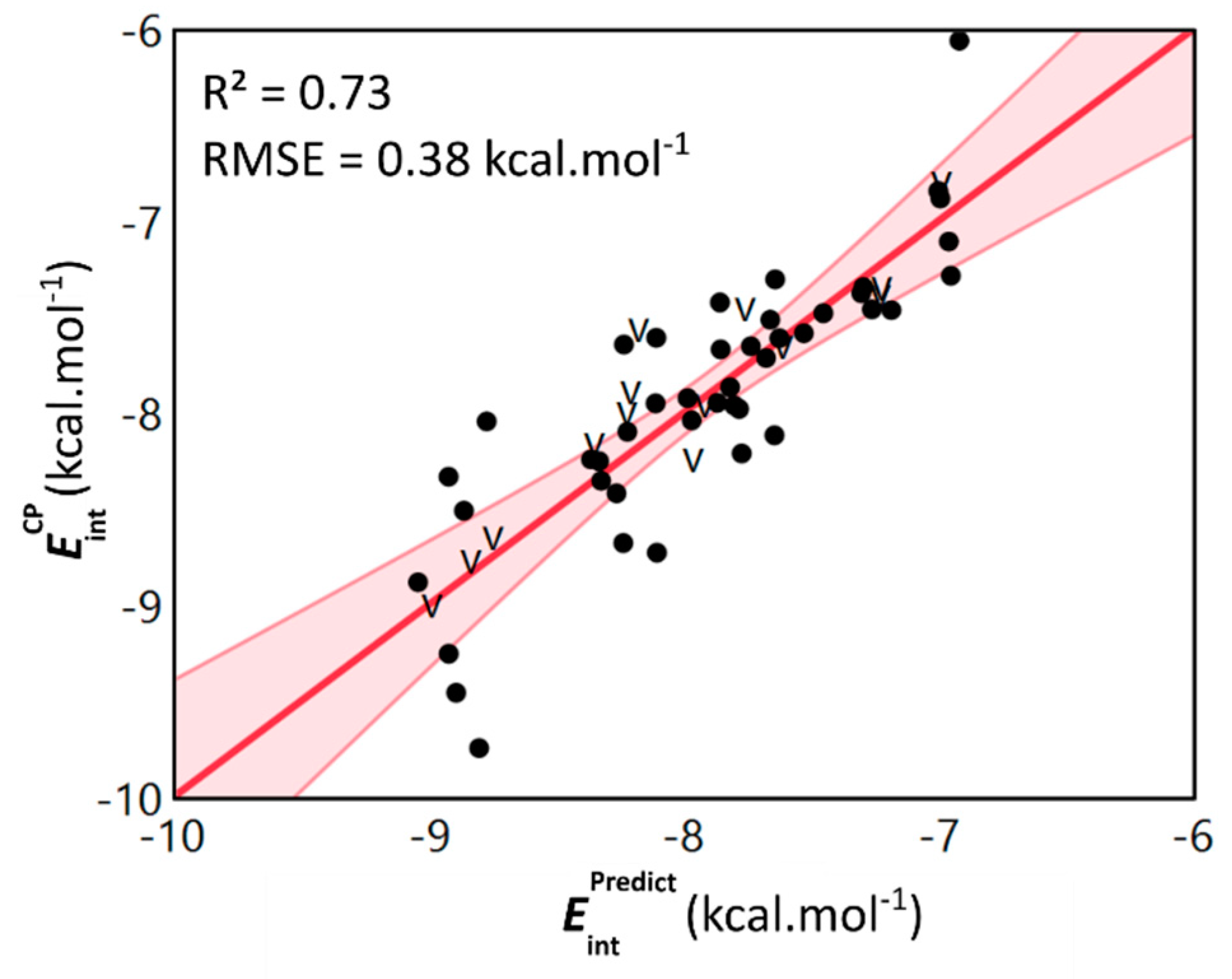
2.2. Linking Interaction Strength to Monomer Properties
2.3. General Model Applicability–Cross-Halogenation and Phenylalanine Stacking
3. Materials and Methods
3.1. Determination of Interaction Energies
3.2. Energy Decomposition Analysis
3.3. Computing the Monomer Descriptors
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Inman, M.; Moody, C.J. Indole synthesis—Something old, something new. Chem. Sci. 2013, 4, 29–41. [Google Scholar] [CrossRef]
- Kumar, S.; Ritika. A brief review of the biological potential of indole derivatives. Future J. Pharm. Sci. 2020, 6, 121. [Google Scholar] [CrossRef]
- Sharma, V.; Kumar, P.; Pathak, D. Biological importance of the indole nucleus in recent years: A comprehensive review. J. Heterocycl. Chem. 2010, 47, 491–502. [Google Scholar] [CrossRef]
- Chadha, N.; Silakari, O. Indoles as therapeutics of interest in medicinal chemistry: Bird’s eye view. Eur. J. Med. Chem. 2017, 134, 159–184. [Google Scholar] [CrossRef]
- Taber, D.F.; Tirunahari, P.K. Indole synthesis: A review and proposed classification. Tetrahedron 2011, 67, 7195–7210. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vitaku, E.; Smith, D.T.; Njardarson, J.T. Analysis of the Structural Diversity, Substitution Patterns, and Frequency of Nitrogen Heterocycles among U.S. FDA Approved Pharmaceuticals. J. Med. Chem. 2014, 57, 10257–10274. [Google Scholar] [CrossRef] [PubMed]
- Fantacuzzi, M.; De Filippis, B.; Gallorini, M.; Ammazzalorso, A.; Giampietro, L.; Maccallini, C.; Aturki, Z.; Donati, E.; Ibrahim, R.S.; Shawky, E.; et al. Synthesis, biological evaluation, and docking study of indole aryl sulfonamides as aromatase inhibitors. Eur. J. Med. Chem. 2020, 185, 111815. [Google Scholar] [CrossRef]
- Razak, S.; Afsar, T.; Bibi, N.; Abulmeaty, M.; Qamar, W.; Almajwal, A.; Inam, A.; Al Disi, D.; Shabbir, M.; Bhat, M.A. Molecular docking, pharmacokinetic studies, and in vivo pharmacological study of indole derivative 2-(5-methoxy-2-methyl-1H-indole-3-yl)-N′-[(E)-(3-nitrophenyl) methylidene] acetohydrazide as a promising chemoprotective agent against cisplatin induced organ damage. Sci. Rep. 2021, 11, 6245. [Google Scholar] [CrossRef]
- McGaughey, G.B.; Gagné, M.; Rappé, A.K. π-Stacking Interactions: Alive and Well in Proteins*. J. Biol. Chem. 1998, 273, 15458–15463. [Google Scholar] [CrossRef] [Green Version]
- Trinquier, G.; Sanejouand, Y.H. Which effective property of amino acids is best preserved by the genetic code? Protein Eng. 1998, 11, 153–169. [Google Scholar] [CrossRef] [Green Version]
- Samanta, U.; Pal, D.; Chakrabarti, P. Environment of tryptophan side chains in proteins. Proteins 2000, 38, 288–300. [Google Scholar] [CrossRef]
- Taverna, S.D.; Li, H.; Ruthenburg, A.J.; Allis, C.D.; Patel, D.J. How chromatin-binding modules interpret histone modifications: Lessons from professional pocket pickers. Nat. Struct. Mol. Biol. 2007, 14, 1025–1040. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rahman, M.M.; Muhseen, T.Z.; Junaid, M.; Zhang, H. The Aromatic Stacking Interactions between Proteins and their Macromolecular Ligands. Curr. Protein Pept. Sci. 2015, 16, 502–512. [Google Scholar] [CrossRef] [PubMed]
- Asensio, J.L.; Ardá, A.; Cañada, F.J.; Jiménez-Barbero, J. Carbohydrate–Aromatic Interactions. Acc. Chem. Res. 2013, 46, 946–954. [Google Scholar] [CrossRef] [Green Version]
- Kumar, K.; Woo, S.M.; Siu, T.; Cortopassi, W.A.; Duarte, F.; Paton, R.S. Cation–π interactions in protein–ligand binding: Theory and data-mining reveal different roles for lysine and arginine. Chem. Sci. 2018, 9, 2655–2665. [Google Scholar] [CrossRef] [Green Version]
- Khemaissa, S.; Sagan, S.; Walrant, A. Tryptophan, an Amino-Acid Endowed with Unique Properties and Its Many Roles in Membrane Proteins. Crystals 2021, 11, 1032. [Google Scholar] [CrossRef]
- Jonasson, P.; Aronsson, G.; Carlsson, U.; Jonsson, B.-H. Tertiary Structure Formation at Specific Tryptophan Side Chains in the Refolding of Human Carbonic Anhydrase II. Biochemistry 1997, 36, 5142–5148. [Google Scholar] [CrossRef]
- Cochran Andrea, G.; Skelton Nicholas, J.; Starovasnik Melissa, A. Tryptophan zippers: Stable, monomeric β-hairpins. Proc. Natl. Acad. Sci. USA 2001, 98, 5578–5583. [Google Scholar] [CrossRef] [Green Version]
- Pace, C.J.; Gao, J. Exploring and Exploiting Polar−π Interactions with Fluorinated Aromatic Amino Acids. Acc. Chem. Res. 2013, 46, 907–915. [Google Scholar] [CrossRef]
- Schneider, H.-J. Interactions in Supramolecular Complexes Involving Arenes: Experimental Studies. Acc. Chem. Res. 2013, 46, 1010–1019. [Google Scholar] [CrossRef]
- Riwar, L.-J.; Trapp, N.; Kuhn, B.; Diederich, F. Substituent Effects in Parallel-Displaced π–π Stacking Interactions: Distance Matters. Angew. Chem. Int. Ed. 2017, 56, 11252–11257. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bettens, T.; Lacanau, V.; Van Lommel, R.; De Maeseneer, T.; Vandeplassche, W.; Bertouille, J.; Brancart, J.; Barlow, T.M.A.; Woller, T.; Van den Brande, N.; et al. Towards the understanding of halogenation in peptide hydrogels: A quantum chemical approach. Mater. Adv. 2021, 2, 4792–4803. [Google Scholar] [CrossRef]
- Hunter, C.A.; Sanders, J.K.M. The nature of π–π interactions. J. Am. Chem. Soc. 1990, 112, 5525–5534. [Google Scholar] [CrossRef]
- Cockroft, S.L.; Hunter, C.A.; Lawson, K.R.; Perkins, J.; Urch, C.J. Electrostatic Control of Aromatic Stacking Interactions. J. Am. Chem. Soc. 2005, 127, 8594–8595. [Google Scholar] [CrossRef]
- Cozzi, F.; Cinquini, M.; Annunziata, R.; Dwyer, T.; Siegel, J.S. Polar/π interactions between stacked aryls in 1,8-diarylnaphthalenes. J. Am. Chem. Soc. 1992, 114, 5729–5733. [Google Scholar] [CrossRef]
- Cozzi, F.; Cinquini, M.; Annuziata, R.; Siegel, J.S. Dominance of polar/.pi. over charge-transfer effects in stacked phenyl interactions. J. Am. Chem. Soc. 1993, 115, 5330–5331. [Google Scholar] [CrossRef]
- Wheeler, S.E.; Houk, K.N. Substituent Effects in the Benzene Dimer are Due to Direct Interactions of the Substituents with the Unsubstituted Benzene. J. Am. Chem. Soc. 2008, 130, 10854–10855. [Google Scholar] [CrossRef] [Green Version]
- Wheeler, S.E. Local Nature of Substituent Effects in Stacking Interactions. J. Am. Chem. Soc. 2011, 133, 10262–10274. [Google Scholar] [CrossRef]
- Bootsma, A.N.; Doney, A.C.; Wheeler, S.E. Predicting the Strength of Stacking Interactions between Heterocycles and Aromatic Amino Acid Side Chains. J. Am. Chem. Soc. 2019, 141, 11027–11035. [Google Scholar] [CrossRef]
- Guvench, O.; Brooks, C.L. Tryptophan Side Chain Electrostatic Interactions Determine Edge-to-Face vs. Parallel-Displaced Tryptophan Side Chain Geometries in the Designed β-Hairpin “trpzip2”. J. Am. Chem. Soc. 2005, 127, 4668–4674. [Google Scholar] [CrossRef]
- Chinnasamy, S.; Muthusamy, K.; Chinnasamy, S. Nanopeptides: Non-Covalent Interactions in Chemistry and Biological Functions. J. Appl. Pharm. 2016, 8, 1000218. [Google Scholar] [CrossRef]
- Zhao, Y.; Truhlar, D.G. The M06 suite of density functionals for main group thermochemistry, thermochemical kinetics, noncovalent interactions, excited states, and transition elements: Two new functionals and systematic testing of four M06-class functionals and 12 other functionals. Theor. Chem. Acc. 2008, 120, 215–241. [Google Scholar] [CrossRef] [Green Version]
- Dunning, T.H. Gaussian basis sets for use in correlated molecular calculations. I. The atoms boron through neon and hydrogen. J. Chem. Phys. 1989, 90, 1007–1023. [Google Scholar] [CrossRef]
- Peterson, K.A.; Figgen, D.; Goll, E.; Stoll, H.; Dolg, M. Systematically convergent basis sets with relativistic pseudopotentials. II. Small-core pseudopotentials and correlation consistent basis sets for the post-d group 16–18 elements. J. Chem. Phys. 2003, 119, 11113–11123. [Google Scholar] [CrossRef] [Green Version]
- Boys, S.F.; Bernardi, F. The calculation of small molecular interactions by the differences of separate total energies. Some procedures with reduced errors. Mol. Phys. 1970, 19, 553–566. [Google Scholar] [CrossRef]
- Waters, M.L. Aromatic interactions in peptides: Impact on structure and function. Pept. Sci. 2004, 76, 435–445. [Google Scholar] [CrossRef]
- Ziegler, T.; Rauk, A. Carbon monoxide, carbon monosulfide, molecular nitrogen, phosphorus trifluoride, and methyl isocyanide as.sigma. donors and.pi. acceptors. A theoretical study by the Hartree-Fock-Slater transition-state method. Inorg. Chem. 1979, 18, 1755–1759. [Google Scholar] [CrossRef]
- Lenthe, E.v.; Baerends, E.J.; Snijders, J.G. Relativistic regular two-component Hamiltonians. J. Chem. Phys. 1993, 99, 4597–4610. [Google Scholar] [CrossRef]
- van Lenthe, E.; Baerends, E.J.; Snijders, J.G. Relativistic total energy using regular approximations. J. Chem. Phys. 1994, 101, 9783–9792. [Google Scholar] [CrossRef]
- Van Lenthe, E.; Baerends, E.J. Optimized Slater-type basis sets for the elements 1–118. J. Comput. Chem. 2003, 24, 1142–1156. [Google Scholar] [CrossRef]
- Bootsma, A.N.; Wheeler, S.E. Converting SMILES to Stacking Interaction Energies. J. Chem. Inf. Modeling 2019, 59, 3413–3421. [Google Scholar] [CrossRef] [PubMed]
- Bootsma, A.N.; Wheeler, S.E. Tuning Stacking Interactions between Asp–Arg Salt Bridges and Heterocyclic Drug Fragments. J. Chem. Inf. Modeling 2019, 59, 149–158. [Google Scholar] [CrossRef] [PubMed]
- Geerlings, P.; De Proft, F.; Langenaeker, W. Conceptual Density Functional Theory. Chem. Rev. 2003, 103, 1793–1874. [Google Scholar] [CrossRef]
- SAS Institute Inc. JMP® 16 Fitting Linear Models; SAS Institute Inc.: Cary, NC, USA, 2020–2021. [Google Scholar]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Petersson, G.A.; Nakatsuji, H.; et al. Gaussian 16 Revision A.03; Gaussian, Inc.: Wallingford, CT, USA, 2016. [Google Scholar]
- Peterson, K.A.; Shepler, B.C.; Figgen, D.; Stoll, H. On the Spectroscopic and Thermochemical Properties of ClO, BrO, IO, and Their Anions. J. Phys. Chem. A 2006, 110, 13877–13883. [Google Scholar] [CrossRef]
- Wheeler, S.E.; Houk, K.N. Integration Grid Errors for Meta-GGA-Predicted Reaction Energies: Origin of Grid Errors for the M06 Suite of Functionals. J. Chem. Theory Comput. 2010, 6, 395–404. [Google Scholar] [CrossRef]
- Sun, X.; Soini, T.M.; Poater, J.; Hamlin, T.A.; Bickelhaupt, F.M. PyFrag 2019—Automating the exploration and analysis of reaction mechanisms. J. Comput. Chem. 2019, 40, 2227–2233. [Google Scholar] [CrossRef] [PubMed]
- te Velde, G.; Bickelhaupt, F.M.; Baerends, E.J.; Fonseca Guerra, C.; van Gisbergen, S.J.A.; Snijders, J.G.; Ziegler, T. Chemistry with ADF. J. Comput. Chem. 2001, 22, 931–967. [Google Scholar] [CrossRef]
- Vermeeren, P.; van der Lubbe, S.C.C.; Fonseca Guerra, C.; Bickelhaupt, F.M.; Hamlin, T.A. Understanding chemical reactivity using the activation strain model. Nat. Protoc. 2020, 15, 649–667. [Google Scholar] [CrossRef]
- Zhao, L.; von Hopffgarten, M.; Andrada, D.M.; Frenking, G. Energy decomposition analysis. WIREs Comput. Mol. Sci. 2018, 8, e1345. [Google Scholar] [CrossRef]
- Tozer, D.J.; De Proft, F. Computation of the Hardness and the Problem of Negative Electron Affinities in Density Functional Theory. J. Phys. Chem. A 2005, 109, 8923–8929. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Dimer | |||||
|---|---|---|---|---|---|
| H-H | −7.6 | −8.2 | −6.6 | 1.4 | −3.0 |
| H-4F | −7.6 | −8.2 | −7.0 | 2.1 | −3.3 |
| H-4Cl | −8.0 | −8.8 | −7.3 | 1.9 | −3.4 |
| H-4Br | −8.0 | −8.8 | −7.3 | 2.0 | −3.5 |
| H-4I | −8.4 | −9.0 | −7.5 | 2.1 | −3.6 |
| H-5F | −7.5 | −8.0 | −6.6 | 1.7 | −3.0 |
| H-5Cl | −7.6 | −8.2 | −6.7 | 1.5 | −3.0 |
| H-5Br | −7.7 | −8.3 | −6.9 | 1.8 | −3.2 |
| H-5I | −7.9 | −8.2 | −7.0 | 1.9 | −3.0 |
| H-6F | −7.4 | −7.9 | −6.9 | 2.2 | −3.2 |
| H-6Cl | −7.7 | −8.3 | −7.4 | 2.4 | −3.3 |
| H-6Br | −7.9 | −8.6 | −7.6 | 2.5 | −3.5 |
| H-6I | −8.4 | −8.8 | −7.6 | 2.2 | −3.4 |
| H-7F | −7.3 | −8.0 | −6.6 | 1.7 | −3.1 |
| H-7Cl | −7.3 | −7.9 | −6.7 | 1.9 | −3.1 |
| H-7Br | −7.4 | −8.0 | −6.4 | 1.4 | −3.1 |
| H-7I | −7.5 | −8.0 | −6.3 | 1.3 | −3.0 |
| 4F-4F | −7.5 | −8.2 | −6.6 | 1.5 | −3.1 |
| 4Cl-4Cl | −8.2 | −8.7 | −7.7 | 2.4 | −3.4 |
| 4Br-4Br | −8.2 | −9.1 | −7.6 | 2.1 | −3.6 |
| 4I-4I | −9.3 | −9.8 | −8.6 | 2.8 | −4.1 |
| 4F-5F | −7.5 | −8.0 | −6.8 | 2.0 | −3.2 |
| 4Cl-5Cl | −8.0 | −8.7 | −7.7 | 2.4 | −3.4 |
| 4Br-5Br | −8.0 | −8.8 | −7.7 | 2.6 | −3.6 |
| 4I-5I | −8.5 | −9.0 | −8.2 | 3.0 | −3.8 |
| 4F-6F | −7.4 | −7.9 | −6.8 | 2.0 | −3.2 |
| 4Cl-6Cl | −7.9 | −8.7 | −7.8 | 2.6 | −3.5 |
| 4Br-6Br | −8.1 | −8.9 | −7.8 | 2.6 | −3.7 |
| 4I-6I | −9.0 | −9.2 | −8.3 | 2.7 | −3.6 |
| 4F-7F | −7.3 | −8.0 | −7.7 | 3.4 | −3.7 |
| 4Cl-7Cl | −7.9 | −8.5 | −7.2 | 1.9 | −3.2 |
| 4Br-7Br | −8.1 | −8.8 | −7.3 | 1.9 | −3.4 |
| 4I-7I | −8.7 | −8.9 | −7.3 | 1.7 | −3.3 |
| 5F-5F | −6.1 | −6.7 | −5.5 | 1.6 | −2.8 |
| 5Cl-5Cl | −8.1 | −8.8 | −6.6 | 1.0 | −3.2 |
| 5Br-5Br | −8.7 | −9.5 | −6.8 | 0.8 | −3.6 |
| 5I-5I | −9.7 | −10.0 | −7.3 | 1.0 | −3.7 |
| 5F-6F | −7.1 | −7.4 | −6.2 | 1.7 | −2.9 |
| 5Cl-6Cl | −7.4 | −8.0 | −7.0 | 2.1 | −3.1 |
| 5Br-6Br | −7.6 | −8.5 | −7.4 | 2.4 | −3.5 |
| 5I-6I | −8.3 | −8.6 | −7.4 | 2.1 | −3.2 |
| 5F-7F | −7.3 | −7.9 | −6.6 | 1.9 | −3.1 |
| 5Cl-7Cl | −7.5 | −8.1 | −6.5 | 1.5 | −3.0 |
| 5Br-7Br | −7.6 | −8.2 | −6.5 | 1.4 | −3.1 |
| 5I-7I | −8.0 | −8.3 | −6.4 | 1.2 | −3.1 |
| 6F-6F | −6.8 | −7.5 | −7.1 | 2.8 | −3.3 |
| 6Cl-6Cl | −7.9 | −8.6 | −7.3 | 2.1 | −3.3 |
| 6Br-6Br | −8.2 | −9.1 | −8.0 | 2.8 | −3.9 |
| 6I-6I | −8.9 | −9.2 | −7.7 | 2.1 | −3.5 |
| 6F-7F | −6.9 | −7.7 | −7.2 | 2.8 | −3.4 |
| 6Cl-7Cl | −8.2 | −9.0 | −8.1 | 2.9 | −3.8 |
| 6Br-7Br | −8.7 | −9.6 | −8.3 | 2.9 | −4.1 |
| 6I-7I | −9.5 | −9.8 | −8.4 | 2.7 | −4.1 |
| 7F-7F | −6.8 | −7.6 | −6.8 | 2.5 | −3.3 |
| 7Cl-7Cl | −7.7 | −8.3 | −6.8 | 1.9 | −3.4 |
| 7Br-7Br | −7.9 | −8.6 | −6.5 | 1.5 | −3.5 |
| 7I-7I | −8.6 | −8.6 | −6.9 | 1.6 | −3.3 |
| Dimer | |||
|---|---|---|---|
| 4F-6I | −8.8 | −8.3 | 6.36% |
| 4I-7Cl | −8.4 | −8.3 | 1.43% |
| 5Cl-6F | −7.2 | −7.3 | 1.10% |
| 5Cl-7Br | −7.6 | −7.9 | 3.28% |
| 6F-6Br | −8.1 | −7.7 | 5.42% |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Van Lommel, R.; Bettens, T.; Barlow, T.M.A.; Bertouille, J.; Ballet, S.; De Proft, F. A Quantum Chemical Deep-Dive into the π-π Interactions of 3-Methylindole and Its Halogenated Derivatives—Towards an Improved Ligand Design and Tryptophan Stacking. Pharmaceuticals 2022, 15, 935. https://doi.org/10.3390/ph15080935
Van Lommel R, Bettens T, Barlow TMA, Bertouille J, Ballet S, De Proft F. A Quantum Chemical Deep-Dive into the π-π Interactions of 3-Methylindole and Its Halogenated Derivatives—Towards an Improved Ligand Design and Tryptophan Stacking. Pharmaceuticals. 2022; 15(8):935. https://doi.org/10.3390/ph15080935
Chicago/Turabian StyleVan Lommel, Ruben, Tom Bettens, Thomas M. A. Barlow, Jolien Bertouille, Steven Ballet, and Frank De Proft. 2022. "A Quantum Chemical Deep-Dive into the π-π Interactions of 3-Methylindole and Its Halogenated Derivatives—Towards an Improved Ligand Design and Tryptophan Stacking" Pharmaceuticals 15, no. 8: 935. https://doi.org/10.3390/ph15080935
APA StyleVan Lommel, R., Bettens, T., Barlow, T. M. A., Bertouille, J., Ballet, S., & De Proft, F. (2022). A Quantum Chemical Deep-Dive into the π-π Interactions of 3-Methylindole and Its Halogenated Derivatives—Towards an Improved Ligand Design and Tryptophan Stacking. Pharmaceuticals, 15(8), 935. https://doi.org/10.3390/ph15080935