Improvement Effect of Metformin on Female and Male Reproduction in Endocrine Pathologies and Its Mechanisms


Abstract
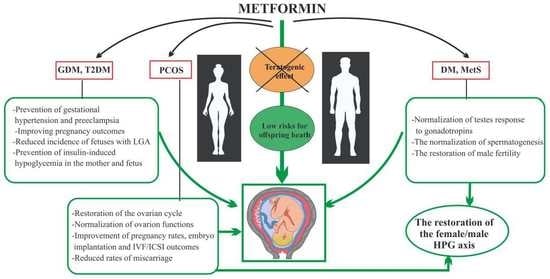
{kind=link}
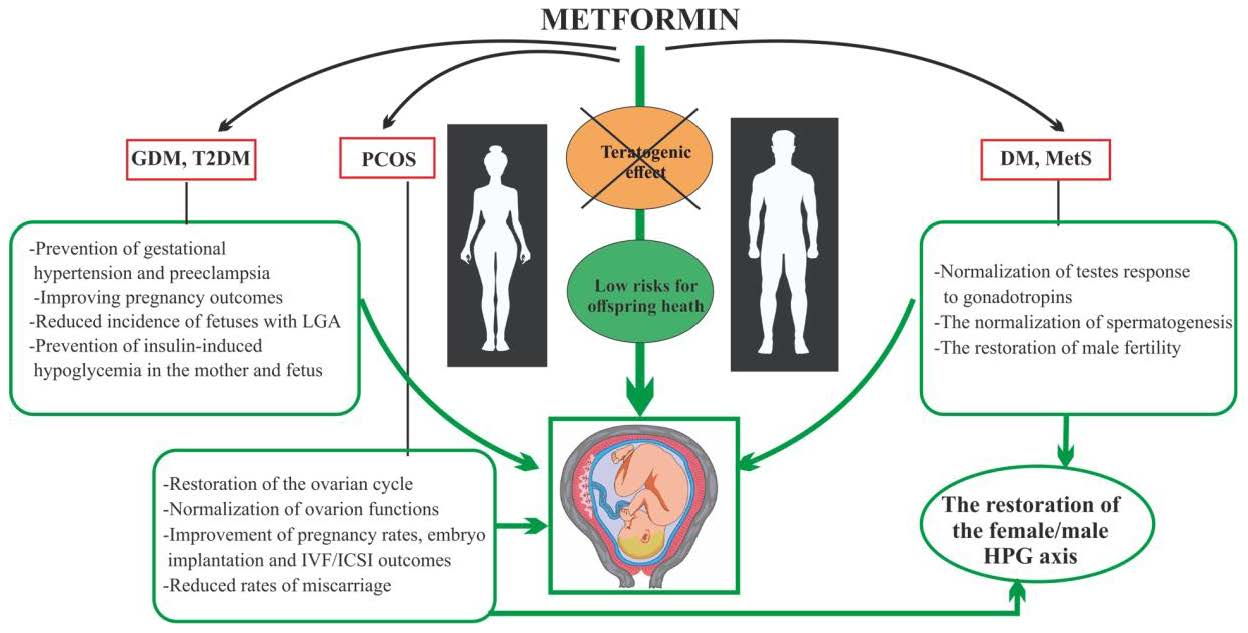
{kind=link}
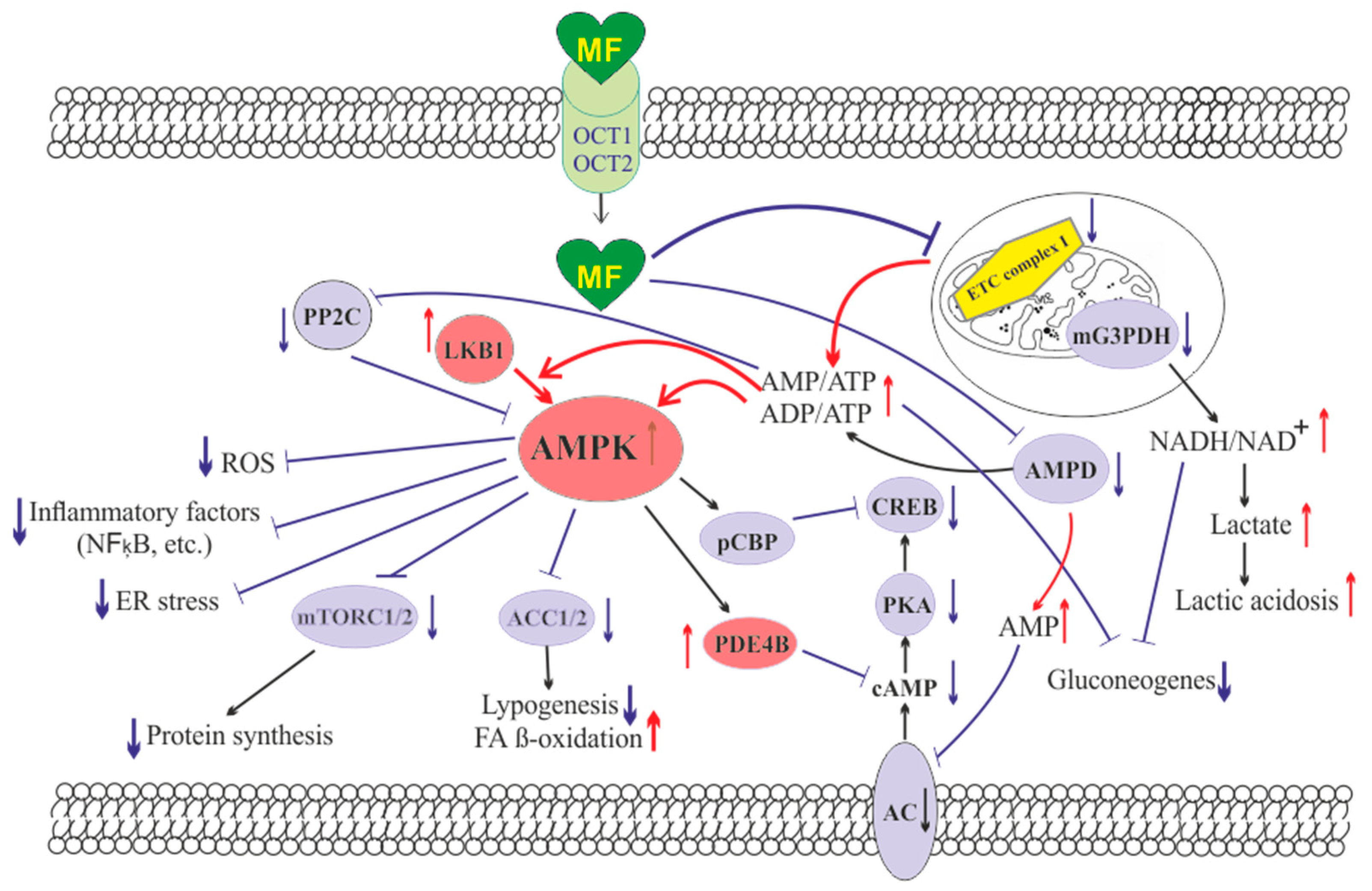
{kind=link}
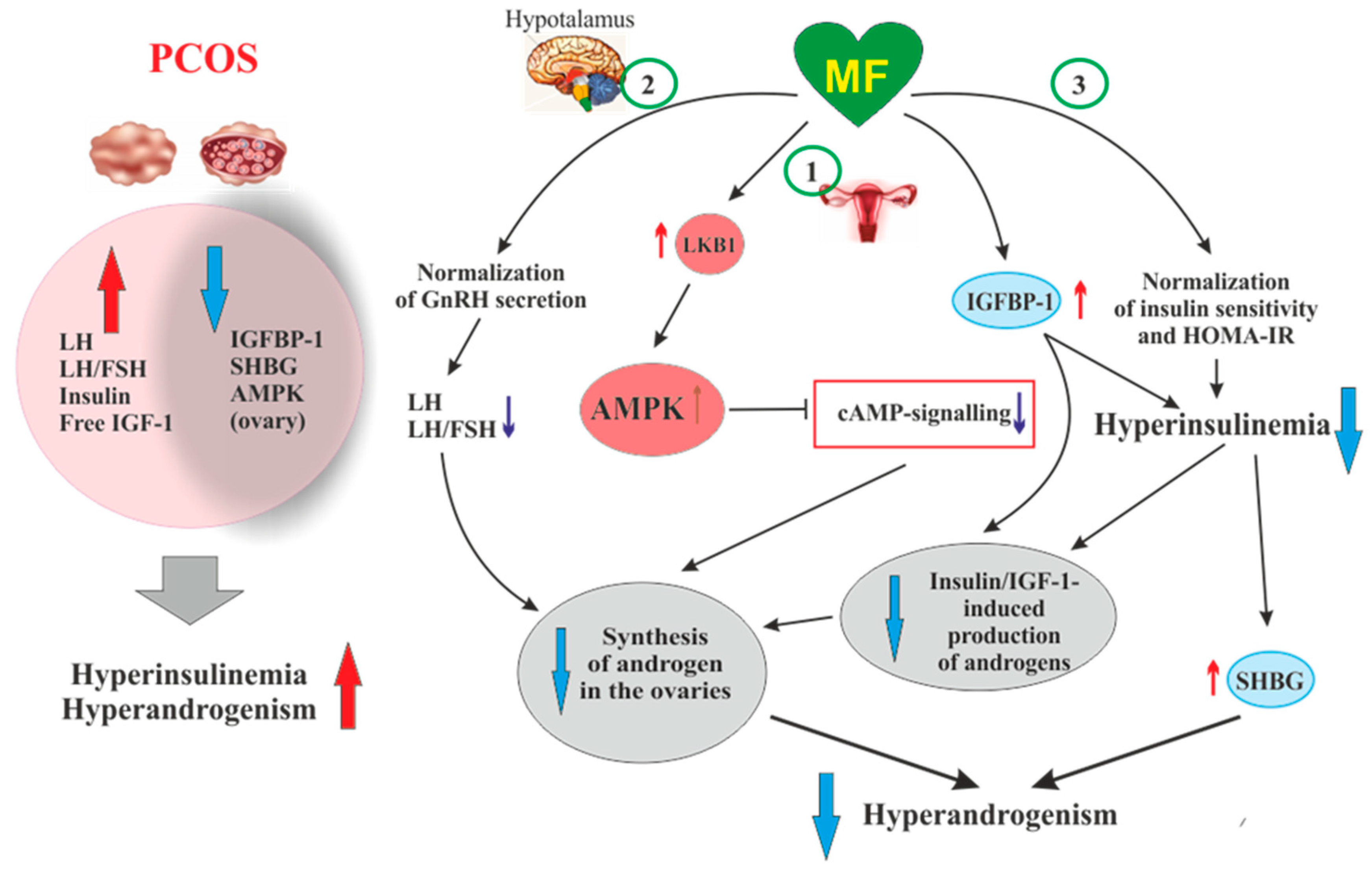
{kind=link}
1. Introduction
2. Summary of Cell Targets and Molecular Mechanisms of Action of Metformin
3. Metformin and Polycystic Ovary Syndrome
3.1. Pathophysiology of Polycystic Ovary Syndrome
3.2. The Use of Metformin in PCOS Women
3.3. Combined Use of Metformin with Clomiphene Citrate, Letrozole, Liraglutide, Saxagliptin, or Oral Contraceptives
3.4. The Mechanisms of Metformin Effects on Reproductive Functions in PCOS
3.4.1. Metformin-Induced Inhibition of Hyperandrogenism and Normalization of the Steroid Hormones Balance
3.4.2. Protective efFect of Metformin against Excess Androgens in PCOS
3.4.3. Effects of Metformin on FSH-Activated Signaling in the PCOS Ovaries
3.4.4. The Effect of Metformin on the Production of Anti-Müllerian Hormone in PCOS
3.4.5. Effect of Metformin on Metalloproteinases in PCOS
3.4.6. Influence of Metformin on Inflammation and Lipid Status in PCOS
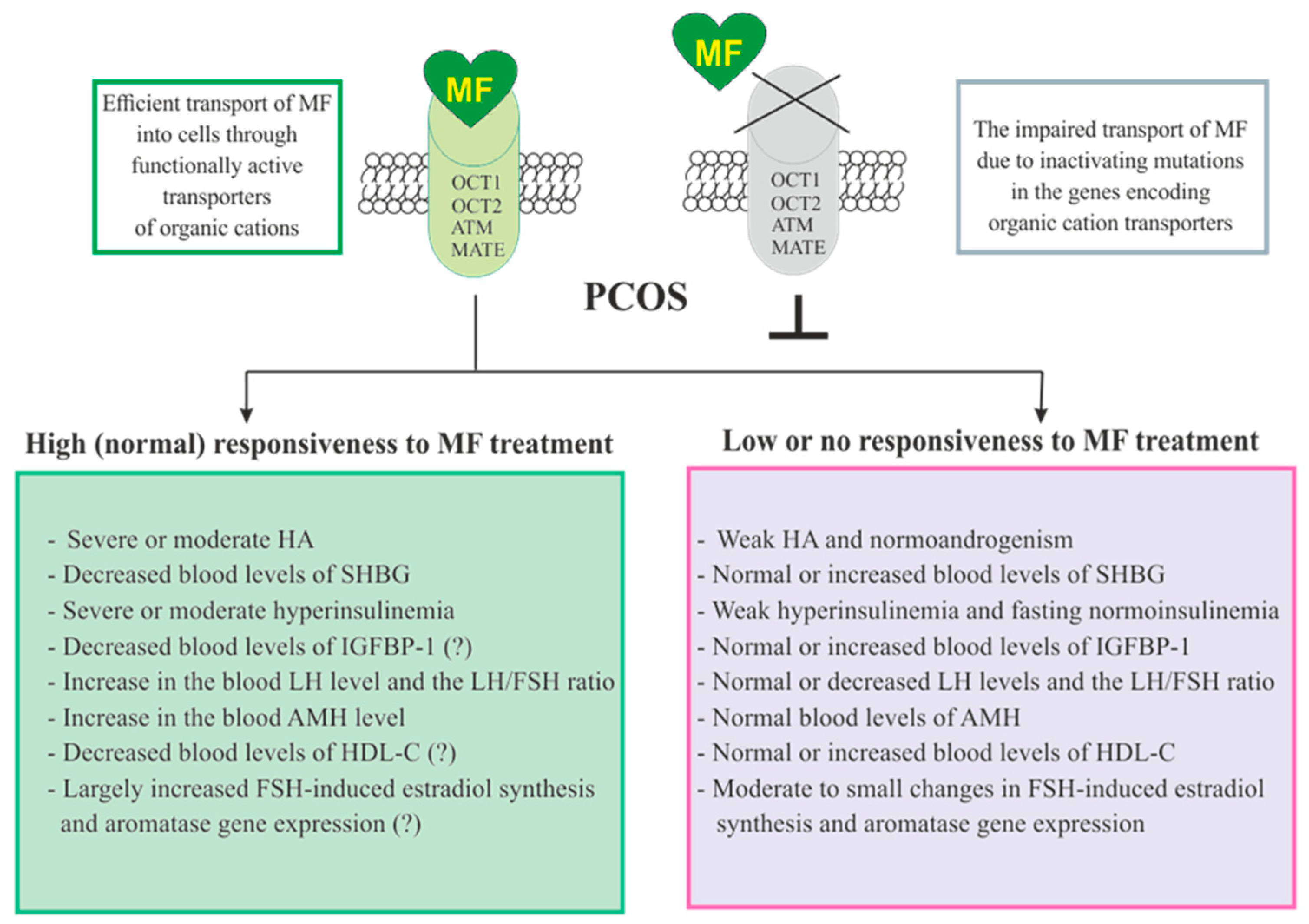
3.5. The Sensitivity of PCOS Women to Metformin Therapy
4. Metformin and Gestational Diabetes Mellitus
5. Metformin Treatment of Women with Diabetes Mellitus and Obesity
6. Metformin and the Male Reproduction
6.1. Effects Metformin on the Male Reproduction in Metabolic Disorders
6.2. The Clinical Studies of the Metformin Efficacy to Treat Reproductive Dysfunctions in Men
6.3. The Experimental Studies of Metformin Effects on Male Reproductive Dysfunctions in Animal Models of Metabolic Diseases
7. Conclusions
Funding
Institutional Review Board Statement
Informed Consent Statement
Conflicts of Interest
Abbreviations
| AICAR | 5-aminoimidazole-4-carboxamide ribonucleotide |
| AMH | anti-Müllerian hormone |
| AMPK | AMP-activated protein kinase |
| ART | assisted reproductive technology |
| CaMKK2 | Ca2+-calmodulin dependent protein kinase kinase 2 |
| CC | clomiphene citrate |
| CREB | cAMP-response element-binding protein |
| CRTC2 | CREB-regulated transcription coactivator 2 |
| DHEA | dehydroepiandrosterone |
| DM | diabetes mellitus |
| ER stress | endoplasmic reticulum stress |
| FSH | follicle-stimulating hormone |
| GDM | gestational diabetes mellitus |
| GLP-1 | glucagon-like peptide-1 |
| GnRH | gonadotropin-releasing hormone |
| HA | hyperandrogenism |
| hCG | human chorionic gonadotropin |
| HDL-C | high-density lipoprotein cholesterol |
| HFD | high-fat diet |
| HOMA-IR | homeostasis model assessment of insulin resistance |
| HPG axis | hypothalamic-pituitary-gonadal axis |
| 3β- and 17β-HSD | 3β- and 17β-hydroxysteroid dehydrogenases |
| ICSI | intracytoplasmic sperm injection |
| IGF-1 | insulin-like growth factor-1 |
| IGFBP-1 | insulin-like growth factor-binding protein-1 |
| IR | insulin resistance |
| IVF | in vitro fertilization |
| LGA | large for gestational-age |
| LH | luteinizing hormone |
| LKB1 | liver kinase B1 |
| MAPK | mitogen-activated protein kinase |
| MF | metformin |
| mG3PDH | mitochondrial glycerol-3-phosphate dehydrogenase |
| MMP | matrix metalloproteinase |
| mTOR | mammalian target of rapamycin |
| mTORC1, mTORC2 | mTOR complexes 1 and 2 |
| NF-κB | nuclear factor κB |
| OCT1, OCT2 | organic cation transporters-1 and 2 |
| OHSS | ovarian hyperstimulation syndrome |
| PCOS | polycystic ovary syndrome |
| PGC1α | peroxisome proliferator-activated receptor γ coactivator 1-α |
| PI 3-K | phosphatidylinositol 3-kinase |
| PKA | cAMP-dependent protein kinase |
| PlGF | placental growth factor |
| PP2C | protein phosphatase 2C |
| PTEN | phosphatase and tensin homolog |
| sFlt-1 | soluble fms-like tyrosine kinase-1 |
| SGA | small for gestational age |
| SHBG | androgen and sex hormone-binding globulin |
| STZ | streptozotocin |
| T1DM | type 1 diabetes mellitus |
| T2DM | type 2 diabetes mellitus |
| TFAM | mitochondrial transcription factor A |
| TGF-β | transforming growth factor β |
| TNF-α | tumor necrosis factor-α |
| VEGF | vascular endothelial growth factor |
References
- Madsen, K.S.; Chi, Y.; Metzendorf, M.I.; Richter, B.; Hemmingsen, B. Metformin for prevention or delay of type 2 diabetes mellitus and its associated complications in persons at increased risk for the development of type 2 diabetes mellitus. Cochrane Database Syst. Rev. 2019, 12, CD008558. [Google Scholar] [CrossRef] [PubMed]
- Gnesin, F.; Thuesen, A.C.B.; Kähler, L.K.A.; Madsbad, S.; Hemmingsen, B. Metformin monotherapy for adults with type 2 diabetes mellitus. Cochrane Database Syst. Rev. 2020, 6, CD012906. [Google Scholar] [CrossRef]
- Lavine, J.E.; Schwimmer, J.B.; Van Natta, M.L.; Molleston, J.P.; Murray, K.F.; Rosenthal, P.; Abrams, S.H.; Scheimann, A.O.; Sanyal, A.J.; Chalasani, N.; et al. Effect of vitamin E or metformin for treatment of nonalcoholic fatty liver disease in children and adolescents: The TONIC randomized controlled trial. JAMA 2011, 305, 1659–1668. [Google Scholar] [CrossRef] [PubMed]
- Dziubak, A.; Wójcicka, G.; Wojtak, A.; Bełtowski, J. Metabolic Effects of Metformin in the Failing Heart. Int. J. Mol. Sci. 2018, 19, 2869. [Google Scholar] [CrossRef]
- Mohan, M.; Al-Talabany, S.; McKinnie, A.; Mordi, I.R.; Singh, J.S.S.; Gandy, S.J.; Baig, F.; Hussain, M.S.; Bhalraam, U.; Khan, F.; et al. A randomized controlled trial of metformin on left ventricular hypertrophy in patients with coronary artery disease without diabetes: The MET-REMODEL trial. Eur. Heart J. 2019, 40, 3409–3417. [Google Scholar] [CrossRef] [PubMed]
- Bell, S.; Farran, B.; McGurnaghan, S.; McCrimmon, R.J.; Leese, G.P.; Petrie, J.R.; McKeigue, P.; Sattar, N.; Wild, S.; McKnight, J.; et al. Risk of acute kidney injury and survival in patients treated with Metformin: An observational cohort study. BMC Nephrol. 2017, 18, 163. [Google Scholar] [CrossRef]
- Lv, Z.; Guo, Y. Metformin and Its Benefits for Various Diseases. Front. Endocrinol. 2020, 11, 191. [Google Scholar] [CrossRef]
- Lee, J.; Yesilkanal, A.E.; Wynne, J.P.; Frankenberger, C.; Liu, J.; Yan, J.; Elbaz, M.; Rabe, D.C.; Rustandy, F.D.; Tiwari, P.; et al. Effective breast cancer combination therapy targeting BACH1 and mitochondrial metabolism. Nature 2019, 568, 254–258. [Google Scholar] [CrossRef]
- Meyerhardt, J.A.; Irwin, M.L.; Jones, L.W.; Zhang, S.; Campbell, N.; Brown, J.C.; Pollak, M.; Sorrentino, A.; Cartmel, B.; Harrigan, M.; et al. Randomized Phase II Trial of Exercise, Metformin, or Both on Metabolic Biomarkers in Colorectal and Breast Cancer Survivors. JNCI Cancer Spectr. 2019, 4, pkz096. [Google Scholar] [CrossRef]
- Ko, E.M.; Walter, P.; Jackson, A.; Clark, L.; Franasiak, J.; Bolac, C.; Havrilesky, L.J.; Secord, A.A.; Moore, D.T.; Gehrig, P.A.; et al. Metformin is associated with improved survival in endometrial cancer. Gynecol. Oncol. 2014, 132, 438–442. [Google Scholar] [CrossRef]
- Tang, Y.L.; Zhu, L.Y.; Li, Y.; Yu, J.; Wang, J.; Zeng, X.X.; Hu, K.X.; Liu, J.Y.; Xu, J.X. Metformin Use Is Associated with Reduced Incidence and Improved Survival of Endometrial Cancer: A Meta-Analysis. Biomed. Res. Int. 2017, 2017, 5905384. [Google Scholar] [CrossRef] [PubMed]
- Mu, N.; Xu, T.; Gao, M.; Dong, M.; Tang, Q.; Hao, L.; Wang, G.; Li, Z.; Wang, W.; Yang, Y.; et al. Therapeutic effect of metformin in the treatment of endometrial cancer. Oncol. Lett. 2020, 20, 156. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.J.; Zheng, Z.J.; Kan, H.; Song, Y.; Cui, W.; Zhao, G.; Kip, K.E. Reduced risk of colorectal cancer with metformin therapy in patients with type 2 diabetes: A meta-analysis. Diabetes Care 2011, 34, 2323–2328. [Google Scholar] [CrossRef] [PubMed]
- Pircher, A.; Zieher, M.; Eigentler, A.; Pichler, R.; Schäfer, G.; Fritz, J.; Puhr, M.; Steiner, E.; Horninger, W.; Klocker, H.; et al. Antidiabetic drugs influence molecular mechanisms in prostate cancer. Cancer Biol. Ther. 2018, 19, 1153–1161. [Google Scholar] [CrossRef]
- Vallianou, N.G.; Evangelopoulos, A.; Kazazis, C. Metformin and cancer. Rev. Diabet. Stud. 2013, 10, 228–235. [Google Scholar] [CrossRef]
- Podhorecka, M.; Ibanez, B.; Dmoszyńska, A. Metformin—Its potential anti-cancer and anti-aging effects. Postepy Hig. Med. Dosw. 2017, 71, 170–175. [Google Scholar] [CrossRef]
- Batandier, C.; Guigas, B.; Detaille, D.; El-Mir, M.Y.; Fontaine, E.; Rigoulet, M.; Leverve, X.M. The ROS production induced by a reverse-electron flux at respiratory-chain complex 1 is hampered by metformin. J. Bioenerg. Biomembr. 2006, 38, 33–42. [Google Scholar] [CrossRef]
- Viollet, B.; Guigas, B.; Sanz Garcia, N.; Leclerc, J.; Foretz, M.; Andreelli, F. Cellular and molecular mechanisms of metformin: An overview. Clin. Sci. 2012, 122, 253–270. [Google Scholar] [CrossRef]
- Foretz, M.; Guigas, B.; Bertrand, L.; Pollak, M.; Viollet, B. Metformin: From mechanisms of action to therapies. Cell Metab. 2014, 20, 953–966. [Google Scholar] [CrossRef]
- Foretz, M.; Guigas, B.; Viollet, B. Understanding the glucoregulatory mechanisms of metformin in type 2 diabetes mellitus. Nat. Rev. Endocrinol. 2019, 15, 569. [Google Scholar] [CrossRef]
- Zhang, C.S.; Li, M.; Ma, T.; Zong, Y.; Cui, J.; Feng, J.W.; Wu, Y.Q.; Lin, S.Y.; Lin, S.C. Metformin activates AMPK through the lysosomal pathway. Cell Metab. 2016, 24, 521–522. [Google Scholar] [CrossRef]
- Rena, G.; Hardie, D.G.; Pearson, E.R. The mechanisms of action of metformin. Diabetologia 2017, 60, 1577–1585. [Google Scholar] [CrossRef] [PubMed]
- Li, M.; Li, X.; Zhang, H.; Lu, Y. Molecular Mechanisms of Metformin for Diabetes and Cancer Treatment. Front. Physiol. 2018, 9, 1039. [Google Scholar] [CrossRef] [PubMed]
- Agius, L.; Ford, B.E.; Chachra, S.S. The Metformin Mechanism on Gluconeogenesis and AMPK Activation: The Metabolite Perspective. Int. J. Mol. Sci. 2020, 21, 3240. [Google Scholar] [CrossRef] [PubMed]
- An, H.; He, L. Current understanding of metformin effect on the control of hyperglycemia in diabetes. J. Endocrinol. 2016, 228, R97–R106. [Google Scholar] [CrossRef]
- He, L. Metformin and Systemic Metabolism. Trends Pharm. Sci. 2020, 41, 868–881. [Google Scholar] [CrossRef] [PubMed]
- Cioce, M.; Pulito, C.; Strano, S.; Blandino, G.; Fazio, V.M. Metformin: Metabolic Rewiring Faces Tumor Heterogeneity. Cells 2020, 9, 2439. [Google Scholar] [CrossRef]
- Chan, P.; Shao, L.; Tomlinson, B.; Zhang, Y.; Liu, Z.M. Metformin transporter pharmacogenomics: Insights into drug disposition-where are we now? Expert Opin. Drug Metab. Toxicol. 2018, 14, 1149–1159. [Google Scholar] [CrossRef]
- Lee, N.; Hebert, M.F.; Wagner, D.J.; Easterling, T.R.; Liang, C.J.; Rice, K.; Wang, J. Organic Cation Transporter 3 Facilitates Fetal Exposure to Metformin during Pregnancy. Mol. Pharmacol. 2018, 94, 1125–1131. [Google Scholar] [CrossRef]
- Hardie, D.G.; Ross, F.A.; Hawley, S.A. AMPK: A nutrient and energy sensor that maintains energy homeostasis. Nat. Rev. Mol. Cell Biol. 2012, 13, 251–262. [Google Scholar] [CrossRef]
- Hardie, D.G.; Schaffer, B.E.; Brunet, A. AMPK: An Energy-Sensing Pathway with Multiple Inputs and Outputs. Trends Cell Biol. 2016, 27, 190–201. [Google Scholar] [CrossRef] [PubMed]
- Lin, S.C.; Hardie, D.G. AMPK: Sensing Glucose as well as Cellular Energy Status. Cell Metab. 2018, 27, 299–313. [Google Scholar] [CrossRef] [PubMed]
- Hardie, D.G. AMPK: A key regulator of energy balance in the single cell and the whole organism. Int. J. Obes. 2008, 32, S7–S12. [Google Scholar] [CrossRef] [PubMed]
- Hardie, D.G. Keeping the home fires burning: AMP-activated protein kinase. J. R. Soc. Interface 2018, 15, 20170774. [Google Scholar] [CrossRef] [PubMed]
- Lizcano, J.M.; Göransson, O.; Toth, R.; Deak, M.; Morrice, N.A.; Boudeau, J.; Hawley, S.A.; Udd, L.; Mäkelä, T.P.; Hardie, D.G.; et al. LKB1 is a master kinase that activates 13 kinases of the AMPK subfamily, including MARK/PAR-1. EMBO J. 2004, 23, 833–843. [Google Scholar] [CrossRef]
- Hawley, S.A.; Pan, D.A.; Mustard, K.J.; Ross, L.; Bain, J.; Edelman, A.M.; Frenguelli, B.G.; Hardie, D.G. Calmodulin-dependent protein kinase kinase-beta is an alternative upstream kinase for AMP-activated protein kinase. Cell Metab. 2005, 2, 9–19. [Google Scholar] [CrossRef]
- Woods, A.; Dickerson, K.; Heath, R.; Hong, S.P.; Momcilovic, M.; Johnstone, S.R.; Carlson, M.; Carling, D. Ca2+/calmodulin-dependent protein kinase kinase-beta acts upstream of AMP-activated protein kinase in mammalian cells. Cell Metab. 2005, 2, 21–33. [Google Scholar] [CrossRef] [PubMed]
- Momcilovic, M.; Hong, S.P.; Carlson, M. Mammalian TAK1 activates Snf1 protein kinase in yeast and phosphorylates AMP-activated protein kinase in vitro. J. Biol. Chem. 2006, 281, 25336–25343. [Google Scholar] [CrossRef]
- Jia, J.; Abudu, Y.P.; Claude-Taupin, A.; Gu, Y.; Kumar, S.; Choi, S.W.; Peters, R.; Mudd, M.H.; Allers, L.; Salemi, M.; et al. Galectins Control mTOR in Response to Endomembrane Damage. Mol. Cell 2018, 70, 120–135. [Google Scholar] [CrossRef]
- Jia, J.; Bissa, B.; Brecht, L.; Allers, L.; Choi, S.W.; Gu, Y.; Zbinden, M.; Burge, M.R.; Timmins, G.; Hallows, K.; et al. AMPK, a Regulator of Metabolism and Autophagy, Is Activated by Lysosomal Damage via a Novel Galectin-Directed Ubiquitin Signal Transduction System. Mol. Cell. 2020, 77, 951–969. [Google Scholar] [CrossRef]
- Zhou, G.; Myers, R.; Li, Y.; Chen, Y.; Shen, X.; Fenyk-Melody, J.; Wu, M.; Ventre, J.; Doebber, T.; Fujii, N.; et al. Role of AMP-activated protein kinase in mechanism of metformin action. J. Clin. Investig. 2001, 108, 1167–1174. [Google Scholar] [CrossRef] [PubMed]
- Woods, A.; Johnstone, S.R.; Dickerson, K.; Leiper, F.C.; Fryer, L.G.; Neumann, D.; Schlattner, U.; Wallimann, T.; Carlson, M.; Carling, D. LKB1 is the upstream kinase in the AMP-activated protein kinase cascade. Curr. Biol. 2003, 13, 2004–2008. [Google Scholar] [CrossRef] [PubMed]
- Cao, J.; Meng, S.; Chang, E.; Beckwith-Fickas, K.; Xiong, L.; Cole, R.N.; Radovick, S.; Wondisford, F.E.; He, L. Low concentrations of metformin suppress glucose production in hepatocytes through AMP-activated protein kinase (AMPK). J. Biol. Chem. 2014, 289, 20435–20446. [Google Scholar] [CrossRef] [PubMed]
- Oakhill, J.S.; Steel, R.; Chen, Z.P.; Scott, J.W.; Ling, N.; Tam, S.; Kemp, B.E. AMPK is a direct adenylate charge-regulated protein kinase. Science 2011, 332, 1433–1435. [Google Scholar] [CrossRef] [PubMed]
- Xiao, B.; Sanders, M.J.; Underwood, E.; Heath, R.; Mayer, F.V.; Carmena, D.; Jing, C.; Walker, P.A.; Eccleston, J.F.; Haire, L.F.; et al. Structure of mammalian AMPK and its regulation by ADP. Nature 2011, 472, 230–233. [Google Scholar] [CrossRef]
- Ross, F.A.; Jensen, T.E.; Hardie, D.G. Differential regulation by AMP and ADP of AMPK complexes containing different γ subunit isoforms. Biochem. J. 2016, 473, 189–199. [Google Scholar] [CrossRef]
- Zhang, C.S.; Hawley, S.A.; Zong, Y.; Li, M.; Wang, Z.; Gray, A.; Ma, T.; Cui, J.; Feng, J.W.; Zhu, M.; et al. Fructose-1,6-bisphosphate and aldolase mediate glucose sensing by AMPK. Nature 2017, 548, 112–116. [Google Scholar] [CrossRef]
- Zong, Y.; Zhang, C.S.; Li, M.; Wang, W.; Wang, Z.; Hawley, S.A.; Ma, T.; Feng, J.W.; Tian, X.; Qi, Q.; et al. Hierarchical activation of compartmentalized pools of AMPK depends on severity of nutrient or energy stress. Cell Res. 2019, 29, 460–473. [Google Scholar] [CrossRef]
- Davies, S.P.; Helps, N.R.; Cohen, P.T.; Hardie, D.G. 5’-AMP inhibits dephosphorylation, as well as promoting phosphorylation, of the AMP-activated protein kinase. Studies using bacterially expressed human protein phosphatase-2C alpha and native bovine protein phosphatase-2AC. FEBS Lett. 1995, 377, 421–425. [Google Scholar] [CrossRef]
- Suter, M.; Riek, U.; Tuerk, R.; Schlattner, U.; Wallimann, T.; Neumann, D. Dissecting the role of 5’-AMP for allosteric stimulation, activation, and deactivation of AMP-activated protein kinase. J. Biol. Chem. 2006, 281, 32207–32216. [Google Scholar] [CrossRef]
- El-Mir, M.Y.; Nogueira, V.; Fontaine, E.; Averet, N.; Rigoulet, M.; Leverve, X. Dimethylbiguanide inhibits cell respiration via an indirect effect targeted on the respiratory chain complex I. J. Biol. Chem. 2000, 275, 223–228. [Google Scholar] [CrossRef]
- Owen, M.R.; Doran, E.; Halestrap, A.P. Evidence that metformin exerts its anti-diabetic effects through inhibition of complex 1 of the mitochondrial respiratory chain. Biochem. J. 2000, 348, 607–614. [Google Scholar] [CrossRef]
- Foretz, M.; Hébrard, S.; Leclerc, J.; Zarrinpashneh, E.; Soty, M.; Mithieux, G.; Sakamoto, K.; Andreelli, F.; Viollet, B. Metformin inhibits hepatic gluconeogenesis in mice independently of the LKB1/AMPK pathway via a decrease in hepatic energy state. J. Clin. Investig. 2010, 120, 2355–2369. [Google Scholar] [CrossRef]
- Bridges, H.R.; Jones, A.J.; Pollak, M.N.; Hirst, J. Effects of metformin and other biguanides on oxidative phosphorylation in mitochondria. Biochem. J. 2014, 462, 475–487. [Google Scholar] [CrossRef] [PubMed]
- Ouyang, J.; Parakhia, R.A.; Ochs, R.S. Metformin activates AMP kinase through inhibition of AMP deaminase. J. Biol. Chem. 2011, 286, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Meng, S.; Cao, J.; He, Q.; Xiong, L.; Chang, E.; Radovick, S.; Wondisford, F.E.; He, L. Metformin activates AMP-activated protein kinase by promoting formation of the αβγ heterotrimeric complex. J. Biol. Chem. 2015, 290, 3793–3802. [Google Scholar] [CrossRef] [PubMed]
- He, L.; Wondisford, F.E. Metformin action: Concentrations matter. Cell Metab. 2015, 21, 159–162. [Google Scholar] [CrossRef] [PubMed]
- Sliwinska, A.; Drzewoski, J. Molecular action of metformin in hepatocytes: An updated insight. Curr. Diabetes Rev. 2015, 11, 175–181. [Google Scholar] [CrossRef] [PubMed]
- Karnewar, S.; Neeli, P.K.; Panuganti, D.; Kotagiri, S.; Mallappa, S.; Jain, N.; Jerald, M.K.; Kotamraju, S. Metformin regulates mitochondrial biogenesis and senescence through AMPK mediated H3K79 methylation: Relevance in age-associated vascular dysfunction. Biochim. Biophys. Acta Mol. Basis Dis. 2018, 1864, 1115–1128. [Google Scholar] [CrossRef] [PubMed]
- Rattan, R.; Giri, S.; Hartmann, L.C.; Shridhar, V. Metformin attenuates ovarian cancer cell growth in an AMP-kinase dispensable manner. J. Cell. Mol. Med. 2011, 15, 166–178. [Google Scholar] [CrossRef]
- Fullerton, M.D.; Galic, S.; Marcinko, K.; Sikkema, S.; Pulinilkunnil, T.; Chen, Z.P.; O’Neill, H.M.; Ford, R.J.; Palanivel, R.; O’Brien, M.; et al. Single phosphorylation sites in Acc1 and Acc2 regulate lipid homeostasis and the insulin-sensitizing effects of metformin. Nat. Med. 2013, 19, 1649–1654. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.; Yang, G.; Kim, Y.; Kim, J.; Ha, J. AMPK activators: Mechanisms of action and physiological activities. Exp. Mol. Med. 2016, 48, e224. [Google Scholar] [CrossRef] [PubMed]
- Motoshima, H.; Goldstein, B.J.; Igata, M.; Araki, E. AMPK and cell proliferation—AMPK as a therapeutic target for atherosclerosis and cancer. J. Physiol. 2006, 574, 63–71. [Google Scholar] [CrossRef]
- Choi, Y.K.; Park, K.G. Metabolic roles of AMPK and metformin in cancer cells. Mol. Cells 2013, 36, 279–287. [Google Scholar] [CrossRef] [PubMed]
- Gao, F.; Chen, J.; Zhu, H. A potential strategy for treating atherosclerosis: Improving endothelial function via AMP-activated protein kinase. Sci. China Life Sci. 2018, 61, 1024–1029. [Google Scholar] [CrossRef] [PubMed]
- Lyons, C.L.; Roche, H.M. Nutritional Modulation of AMPK-Impact upon Metabolic-Inflammation. Int. J. Mol. Sci. 2018, 19, 3092. [Google Scholar] [CrossRef]
- Viollet, B.; Foretz, M. Revisiting the mechanisms of metformin action in the liver. Ann. Endocrinol. 2013, 74, 123–129. [Google Scholar] [CrossRef]
- Johanns, M.; Lai, Y.C.; Hsu, M.F.; Jacobs, R.; Vertommen, D.; Van Sande, J.; Dumont, J.E.; Woods, A.; Carling, D.; Hue, L.; et al. AMPK antagonizes hepatic glucagon-stimulated cyclic AMP signalling via phosphorylation-induced activation of cyclic nucleotide phosphodiesterase 4B. Nat. Commun. 2016, 7, 10856. [Google Scholar] [CrossRef]
- Miller, R.A.; Chu, Q.; Xie, J.; Foretz, M.; Viollet, B.; Birnbaum, M.J. Biguanides suppress hepatic glucagon signalling by decreasing production of cyclic AMP. Nature 2013, 494, 256–260. [Google Scholar] [CrossRef]
- He, L.; Sabet, A.; Djedjos, S.; Miller, R.; Sun, X.; Hussain, M.A.; Radovick, S.; Wondisford, F.E. Metformin and insulin suppress hepatic gluconeogenesis through phosphorylation of CREB binding protein. Cell 2009, 137, 635–646. [Google Scholar] [CrossRef]
- Madiraju, A.K.; Erion, D.M.; Rahimi, Y.; Zhang, X.M.; Braddock, D.T.; Albright, R.A.; Prigaro, B.J.; Wood, J.L.; Bhanot, S.; MacDonald, M.J.; et al. Metformin suppresses gluconeogenesis by inhibiting mitochondrial glycerophosphate dehydrogenase. Nature 2014, 510, 542–546. [Google Scholar] [CrossRef] [PubMed]
- Madiraju, A.K.; Qiu, Y.; Perry, R.J.; Rahimi, Y.; Zhang, X.M.; Zhang, D.; Camporez, J.G.; Cline, G.W.; Butrico, G.M.; Kemp, B.E.; et al. Metformin inhibits gluconeogenesis via a redox-dependent mechanism in vivo. Nat. Med. 2018, 24, 1384–1394. [Google Scholar] [CrossRef] [PubMed]
- Cuyàs, E.; Verdura, S.; Llorach-Pares, L.; Fernández-Arroyo, S.; Luciano-Mateo, F.; Cabré, N.; Stursa, J.; Werner, L.; Martin-Castillo, B.; Viollet, B.; et al. Metformin directly targets the H3K27me3 demethylase KDM6A/UTX. Aging Cell. 2018, 17, e12772. [Google Scholar] [CrossRef] [PubMed]
- Wu, H.; Esteve, E.; Tremaroli, V.; Khan, M.T.; Caesar, R.; Mannerås-Holm, L.; Ståhlman, M.; Olsson, L.M.; Serino, M.; Planas-Fèlix, M.; et al. Metformin alters the gut microbiome of individuals with treatment-naive type 2 diabetes, contributing to the therapeutic effects of the drug. Nat. Med. 2017, 23, 850–858. [Google Scholar] [CrossRef]
- Shin, N.R.; Lee, J.C.; Lee, H.Y.; Kim, M.S.; Whon, T.W.; Lee, M.S.; Bae, J.W. An increase in the Akkermansia spp. population induced by metformin treatment improves glucose homeostasis in diet-induced obese mice. Gut 2014, 63, 727–735. [Google Scholar] [CrossRef]
- Duca, F.A.; Côté, C.D.; Rasmussen, B.A.; Zadeh-Tahmasebi, M.; Rutter, G.A.; Filippi, B.M.; Lam, T.K. Metformin activates a duodenal Ampk-dependent pathway to lower hepatic glucose production in rats. Nat. Med. 2015, 21, 506–511. [Google Scholar] [CrossRef]
- Hattori, Y.; Suzuki, K.; Hattori, S.; Kasai, K. Metformin inhibits cytokine-induced nuclear factor kappaB activation via AMP-activated protein kinase activation in vascular endothelial cells. Hypertension 2006, 47, 1183–1188. [Google Scholar] [CrossRef]
- Huang, N.L.; Chiang, S.H.; Hsueh, C.H.; Liang, Y.J.; Chen, Y.J.; Lai, L.P. Metformin inhibits TNF-alpha-induced IkappaB kinase phosphorylation, IkappaB-alpha degradation and IL-6 production in endothelial cells through PI3K-dependent AMPK phosphorylation. Int. J. Cardiol. 2009, 134, 169–175. [Google Scholar] [CrossRef]
- Okamura, H.; Yoshida, K.; Sasaki, E.; Qiu, L.; Amorim, B.R.; Morimoto, H.; Haneji, T. Expression of PTEN and Akt phosphorylation in lipopolysaccharide-treated NIH3T3 cells. Cell Biol. Int. 2007, 31, 119–125. [Google Scholar] [CrossRef]
- Lee, S.K.; Lee, J.O.; Kim, J.H.; Kim, S.J.; You, G.Y.; Moon, J.W.; Jung, J.H.; Park, S.H.; Uhm, K.O.; Park, J.M.; et al. Metformin sensitizes insulin signaling through AMPK-mediated PTEN down-regulation in preadipocyte 3T3-L1 cells. J. Cell. Biochem. 2011, 112, 1259–1267. [Google Scholar] [CrossRef]
- Balen, A.H.; Morley, L.C.; Misso, M.; Franks, S.; Legro, R.S.; Wijeyaratne, C.N.; Stener-Victorin, E.; Fauser, B.C.; Norman, R.J.; Teede, H. The management of anovulatory infertility in women with polycystic ovary syndrome: An analysis of the evidence to support the development of global WHO guidance. Hum. Reprod. Update 2016, 22, 687–708. [Google Scholar] [CrossRef] [PubMed]
- Norman, R.J.; Dewailly, D.; Legro, R.S.; Hickey, T.E. Polycystic ovary syndrome. Lancet 2007, 370, 685–697. [Google Scholar] [CrossRef]
- Fauser, B.C.; Tarlatzis, B.C.; Rebar, R.W.; Legro, R.S.; Balen, A.H.; Lobo, R.; Carmina, E.; Chang, J.; Yildiz, B.O.; Laven, J.S.; et al. Consensus on women’s health aspects of polycystic ovary syndrome (PCOS): The Amsterdam ESHRE/ASRM-Sponsored 3rd PCOS Consensus Workshop Group. Fertil. Steril. 2012, 97, 28–38.e25. [Google Scholar] [CrossRef] [PubMed]
- Abbott, D.H.; Dumesic, D.A.; Levine, J.E. Hyperandrogenic origins of polycystic ovary syndrome—Implications for pathophysiology and therapy. Expert Rev. Endocrinol. Metab. 2019, 14, 131–143. [Google Scholar] [CrossRef]
- Witchel, S.F.; Oberfield, S.E.; Peña, A.S. Polycystic Ovary Syndrome: Pathophysiology, Presentation, and Treatment with Emphasis on Adolescent Girls. J. Endocr. Soc. 2019, 3, 1545–1573. [Google Scholar] [CrossRef] [PubMed]
- Alvarez-Blasco, F.; Botella-Carretero, J.I.; San Millán, J.L.; Escobar-Morreale, H.F. Prevalence and characteristics of the polycystic ovary syndrome in overweight and obese women. Arch. Intern. Med. 2006, 166, 2081–2086. [Google Scholar] [CrossRef]
- Legro, R.S.; Barnhart, H.X.; Schlaff, W.D.; Carr, B.R.; Diamond, M.P.; Carson, S.A.; Steinkampf, M.P.; Coutifaris, C.; McGovern, P.G.; Cataldo, N.A.; et al. Clomiphene, metformin, or both for infertility in the polycystic ovary syndrome. N. Engl. J. Med. 2007, 356, 551–566. [Google Scholar] [CrossRef]
- Azziz, R.; Carmina, E.; Dewailly, D.; Diamanti-Kandarakis, E.; Escobar-Morreale, H.F.; Futterweit, W.; Janssen, O.E.; Legro, R.S.; Norman, R.J.; Taylor, A.E.; et al. The Androgen Excess and PCOS Society criteria for the polycystic ovary syndrome: The complete task force report. Fertil. Steril. 2009, 91, 456–488. [Google Scholar] [CrossRef]
- Li, R.; Zhang, Q.; Yang, D.; Li, S.; Lu, S.; Wu, X.; Wei, Z.; Song, X.; Wang, X.; Fu, S.; et al. Prevalence of polycystic ovary syndrome in women in China: A large community-based study. Hum. Reprod. 2013, 28, 2562–2569. [Google Scholar] [CrossRef]
- Kollmann, M.; Klaritsch, P.; Martins, W.P.; Guenther, F.; Schneider, V.; Herzog, S.A.; Craciunas, L.; Lang, U.; Obermayer-Pietsch, B.; Lerchbaum, E.; et al. Maternal and neonatal outcomes in pregnant women with PCOS: Comparison of different diagnostic definitions. Hum. Reprod. 2015, 30, 2396–2403. [Google Scholar] [CrossRef]
- Cassar, S.; Misso, M.L.; Hopkins, W.G.; Shaw, C.S.; Teede, H.J.; Stepto, N.K. Insulin resistance in polycystic ovary syndrome: A systematic review and meta-analysis of euglycaemic-hyperinsulinaemic clamp studies. Hum. Reprod. 2016, 31, 2619–2631. [Google Scholar] [CrossRef] [PubMed]
- Spritzer, P.M. Polycystic ovary syndrome: Reviewing diagnosis and management of metabolic disturbances. Arq. Bras. Endocrinol. Metabol. 2014, 58, 182–187. [Google Scholar] [CrossRef] [PubMed]
- Dumesic, D.A.; Oberfield, S.E.; Stener-Victorin, E.; Marshall, J.C.; Laven, J.S.; Legro, R.S. Scientific Statement on the Diagnostic Criteria, Epidemiology, Pathophysiology, and Molecular Genetics of Polycystic Ovary Syndrome. Endocr. Rev. 2015, 36, 487–525. [Google Scholar] [CrossRef] [PubMed]
- Faure, M.; Bertoldo, M.J.; Khoueiry, R.; Bongrani, A.; Brion, F.; Giulivi, C.; Dupont, J.; Froment, P. Metformin in Reproductive Biology. Front. Endocrinol. 2018, 9, 675. [Google Scholar] [CrossRef]
- Jones, M.R.; Goodarzi, M.O. Genetic determinants of polycystic ovary syndrome: Progress and future directions. Fertil. Steril. 2016, 106, 25–32. [Google Scholar] [CrossRef]
- Liu, H.; Zhao, H.; Chen, Z.J. Genome-Wide Association Studies for Polycystic Ovary Syndrome. Semin. Reprod. Med. 2016, 34, 224–229. [Google Scholar] [CrossRef]
- Khan, M.J.; Ullah, A.; Basit, S. Genetic Basis of Polycystic Ovary Syndrome (PCOS): Current Perspectives. Appl. Clin. Genet. 2019, 12, 249–260. [Google Scholar] [CrossRef]
- Dapas, M.; Lin, F.T.J.; Nadkarni, G.N.; Sisk, R.; Legro, R.S.; Urbanek, M.; Hayes, M.G.; Dunaif, A. Distinct subtypes of polycystic ovary syndrome with novel genetic associations: An unsupervised, phenotypic clustering analysis. PLoS Med. 2020, 17, e1003132. [Google Scholar] [CrossRef]
- Xita, N.; Tsatsoulis, A. Review: Fetal programming of polycystic ovary syndrome by androgen excess: Evidence from experimental, clinical, and genetic association studies. J. Clin. Endocrinol. Metab. 2006, 91, 1660–1666. [Google Scholar] [CrossRef]
- Ilie, I.R.; Georgescu, C.E. Polycystic Ovary Syndrome-Epigenetic Mechanisms and Aberrant MicroRNA. Adv. Clin. Chem. 2015, 71, 25–45. [Google Scholar] [CrossRef]
- Yu, Y.Y.; Sun, C.X.; Liu, Y.K.; Li, Y.; Wang, L.; Zhang, W. Genome-wide screen of ovary-specific DNA methylation in polycystic ovary syndrome. Fertil. Steril. 2015, 104, 145–153. [Google Scholar] [CrossRef]
- Wang, S.; Alvero, R. Racial and ethnic differences in physiology and clinical symptoms of polycystic ovary syndrome. Semin. Reprod. Med. 2013, 31, 365–369. [Google Scholar] [CrossRef]
- Merkin, S.S.; Phy, J.L.; Sites, C.K.; Yang, D. Environmental determinants of polycystic ovary syndrome. Fertil. Steril. 2016, 106, 16–24. [Google Scholar] [CrossRef] [PubMed]
- Abdolahian, S.; Tehrani, F.R.; Amiri, M.; Ghodsi, D.; Yarandi, R.B.; Jafari, M.; Majd, H.A.; Nahidi, F. Effect of lifestyle modifications on anthropometric, clinical, and biochemical parameters in adolescent girls with polycystic ovary syndrome: A systematic review and meta-analysis. BMC Endocr. Disord. 2020, 20, 71. [Google Scholar] [CrossRef]
- Abbott, D.H.; Barnett, D.K.; Bruns, C.M.; Dumesic, D.A. Androgen excess fetal programming of female reproduction: A developmental etiology for polycystic ovary syndrome? Hum. Reprod. Update 2005, 11, 357–374. [Google Scholar] [CrossRef]
- Abbott, D.H.; Bacha, F. Ontogeny of polycystic ovary syndrome and insulin resistance in utero and early childhood. Fertil. Steril. 2013, 100, 2–11. [Google Scholar] [CrossRef]
- De Melo, A.S.; Dias, S.V.; Cavalli, R.d.C.; Cardoso, V.C.; Bettiol, H.; Barbieri, M.A.; Ferriani, R.A.; Vieira, C.S. Pathogenesis of polycystic ovary syndrome: Multifactorial assessment from the foetal stage to menopause. Reproduction 2015, 150, 1–24. [Google Scholar] [CrossRef]
- Morley, L.C.; Tang, T.; Yasmin, E.; Norman, R.J.; Balen, A.H. Insulin-sensitising drugs (metformin, rosiglitazone, pioglitazone, D-chiro-inositol) for women with polycystic ovary syndrome, oligo amenorrhoea and subfertility. Cochrane Database Syst. Rev. 2017, 11, CD003053. [Google Scholar] [CrossRef]
- Practice Committee of the American Society for Reproductive Medicine. Electronic address: ASRM@asrm.org; Practice Committee of the American Society for Reproductive Medicine. Role of metformin for ovulation induction in infertile patients with polycystic ovary syndrome (PCOS): A guideline. Fertil. Steril. 2017, 108, 426–441. [Google Scholar] [CrossRef]
- Abdalla, M.A.; Deshmukh, H.; Atkin, S.; Sathyapalan, T. A review of therapeutic options for managing the metabolic aspects of polycystic ovary syndrome. Ther. Adv. Endocrinol. Metab. 2020, 11. [Google Scholar] [CrossRef]
- Bordewijk, E.M.; Nahuis, M.; Costello, M.F.; Van der Veen, F.; Tso, L.O.; Mol, B.W.; van Wely, M. Metformin during ovulation induction with gonadotrophins followed by timed intercourse or intrauterine insemination for subfertility associated with polycystic ovary syndrome. Cochrane Database Syst. Rev. 2017, 1, CD009090. [Google Scholar] [CrossRef] [PubMed]
- Sharpe, A.; Morley, L.C.; Tang, T.; Norman, R.J.; Balen, A.H. Metformin for ovulation induction (excluding gonadotrophins) in women with polycystic ovary syndrome. Cochrane Database Syst. Rev. 2019, 12, CD013505. [Google Scholar] [CrossRef]
- Gadalla, M.A.; Norman, R.J.; Tay, C.T.; Hiam, D.S.; Melder, A.; Pundir, J.; Thangaratinam, S.; Teede, H.J.; Mol, B.W.J.; Moran, L.J. Medical and Surgical Treatment of Reproductive Outcomes in Polycystic Ovary Syndrome: An Overview of Systematic Reviews. Int. J. Fertil. Steril. 2020, 13, 257–270. [Google Scholar] [CrossRef]
- Wu, Y.; Tu, M.; Huang, Y.; Liu, Y.; Zhang, D. Association of Metformin with Pregnancy Outcomes in Women With Polycystic Ovarian Syndrome Undergoing In Vitro Fertilization: A Systematic Review and Meta-analysis. JAMA Netw. Open 2020, 3, e2011995. [Google Scholar] [CrossRef]
- Rojas, J.; Chávez-Castillo, M.; Bermúdez, V. The Role of Metformin in Metabolic Disturbances during Pregnancy: Polycystic Ovary Syndrome and Gestational Diabetes Mellitus. Int. J. Reprod. Med. 2014, 2014, 797681. [Google Scholar] [CrossRef]
- Teede, H.J.; Misso, M.L.; Costello, M.F.; Dokras, A.; Laven, J.; Moran, L.; Piltonen, T.; Norman, R.J.; International PCOS Network. Recommendations from the international evidence-based guideline for the assessment and management of polycystic ovary syndrome. Clin. Endocrinol. 2018, 89, 251–268. [Google Scholar] [CrossRef]
- Gormsen, L.C.; Søndergaard, E.; Christensen, N.L.; Brøsen, K.; Jessen, N.; Nielsen, S. Metformin increases endogenous glucose production in non-diabetic individuals and individuals with recent-onset type 2 diabetes. Diabetologia 2019, 62, 1251–1256. [Google Scholar] [CrossRef]
- McCreight, L.J.; Mari, A.; Coppin, L.; Jackson, N.; Umpleby, A.M.; Pearson, E.R. Metformin increases fasting glucose clearance and endogenous glucose production in non-diabetic individuals. Diabetologia 2020, 63, 444–447. [Google Scholar] [CrossRef]
- Bryrup, T.; Thomsen, C.W.; Kern, T.; Allin, K.H.; Brandslund, I.; Jørgensen, N.R.; Vestergaard, H.; Hansen, T.; Hansen, T.H.; Pedersen, O.; et al. Metformin-induced changes of the gut microbiota in healthy young men: Results of a non-blinded, one-armed intervention study. Diabetologia 2019, 62, 1024–1035. [Google Scholar] [CrossRef]
- Derkach, K.V.; Kuznetsova, L.A.; Sharova, T.S.; Ignat’eva, P.A.; Bondareva, V.M.; Shpakov, A.O. The effect of prolonged metformin treatment on the activity of the adenylate cyclase system and NO-synthase in the brain and myocardium of obese rats. Cell Tissue Biol. 2015, 9, 385–394. [Google Scholar] [CrossRef]
- Thessaloniki ESHRE/ASRM-Sponsored PCOS Consensus Workshop Group. Consensus on infertility treatment related to polycystic ovary syndrome. Fertil. Steril. 2008, 89, 505–522. [Google Scholar] [CrossRef]
- Lautatzis, M.E.; Goulis, D.G.; Vrontakis, M. Efficacy and safety of metformin during pregnancy in women with gestational diabetes mellitus or polycystic ovary syndrome: A systematic review. Metabolism 2013, 62, 1522–1534. [Google Scholar] [CrossRef]
- Sivalingam, V.N.; Myers, J.; Nicholas, S.; Balen, A.H.; Crosbie, E.J. Metformin in reproductive health, pregnancy and gynaecological cancer: Established and emerging indications. Hum. Reprod. Update 2014, 20, 853–868. [Google Scholar] [CrossRef]
- Feng, L.; Lin, X.F.; Wan, Z.H.; Hu, D.; Du, Y.K. Efficacy of metformin on pregnancy complications in women with polycystic ovary syndrome: A meta-analysis. Gynecol. Endocrinol. 2015, 31, 833–839. [Google Scholar] [CrossRef]
- Sinai Talaulikar, V.; Tang, T.; Yasmin, E. Role of Metformin in Women’s Health: Review of Its Current Place in Clinical Practice and Emerging Indications for Future. Obstet. Gynecol. Surv. 2016, 71, 307–317. [Google Scholar] [CrossRef]
- Tan, X.; Li, S.; Chang, Y.; Fang, C.; Liu, H.; Zhang, X.; Wang, Y. Effect of metformin treatment during pregnancy on women with PCOS: A systematic review and meta-analysis. Clin. Investig. Med. 2016, 39, 120–131. [Google Scholar] [CrossRef]
- Zeng, X.L.; Zhang, Y.F.; Tian, Q.; Xue, Y.; An, R.F. Effects of metformin on pregnancy outcomes in women with polycystic ovary syndrome: A meta-analysis. Medicine 2016, 95, e4526. [Google Scholar] [CrossRef]
- Løvvik, T.S.; Carlsen, S.M.; Salvesen, Ø.; Steffensen, B.; Bixo, M.; Gómez-Real, F.; Lønnebotn, M.; Hestvold, K.V.; Zabielska, R.; Hirschberg, A.L.; et al. Use of metformin to treat pregnant women with polycystic ovary syndrome (PregMet2): A randomised, double-blind, placebo-controlled trial. Lancet Diabetes Endocrinol. 2019, 7, 256–266. [Google Scholar] [CrossRef]
- Baillargeon, J.P.; Jakubowicz, D.J.; Iuorno, M.J.; Jakubowicz, S.; Nestler, J.E. Effects of metformin and rosiglitazone, alone and in combination, in nonobese women with polycystic ovary syndrome and normal indices of insulin sensitivity. Fertil. Steril. 2004, 82, 893–902. [Google Scholar] [CrossRef]
- Carmina, E.; Lobo, R.A. Does metformin induce ovulation in normoandrogenic anovulatory women? Am. J. Obstet. Gynecol. 2004, 191, 1580–1584. [Google Scholar] [CrossRef]
- Tang, T.; Lord, J.M.; Norman, R.J.; Yasmin, E.; Balen, A.H. Insulin-sensitising drugs (metformin, rosiglitazone, pioglitazone, D-chiro-inositol) for women with polycystic ovary syndrome, oligo amenorrhoea and subfertility. Cochrane Database Syst. Rev. 2010, 1, CD003053. [Google Scholar] [CrossRef]
- Kjøtrød, S.B.; Carlsen, S.M.; Rasmussen, P.E.; Holst-Larsen, T.; Mellembakken, J.; Thurin-Kjellberg, A.; Haapaniemikouru, K.; Morin-Papunen, L.; Humaidan, P.; Sunde, A.; et al. Use of metformin before and during assisted reproductive technology in non-obese young infertile women with polycystic ovary syndrome: A prospective, randomized, double-blind, multi-centre study. Hum. Reprod. 2011, 26, 2045–2053. [Google Scholar] [CrossRef] [PubMed]
- Morin-Papunen, L.; Rantala, A.S.; Unkila-Kallio, L.; Tiitinen, A.; Hippeläinen, M.; Perheentupa, A.; Tinkanen, H.; Bloigu, R.; Puukka, K.; Ruokonen, A.; et al. Metformin improves pregnancy and live-birth rates in women with polycystic ovary syndrome (PCOS): A multicenter, double-blind, placebo-controlled randomized trial. J. Clin. Endocrinol. Metab. 2012, 97, 1492–1500. [Google Scholar] [CrossRef] [PubMed]
- Tang, T.; Lord, J.M.; Norman, R.J.; Yasmin, E.; Balen, A.H. Insulin-sensitising drugs (metformin, rosiglitazone, pioglitazone, D-chiro-inositol) for women with polycystic ovary syndrome, oligo amenorrhoea and subfertility. Cochrane Database Syst. Rev. 2012, 5, CD003053. [Google Scholar] [CrossRef] [PubMed]
- Tso, L.O.; Costello, M.F.; Albuquerque, L.E.; Andriolo, R.B.; Macedo, C.R. Metformin treatment before and during IVF or ICSI in women with polycystic ovary syndrome. Cochrane Database Syst. Rev. 2014, 2014, CD006105. [Google Scholar] [CrossRef] [PubMed]
- Kollmann, M.; Martins, W.P.; Lima, M.L.; Craciunas, L.; Nastri, C.O.; Richardson, A.; Raine-Fenning, N. Strategies for improving outcome of assisted reproduction in women with polycystic ovary syndrome: Systematic review and meta-analysis. Ultrasound Obstet. Gynecol. 2016, 48, 709–718. [Google Scholar] [CrossRef]
- Palomba, S.; Falbo, A.; Carrillo, L.; Villani, M.T.; Orio, F.; Russo, T.; Di Cello, A.; Cappiello, F.; Capasso, S.; Tolino, A.; et al. Metformin reduces risk of ovarian hyperstimulation syndrome in patients with polycystic ovary syndrome during gonadotropin-stimulated in vitro fertilization cycles: A randomized, controlled trial. Fertil. Steril. 2011, 96, 1384–1390. [Google Scholar] [CrossRef]
- Palomba, S.; Falbo, A.; La Sala, G.B. Effects of metformin in women with polycystic ovary syndrome treated with gonadotrophins for in vitro fertilisation and intracytoplasmic sperm injection cycles: A systematic review and meta-analysis of randomised controlled trials. BJOG 2013, 120, 267–276. [Google Scholar] [CrossRef] [PubMed]
- Costello, M.F.; Chapman, M.; Conway, U. A systematic review and meta-analysis of randomized controlled trials on metformin co-administration during gonadotrophin ovulation induction or IVF in women with polycystic ovary syndrome. Hum. Reprod. 2006, 21, 1387–1399. [Google Scholar] [CrossRef] [PubMed]
- Tso, L.O.; Costello, M.F.; Albuquerque, L.E.; Andriolo, R.B.; Freitas, V. Metformin treatment before and during IVF or ICSI in women with polycystic ovary syndrome. Cochrane Database Syst. Rev. 2009, 2, CD006105. [Google Scholar] [CrossRef]
- Palomba, S.; Falbo, A.; Zullo, F.; Orio, F., Jr. Evidence-based and potential benefits of metformin in the polycystic ovary syndrome: A comprehensive review. Endocr. Rev. 2009, 30, 1–50. [Google Scholar] [CrossRef]
- Abdalmageed, O.S.; Farghaly, T.A.; Abdelaleem, A.A.; Abdelmagied, A.E.; Ali, M.K.; Abbas, A.M. Impact of Metformin on IVF Outcomes in Overweight and Obese Women With Polycystic Ovary Syndrome: A Randomized Double-Blind Controlled Trial. Reprod. Sci. 2019, 26, 1336–1342. [Google Scholar] [CrossRef]
- Barbieri, R.L. Metformin for the treatment of polycystic ovary syndrome. Obstet. Gynecol. 2003, 101, 785–793. [Google Scholar] [CrossRef]
- Abu Hashim, H.; Shokeir, T.; Badawy, A. Letrozole versus combined metformin and clomiphene citrate for ovulation induction in clomiphene-resistant women with polycystic ovary syndrome: A randomized controlled trial. Fertil. Steril. 2010, 94, 1405–1409. [Google Scholar] [CrossRef]
- Bjelica, A.; Trninić-Pjević, A.; Mladenović-Segedi, L.; Cetković, N.; Petrović, D. Comparison of the efficiency of clomiphene citrate and letrozole in combination with metformin in moderately obese clomiphene citrate-resistant polycystic ovarian syndrome patients. Srp. Arh. Celok. Lek. 2016, 144, 146–150. [Google Scholar] [CrossRef]
- Nemati, M.; Nemati, S.; Taheri, A.M.; Heidari, B. Comparison of metformin and N-acetyl cysteine, as an adjuvant to clomiphene citrate, in clomiphene-resistant women with polycystic ovary syndrome. J. Gynecol. Obstet. Hum. Reprod. 2017, 46, 579–585. [Google Scholar] [CrossRef]
- Yu, Y.; Fang, L.; Zhang, R.; He, J.; Xiong, Y.; Guo, X.; Du, Q.; Huang, Y.; Sun, Y. Comparative effectiveness of 9 ovulation-induction therapies in patients with clomiphene citrate-resistant polycystic ovary syndrome: A network meta-analysis. Sci. Rep. 2017, 7, 3812. [Google Scholar] [CrossRef]
- Sawant, S.; Bhide, P. Fertility Treatment Options for Women with Polycystic Ovary Syndrome. Clin. Med. Insights Reprod. Health 2019, 13, 1179558119890867. [Google Scholar] [CrossRef]
- Maged, A.M.; Elsawah, H.; Abdelhafez, A.; Bakry, A.; Mostafa, W.A. The adjuvant effect of metformin and N-acetylcysteine to clomiphene citrate in induction of ovulation in patients with Polycystic Ovary Syndrome. Gynecol. Endocrinol. 2015, 31, 635–638. [Google Scholar] [CrossRef]
- Rezk, M.; Shaheen, A.E.; Saif El-Nasr, I. Clomiphene citrate combined with metformin versus letrozole for induction of ovulation in clomiphene-resistant polycystic ovary syndrome: A randomized clinical trial. Gynecol. Endocrinol. 2018, 34, 298–300. [Google Scholar] [CrossRef]
- Chang, H.H.; Hsueh, Y.S.; Cheng, Y.W.; Ou, H.T.; Wu, M.H. Association between Polymorphisms of OCT1 and Metabolic Response to Metformin in Women with Polycystic Ovary Syndrome. Int. J. Mol. Sci. 2019, 20, 1720. [Google Scholar] [CrossRef]
- Hoeger, K.; Davidson, K.; Kochman, L.; Cherry, T.; Kopin, L.; Guzick, D.S. The impact of metformin, oral contraceptives, and lifestyle modification on polycystic ovary syndrome in obese adolescent women in two randomized, placebo-controlled clinical trials. J. Clin. Endocrinol. Metab. 2008, 93, 4299–4306. [Google Scholar] [CrossRef] [PubMed]
- Palomba, S.; Materazzo, C.; Falbo, A.; Orio, F.; La Sala, G.B.; Sultan, C. Metformin, oral contraceptives or both to manage oligo-amenorrhea in adolescents with polycystic ovary syndrome? A clinical review. Gynecol. Endocrinol. 2014, 30, 335–340. [Google Scholar] [CrossRef] [PubMed]
- Stefanaki, C.; Bacopoulou, F.; Kandaraki, E.; Boschiero, D.; Diamandi-Kandarakis, E. Lean Women on Metformin and Oral Contraceptives for Polycystic Ovary Syndrome Demonstrate a Dehydrated Osteosarcopenic Phenotype: A Pilot Study. Nutrients 2019, 11, 2055. [Google Scholar] [CrossRef]
- Douchi, T.; Yamamoto, S.; Oki, T.; Maruta, K.; Kuwahata, R.; Nagata, Y. Serum androgen levels and muscle mass in women with polycystic ovary syndrome. Obstet. Gynecol. 1999, 94, 337–340. [Google Scholar] [CrossRef] [PubMed]
- Cetrone, M.; Mele, A.; Tricarico, D. Effects of the antidiabetic drugs on the age-related atrophy and sarcopenia associated with diabetes type II. Curr. Diabetes Rev. 2014, 10, 231–237. [Google Scholar] [CrossRef] [PubMed]
- Kim, K.H.; Lee, M.S. FGF21 as a mediator of adaptive responses to stress and metabolic benefits of anti-diabetic drugs. J. Endocrinol. 2015, 226, 1–16. [Google Scholar] [CrossRef]
- Gupta, A.; Jelinek, H.F.; Al-Aubaidy, H. Glucagon like peptide-1 and its receptor agonists: Their roles in management of Type 2 diabetes mellitus. Diabetes Metab. Syndr. 2017, 11, 225–230. [Google Scholar] [CrossRef]
- Drucker, D.J. Mechanisms of Action and Therapeutic Application of Glucagon-like Peptide-1. Cell Metab. 2018, 27, 740–756. [Google Scholar] [CrossRef]
- Gentilella, R.; Pechtner, V.; Corcos, A.; Consoli, A. Glucagon-like peptide-1 receptor agonists in type 2 diabetes treatment: Are they all the same? Diabetes Metab. Res. Rev. 2019, 35, e3070. [Google Scholar] [CrossRef]
- Gillani, S.W.; Moosvi, A.F. Clinical Review: Safety and Efficacy Comparison between Sulfonylureas and Dipeptidyl Peptidase-4 Inhibitors as Second-Line Therapies in Type 2 Diabetes Mellitus. Curr. Pharm. Des. 2020, 26, 4315–4322. [Google Scholar] [CrossRef] [PubMed]
- Pani, A.; Gironi, I.; Di Vieste, G.; Mion, E.; Bertuzzi, F.; Pintaudi, B. From Prediabetes to Type 2 Diabetes Mellitus in Women with Polycystic Ovary Syndrome: Lifestyle and Pharmacological Management. Int. J. Endocrinol. 2020, 2020, 6276187. [Google Scholar] [CrossRef] [PubMed]
- Romualdi, D.; Versace, V.; Lanzone, A. What is new in the landscape of insulin-sensitizing agents for polycystic ovary syndrome treatment. Ther. Adv. Reprod. Health 2020, 14, 2633494120908709. [Google Scholar] [CrossRef] [PubMed]
- Jensterle, M.; Kravos, N.A.; Goričar, K.; Janez, A. Short-term effectiveness of low dose liraglutide in combination with metformin versus high dose liraglutide alone in treatment of obese PCOS: Randomized trial. BMC Endocr. Disord. 2017, 17, 5. [Google Scholar] [CrossRef]
- Tao, T.; Wu, P.; Wang, Y.; Liu, W. Comparison of glycemic control and β-cell function in new onset T2DM patients with PCOS of metformin and saxagliptin monotherapy or combination treatment. BMC Endocr. Disord. 2018, 18, 14. [Google Scholar] [CrossRef]
- Elkind-Hirsch, K.E.; Paterson, M.S.; Seidemann, E.L.; Gutowski, H.C. Short-term therapy with combination dipeptidyl peptidase-4 inhibitor saxagliptin/metformin extended release (XR) is superior to saxagliptin or metformin XR monotherapy in prediabetic women with polycystic ovary syndrome: A single-blind, randomized, pilot study. Fertil. Steril. 2017, 107, 253–260. [Google Scholar] [CrossRef]
- Lamos, E.M.; Malek, R.; Davis, S.N. GLP-1 receptor agonists in the treatment of polycystic ovary syndrome. Expert. Rev. Clin. Pharmacol. 2017, 10, 401–408. [Google Scholar] [CrossRef]
- Tzotzas, T.; Karras, S.N.; Katsiki, N. Glucagon-Like Peptide-1 (GLP-1) Receptor Agonists in the Treatment of Obese Women with Polycystic Ovary Syndrome. Curr. Vasc. Pharmacol. 2017, 15, 218–229. [Google Scholar] [CrossRef]
- Cena, H.; Chiovato, L.; Nappi, R.E. Obesity, Polycystic Ovary Syndrome, and Infertility: A New Avenue for GLP-1 Receptor Agonists. J. Clin. Endocrinol. Metab. 2020, 105, e2695–e2709. [Google Scholar] [CrossRef]
- Livadas, S.; Androulakis, I.; Angelopoulos, N.; Lytras, A.; Papagiannopoulos, F.; Kassi, G. Liraglutide administration improves hormonal/metabolic profile and reproductive features in women with HAIR-AN syndrome. Endocrinol. Diabetes Metab. Case Rep. 2020, 2020, 19–0150. [Google Scholar] [CrossRef]
- Nestler, J.E.; Jakubowicz, D.J. Decreases in ovarian cytochrome P450c17 alpha activity and serum free testosterone after reduction of insulin secretion in polycystic ovary syndrome. N. Engl. J. Med. 1996, 335, 617–623. [Google Scholar] [CrossRef]
- Chou, K.H.; von Eye Corleta, H.; Capp, E.; Spritzer, P.M. Clinical, metabolic and endocrine parameters in response to metformin in obese women with polycystic ovary syndrome: A randomized, double-blind and placebo-controlled trial. Horm. Metab. Res. 2003, 35, 86–91. [Google Scholar] [CrossRef]
- Allen, H.F.; Mazzoni, C.; Heptulla, R.A.; Murray, M.A.; Miller, N.; Koenigs, L.; Reiter, E.O. Randomized controlled trial evaluating response to metformin versus standard therapy in the treatment of adolescents with polycystic ovary syndrome. J. Pediatr. Endocrinol. Metab. 2005, 18, 761–768. [Google Scholar] [CrossRef]
- Bridger, T.; MacDonald, S.; Baltzer, F.; Rodd, C. Randomized placebo-controlled trial of metformin for adolescents with polycystic ovary syndrome. Arch. Pediatr. Adolesc. Med. 2006, 160, 241–246. [Google Scholar] [CrossRef]
- Diamanti-Kandarakis, E.; Christakou, C.D.; Kandaraki, E.; Economou, F.N. Metformin: An old medication of new fashion: Evolving new molecular mechanisms and clinical implications in polycystic ovary syndrome. Eur. J. Endocrinol. 2010, 162, 193–212. [Google Scholar] [CrossRef] [PubMed]
- Al-Zubeidi, H.; Klein, K.O. Randomized clinical trial evaluating metformin versus oral contraceptive pills in the treatment of adolescents with polycystic ovarian syndrome. J. Pediatr. Endocrinol. Metab. 2015, 28, 853–858. [Google Scholar] [CrossRef] [PubMed]
- Al Khalifah, R.A.; Florez, I.D.; Dennis, B.; Thabane, L.; Bassilious, E. Metformin or Oral Contraceptives for Adolescents With Polycystic Ovarian Syndrome: A Meta-analysis. Pediatrics 2016, 137, e20154089. [Google Scholar] [CrossRef] [PubMed]
- Sam, S.; Ehrmann, D.A. Metformin therapy for the reproductive and metabolic consequences of polycystic ovary syndrome. Diabetologia 2017, 60, 1656–1661. [Google Scholar] [CrossRef]
- Kupreeva, M.; Diane, A.; Lehner, R.; Watts, R.; Ghosh, M.; Proctor, S.; Vine, D. Effect of metformin and flutamide on insulin, lipogenic and androgen-estrogen signaling, and cardiometabolic risk in a PCOS-prone metabolic syndrome rodent model. Am. J. Physiol. Endocrinol. Metab. 2019, 316, E16–E33. [Google Scholar] [CrossRef]
- Diri, H.; Bayram, F.; Simsek, Y.; Caliskan, Z.; Kocer, D. Comparison of finasteride, metformin, and finasteride plus metformin in PCOS. Acta Endocrinol. 2017, 13, 84–89. [Google Scholar] [CrossRef]
- Kurzthaler, D.; Hadziomerovic-Pekic, D.; Wildt, L.; Seeber, B.E. Metformin induces a prompt decrease in LH-stimulated testosterone response in women with PCOS independent of its insulin-sensitizing effects. Reprod. Biol. Endocrinol. 2014, 12, 98. [Google Scholar] [CrossRef] [PubMed]
- Barber, T.M.; Wass, J.A.; McCarthy, M.I.; Franks, S. Metabolic characteristics of women with polycystic ovaries and oligo-amenorrhoea but normal androgen levels: Implications for the management of polycystic ovary syndrome. Clin. Endocrinol. 2007, 66, 513–517. [Google Scholar] [CrossRef] [PubMed]
- Adams, J.M.; Taylor, A.E.; Crowley, W.F., Jr.; Hall, J.E. Polycystic ovarian morphology with regular ovulatory cycles: Insights into the pathophysiology of polycystic ovarian syndrome. J. Clin. Endocrinol. Metab. 2004, 89, 4343–4350. [Google Scholar] [CrossRef]
- Attia, G.R.; Rainey, W.E.; Carr, B.R. Metformin directly inhibits androgen production in human thecal cells. Fertil. Steril. 2001, 76, 517–524. [Google Scholar] [CrossRef]
- Mansfield, R.; Galea, R.; Brincat, M.; Hole, D.; Mason, H. Metformin has direct effects on human ovarian steroidogenesis. Fertil. Steril. 2003, 79, 956–962. [Google Scholar] [CrossRef]
- Tosca, L.; Chabrolle, C.; Uzbekova, S.; Dupont, J. Effects of metformin on bovine granulosa cells steroidogenesis: Possible involvement of adenosine 5’ monophosphate-activated protein kinase (AMPK). Biol. Reprod. 2007, 76, 368–378. [Google Scholar] [CrossRef]
- Tosca, L.; Froment, P.; Solnais, P.; Ferré, P.; Foufelle, F.; Dupont, J. Adenosine 5′-monophosphate-activated protein kinase regulates progesterone secretion in rat granulosa cells. Endocrinology 2005, 146, 4500–4513. [Google Scholar] [CrossRef]
- Fontaine, E. Metformin-Induced Mitochondrial Complex I Inhibition: Facts, Uncertainties, and Consequences. Front. Endocrinol. 2018, 9, 753. [Google Scholar] [CrossRef]
- Tosca, L.; Crochet, S.; Ferré, P.; Foufelle, F.; Tesseraud, S.; Dupont, J. AMP-activated protein kinase activation modulates progesterone secretion in granulosa cells from hen preovulatory follicles. J. Endocrinol. 2006, 190, 85–97. [Google Scholar] [CrossRef] [PubMed]
- Downs, S.M.; Chen, J. Induction of meiotic maturation in mouse oocytes by adenosine analogs. Mol. Reprod. Dev. 2006, 73, 1159–1168. [Google Scholar] [CrossRef] [PubMed]
- LaRosa, C.; Downs, S.M. Meiotic induction by heat stress in mouse oocytes: Involvement of AMP-activated protein kinase and MAPK family members. Biol. Reprod. 2007, 76, 476–486. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Mayes, M.A.; Laforest, M.F.; Guillemette, C.; Gilchrist, R.B.; Richard, F.J. Adenosine 5′-monophosphate kinase-activated protein kinase (PRKA) activators delay meiotic resumption in porcine oocytes. Biol. Reprod. 2007, 76, 589–597. [Google Scholar] [CrossRef] [PubMed]
- Bilodeau-Goeseels, S. Cows are not mice: The role of cyclic AMP, phosphodiesterases, and adenosine monophosphate-activated protein kinase in the maintenance of meiotic arrest in bovine oocytes. Mol. Reprod. Dev. 2011, 78, 734–743. [Google Scholar] [CrossRef] [PubMed]
- Bertoldo, M.J.; Faure, M.; Dupont, J.; Froment, P. AMPK: A master energy regulator for gonadal function. Front. Neurosci. 2015, 9, 235. [Google Scholar] [CrossRef] [PubMed]
- Reverchon, M.; Cornuau, M.; Cloix, L.; Ramé, C.; Guerif, F.; Royère, D.; Dupont, J. Visfatin is expressed in human granulosa cells: Regulation by metformin through AMPK/SIRT1 pathways and its role in steroidogenesis. Mol. Hum. Reprod. 2013, 19, 313–326. [Google Scholar] [CrossRef] [PubMed]
- Bertoldo, M.J.; Guibert, E.; Faure, M.; Ramé, C.; Foretz, M.; Viollet, B.; Dupont, J.; Froment, P. Specific deletion of AMP-activated protein kinase (α1AMPK) in murine oocytes alters junctional protein expression and mitochondrial physiology. PLoS ONE 2015, 10, e0119680. [Google Scholar] [CrossRef]
- Tosca, L.; Solnais, P.; Ferré, P.; Foufelle, F.; Dupont, J. Metformin-induced stimulation of adenosine 5’ monophosphate-activated protein kinase (PRKA) impairs progesterone secretion in rat granulosa cells. Biol. Reprod. 2006, 75, 342–351. [Google Scholar] [CrossRef]
- Tosca, L.; Ramé, C.; Chabrolle, C.; Tesseraud, S.; Dupont, J. Metformin decreases IGF1-induced cell proliferation and protein synthesis through AMP-activated protein kinase in cultured bovine granulosa cells. Reproduction 2010, 139, 409–418. [Google Scholar] [CrossRef]
- Xu, Y.; Gao, Y.; Huang, Z.; Zheng, Y.; Teng, W.; Zheng, D.; Zheng, X. LKB1 suppresses androgen synthesis in a mouse model of hyperandrogenism via IGF-1 signaling. FEBS Open Bio 2019, 9, 1817–1825. [Google Scholar] [CrossRef]
- Jiang, Z.Z.; Hu, M.W.; Ma, X.S.; Schatten, H.; Fan, H.Y.; Wang, Z.B.; Sun, Q.Y. LKB1 acts as a critical gatekeeper of ovarian primordial follicle pool. Oncotarget 2016, 7, 5738–5753. [Google Scholar] [CrossRef]
- Morris, D.V.; Falcone, T. The relationship between insulin sensitivity and insulin-like growth factor-binding protein-1. Gynecol. Endocrinol. 1996, 10, 407–412. [Google Scholar] [CrossRef]
- Landay, M.; Huang, A.; Azziz, R. Degree of hyperinsulinemia, independent of androgen levels, is an important determinant of the severity of hirsutism in PCOS. Fertil. Steril. 2009, 92, 643–647. [Google Scholar] [CrossRef]
- Firmansyah, A.; Chalid, M.T.; Farid, R.B.; Nusratuddin, N. The correlation between insulin-like growth factor binding protein 1 (IGFBP-1) and homeostasis model assessment of insulin resistance (HOMA-IR) in polycystic ovarian syndrome with insulin resistance. Int. J. Reprod. Biomed. 2018, 16, 679–682. [Google Scholar]
- Barbieri, R.L. Hyperandrogenism, insulin resistance and acanthosis nigricans. 10 years of progress. J. Reprod. Med. 1994, 39, 327–336. [Google Scholar]
- Kelly, C.J.; Stenton, S.R.; Lashen, H. Insulin-like growth factor binding protein-1 in PCOS: A systematic review and meta-analysis. Hum. Reprod. Update 2011, 17, 4–16. [Google Scholar] [CrossRef]
- Bergh, C.; Carlsson, B.; Olsson, J.H.; Selleskog, U.; Hillensjö, T. Regulation of androgen production in cultured human thecal cells by insulin-like growth factor I and insulin. Fertil. Steril. 1993, 59, 323–331. [Google Scholar] [CrossRef]
- Mason, H.D.; Margara, R.; Winston, R.M.; Seppala, M.; Koistinen, R.; Franks, S. Insulin-like growth factor-I (IGF-I) inhibits production of IGF-binding protein-1 while stimulating estradiol secretion in granulosa cells from normal and polycystic human ovaries. J. Clin. Endocrinol. Metab. 1993, 76, 1275–1279. [Google Scholar] [CrossRef]
- Huhtala, M.S.; Tertti, K.; Juhila, J.; Sorsa, T.; Rönnemaa, T. Metformin and insulin treatment of gestational diabetes: Effects on inflammatory markers and IGF-binding protein-1—Secondary analysis of a randomized controlled trial. BMC Pregnancy Childbirth 2020, 20, 401. [Google Scholar] [CrossRef]
- Nestler, J.E.; Powers, L.P.; Matt, D.W.; Steingold, K.A.; Plymate, S.R.; Rittmaster, R.S.; Clore, J.N.; Blackard, W.G. A direct effect of hyperinsulinemia on serum sex hormone-binding globulin levels in obese women with the polycystic ovary syndrome. J. Clin. Endocrinol. Metab. 1991, 72, 83–89. [Google Scholar] [CrossRef]
- Botwood, N.; Hamilton-Fairley, D.; Kiddy, D.; Robinson, S.; Franks, S. Sex hormone-binding globulin and female reproductive function. J. Steroid Biochem. Mol. Biol. 1995, 53, 529–531. [Google Scholar] [CrossRef]
- Franks, S.; Kiddy, D.S.; Hamilton-Fairley, D.; Bush, A.; Sharp, P.S.; Reed, M.J. The role of nutrition and insulin in the regulation of sex hormone binding globulin. J. Steroid Biochem. Mol. Biol. 1991, 39, 835–838. [Google Scholar] [CrossRef]
- Cibula, D.; Fanta, M.; Vrbikova, J.; Stanicka, S.; Dvorakova, K.; Hill, M.; Skrha, J.; Zivny, J.; Skrenkova, J. The effect of combination therapy with metformin and combined oral contraceptives (COC) versus COC alone on insulin sensitivity, hyperandrogenaemia, SHBG and lipids in PCOS patients. Hum. Reprod. 2005, 20, 180–184. [Google Scholar] [CrossRef]
- Wei, W.; Zhao, H.; Wang, A.; Sui, M.; Liang, K.; Deng, H.; Ma, Y.; Zhang, Y.; Zhang, H.; Guan, Y. A clinical study on the short-term effect of berberine in comparison to metformin on the metabolic characteristics of women with polycystic ovary syndrome. Eur. J. Endocrinol. 2012, 166, 99–105. [Google Scholar] [CrossRef]
- Pasquali, R.; Gambineri, A.; Biscotti, D.; Vicennati, V.; Gagliardi, L.; Colitta, D.; Fiorini, S.; Cognigni, G.E.; Filicori, M.; Morselli-Labate, A.M. Effect of long-term treatment with metformin added to hypocaloric diet on body composition, fat distribution, and androgen and insulin levels in abdominally obese women with and without the polycystic ovary syndrome. J. Clin. Endocrinol. Metab. 2000, 85, 2767–2774. [Google Scholar] [CrossRef]
- Wassell, J.; Michail, M.; Soliman, N.; Wardle, P.G. The value of sex hormone binding globulin (SHBG) in predicting treatment response in polycystic ovary syndrome (PCOS). Clin. Lab. 2011, 57, 95–98. [Google Scholar]
- Zhang, J.; Si, Q.; Li, J. Therapeutic effects of metformin and clomiphene in combination with lifestyle intervention on infertility in women with obese polycystic ovary syndrome. Pak. J. Med. Sci. 2017, 33, 8–12. [Google Scholar] [CrossRef]
- Furat Rencber, S.; Kurnaz Ozbek, S.; Eraldemır, C.; Sezer, Z.; Kum, T.; Ceylan, S.; Guzel, E. Effect of resveratrol and metformin on ovarian reserve and ultrastructure in PCOS: An experimental study. J. Ovarian Res. 2018, 11, 55. [Google Scholar] [CrossRef]
- Kadoura, S.; Alhalabi, M.; Nattouf, A.H. Effect of Calcium and Vitamin D Supplements as an Adjuvant Therapy to Metformin on Menstrual Cycle Abnormalities, Hormonal Profile, and IGF-1 System in Polycystic Ovary Syndrome Patients: A Randomized, Placebo-Controlled Clinical Trial. Adv. Pharmacol. Sci. 2019, 2019, 9680390. [Google Scholar] [CrossRef]
- Song, Y.; Wang, H.; Huang, H.; Zhu, Z. Comparison of the efficacy between NAC and metformin in treating PCOS patients: A meta-analysis. Gynecol. Endocrinol. 2020, 36, 204–210. [Google Scholar] [CrossRef]
- Morales, A.J.; Laughlin, G.A.; Bützow, T.; Maheshwari, H.; Baumann, G.; Yen, S.S. Insulin, somatotropic, and luteinizing hormone axes in lean and obese women with polycystic ovary syndrome: Common and distinct features. J. Clin. Endocrinol. Metab. 1996, 81, 2854–2864. [Google Scholar] [CrossRef][Green Version]
- Chang, R.J. The reproductive phenotype in polycystic ovary syndrome. Nat. Clin. Pract. Endocrinol. Metab. 2007, 3, 688–695. [Google Scholar] [CrossRef]
- Sinha, P.; Chitra, T.; Papa, D.; Nandeesha, H. Laparoscopic Ovarian Drilling Reduces Testosterone and Luteinizing Hormone/Follicle-Stimulating Hormone Ratio and Improves Clinical Outcome in Women with Polycystic Ovary Syndrome. J. Hum. Reprod. Sci. 2019, 12, 224–228. [Google Scholar] [CrossRef]
- Dulka, E.A.; Burger, L.L.; Moenter, S.M. Ovarian Androgens Maintain High GnRH Neuron Firing Rate in Adult Prenatally-Androgenized Female Mice. Endocrinology 2020, 161, bqz038. [Google Scholar] [CrossRef] [PubMed]
- McCartney, C.R.; Campbell, R.E. Abnormal GnRH Pulsatility in Polycystic Ovary Syndrome: Recent Insights. Curr. Opin. Endocr. Metab. Res. 2020, 12, 78–84. [Google Scholar] [CrossRef] [PubMed]
- Woo, I.; Tobler, K.; Khafagy, A.; Christianson, M.S.; Yates, M.; Garcia, J. Predictive Value of Elevated LH/FSH Ratio for Ovulation Induction in Patients with Polycystic Ovary Syndrome. J. Reprod. Med. 2015, 60, 495–500. [Google Scholar] [PubMed]
- Maciel, G.A.; Hayashida, S.A.; da Costa, L.C.; Marcondes, J.A.; da Fonseca, A.M.; Soares, J.M., Jr.; Baracat, E.C. Influence of LH and high-density lipoprotein cholesterol (HDL-C) on metformin response in women with polycystic ovary syndrome. Eur. J. Obstet. Gynecol. Reprod. Biol. 2011, 157, 180–184. [Google Scholar] [CrossRef]
- Roland, A.V.; Moenter, S.M. Prenatal androgenization of female mice programs an increase in firing activity of gonadotropin-releasing hormone (GnRH) neurons that is reversed by metformin treatment in adulthood. Endocrinology 2011, 152, 618–628. [Google Scholar] [CrossRef] [PubMed]
- Lv, W.S.; Wen, J.P.; Li, L.; Sun, R.X.; Wang, J.; Xian, Y.X.; Cao, C.X.; Wang, Y.L.; Gao, Y.Y. The effect of metformin on food intake and its potential role in hypothalamic regulation in obese diabetic rats. Brain Res. 2012, 1444, 11–19. [Google Scholar] [CrossRef]
- McIlwraith, E.K.; Belsham, D.D. Hypothalamic reproductive neurons communicate through signal transduction to control reproduction. Mol. Cell. Endocrinol. 2020, 110971. [Google Scholar] [CrossRef]
- Malin, S.K.; Kashyap, S.R. Effects of metformin on weight loss: Potential mechanisms. Curr. Opin. Endocrinol. Diabetes Obes. 2014, 21, 323–329. [Google Scholar] [CrossRef]
- Derkach, K.V.; Zakharova, I.O.; Romanova, I.V.; Zorina, I.I.; Mikhrina, A.L.; Shpakov, A.O. Metabolic parameters and functional state of hypothalamic signaling systems in AY/a mice with genetic predisposition to obesity and the effect of metformin. Dokl. Biochem. Biophys. 2017, 477, 377–381. [Google Scholar] [CrossRef] [PubMed]
- Derkach, K.V.; Zakharova, I.O.; Zorina, I.I.; Bakhtyukov, A.A.; Romanova, I.V.; Bayunova, L.V.; Shpakov, A.O. The evidence of metabolic-improving effect of metformin in Ay/a mice with genetically-induced melanocortin obesity and the contribution of hypothalamic mechanisms to this effect. PLoS ONE 2019, 14, e0213779. [Google Scholar] [CrossRef] [PubMed]
- Yerevanian, A.; Soukas, A.A. Metformin: Mechanisms in Human Obesity and Weight Loss. Curr. Obes. Rep. 2019, 8, 156–164. [Google Scholar] [CrossRef] [PubMed]
- Hausman, G.J.; Barb, C.R.; Lents, C.A. Leptin and reproductive function. Biochimie 2012, 94, 2075–2081. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Gong, M. Review of the role of leptin in the regulation of male reproductive function. Andrologia 2018. [Google Scholar] [CrossRef]
- Huang, Y.; Yu, Y.; Gao, J.; Li, R.; Zhang, C.; Zhao, H.; Zhao, Y.; Qiao, J. Impaired oocyte quality induced by dehydroepiandrosterone is partially rescued by metformin treatment. PLoS ONE 2015, 10, e0122370. [Google Scholar] [CrossRef]
- Jin, J.; Ma, Y.; Tong, X.; Yang, W.; Dai, Y.; Pan, Y.; Ren, P.; Liu, L.; Fan, H.Y.; Zhang, Y.; et al. Metformin inhibits testosterone-induced endoplasmic reticulum stress in ovarian granulosa cells via inactivation of p38 MAPK. Hum. Reprod. 2020, 35, 1145–1158. [Google Scholar] [CrossRef]
- Wu, L.L.; Russell, D.L.; Norman, R.J.; Robker, R.L. Endoplasmic reticulum (ER) stress in cumulus-oocyte complexes impairs pentraxin-3 secretion, mitochondrial membrane potential (DeltaPsi m), and embryo development. Mol. Endocrinol. 2012, 26, 562–573. [Google Scholar] [CrossRef]
- Harada, M.; Nose, E.; Takahashi, N.; Hirota, Y.; Hirata, T.; Yoshino, O.; Koga, K.; Fujii, T.; Osuga, Y. Evidence of the activation of unfolded protein response in granulosa and cumulus cells during follicular growth and maturation. Gynecol. Endocrinol. 2015, 31, 783–787. [Google Scholar] [CrossRef]
- Park, H.J.; Park, J.Y.; Kim, J.W.; Yang, S.G.; Jung, J.M.; Kim, M.J.; Kang, M.J.; Cho, Y.H.; Wee, G.; Yang, H.Y.; et al. Melatonin improves the meiotic maturation of porcine oocytes by reducing endoplasmic reticulum stress during in vitro maturation. J. Pineal Res. 2018, 64, e12458. [Google Scholar] [CrossRef]
- Tobiume, K.; Matsuzawa, A.; Takahashi, T.; Nishitoh, H.; Morita, K.; Takeda, K.; Minowa, O.; Miyazono, K.; Noda, T.; Ichijo, H. ASK1 is required for sustained activations of JNK/p38 MAP kinases and apoptosis. EMBO Rep. 2001, 2, 222–228. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.S.; Kim, Y.; Lim, M.J.; Park, Y.G.; Park, S.I.; Sohn, J. The p38-activated ER stress-ATF6α axis mediates cellular senescence. FASEB J. 2019, 33, 2422–2434. [Google Scholar] [CrossRef] [PubMed]
- Azhary, J.M.K.; Harada, M.; Takahashi, N.; Nose, E.; Kunitomi, C.; Koike, H.; Hirata, T.; Hirota, Y.; Koga, K.; Wada-Hiraike, O.; et al. Endoplasmic Reticulum Stress Activated by Androgen Enhances Apoptosis of Granulosa Cells via Induction of Death Receptor 5 in PCOS. Endocrinology 2019, 160, 119–132. [Google Scholar] [CrossRef] [PubMed]
- Rice, S.; Pellatt, L.; Ramanathan, K.; Whitehead, S.A.; Mason, H.D. Metformin inhibits aromatase via an extracellular signal-regulated kinase-mediated pathway. Endocrinology 2009, 150, 4794–4801. [Google Scholar] [CrossRef] [PubMed]
- Rice, S.; Elia, A.; Jawad, Z.; Pellatt, L.; Mason, H.D. Metformin inhibits follicle-stimulating hormone (FSH) action in human granulosa cells: Relevance to polycystic ovary syndrome. J. Clin. Endocrinol. Metab. 2013, 98, 1491–1500. [Google Scholar] [CrossRef] [PubMed]
- Fuhrmeister, I.P.; Branchini, G.; Pimentel, A.M.; Ferreira, G.D.; Capp, E.; Brum, I.S.; von Eye Corleta, H. Human granulosa cells: Insulin and insulin-like growth factor-1 receptors and aromatase expression modulation by metformin. Gynecol. Obstet. Investig. 2014, 77, 156–162. [Google Scholar] [CrossRef] [PubMed]
- Catteau-Jonard, S.; Jamin, S.P.; Leclerc, A.; Gonzalès, J.; Dewailly, D.; di Clemente, N. Anti-Mullerian hormone, its receptor, FSH receptor, and androgen receptor genes are overexpressed by granulosa cells from stimulated follicles in women with polycystic ovary syndrome. J. Clin. Endocrinol. Metab. 2008, 93, 4456–4461. [Google Scholar] [CrossRef] [PubMed]
- González-Fernández, R.; Peña, Ó.; Hernández, J.; Martín-Vasallo, P.; Palumbo, A.; Ávila, J. Patients with endometriosis and patients with poor ovarian reserve have abnormal follicle-stimulating hormone receptor signaling pathways. Fertil. Steril. 2011, 95, 2373–2378. [Google Scholar] [CrossRef]
- Dewailly, D.; Robin, G.; Peigne, M.; Decanter, C.; Pigny, P.; Catteau-Jonard, S. Interactions between androgens, FSH, anti-Müllerian hormone and estradiol during folliculogenesis in the human normal and polycystic ovary. Hum. Reprod. Update 2016, 22, 709–724. [Google Scholar] [CrossRef]
- Geng, Y.; Sui, C.; Xun, Y.; Lai, Q.; Jin, L. MiRNA-99a can regulate proliferation and apoptosis of human granulosa cells via targeting IGF-1R in polycystic ovary syndrome. J. Assist. Reprod. Genet. 2019, 36, 211–221. [Google Scholar] [CrossRef]
- He, T.; Liu, Y.; Zhao, S.; Liu, H.; Wang, Z.; Shi, Y. Comprehensive assessment the expression of core elements related to IGFIR/PI3K pathway in granulosa cells of women with polycystic ovary syndrome. Eur. J. Obstet. Gynecol. Reprod. Biol. 2019, 233, 134–140. [Google Scholar] [CrossRef]
- He, T.; Sun, Y.; Zhang, Y.; Zhao, S.; Zheng, Y.; Hao, G.; Shi, Y. MicroRNA-200b and microRNA-200c are up-regulated in PCOS granulosa cell and inhibit KGN cell proliferation via targeting PTEN. Reprod. Biol. Endocrinol. 2019, 17, 68. [Google Scholar] [CrossRef]
- Mason, H.D.; Willis, D.S.; Holly, J.M.; Franks, S. Insulin preincubation enhances insulin-like growth factor-II (IGF-II) action on steroidogenesis in human granulosa cells. J. Clin. Endocrinol. Metab. 1994, 78, 1265–1267. [Google Scholar] [CrossRef]
- Willis, D.S.; Mason, H.D.; Watson, H.; Franks, S. Developmentally regulated responses of human granulosa cells to insulin-like growth factors (IGFs): IGF-I and IGF-II action mediated via the type-I IGF receptor. J. Clin. Endocrinol. Metab. 1998, 83, 1256–1259. [Google Scholar] [CrossRef][Green Version]
- Palomba, S.; Falbo, A.; Orio, F., Jr.; Manguso, F.; Russo, T.; Tolino, A.; Annamaria, C.; Dale, B.; Zullo, F. A randomized controlled trial evaluating metformin pre-treatment and co-administration in non-obese insulin-resistant women with polycystic ovary syndrome treated with controlled ovarian stimulation plus timed intercourse or intrauterine insemination. Hum. Reprod. 2005, 20, 2879–2886. [Google Scholar] [CrossRef]
- Huang, X.; Wang, P.; Tal, R.; Lv, F.; Li, Y.; Zhang, X. A systematic review and meta-analysis of metformin among patients with polycystic ovary syndrome undergoing assisted reproductive technology procedures. Int. J. Gynaecol. Obstet. 2015, 131, 111–116. [Google Scholar] [CrossRef]
- Laven, J.S.E. Follicle Stimulating Hormone Receptor (FSHR) Polymorphisms and Polycystic Ovary Syndrome (PCOS). Front. Endocrinol. 2019, 10, 23. [Google Scholar] [CrossRef]
- Dolfin, E.; Guani, B.; Lussiana, C.; Mari, C.; Restagno, G.; Revelli, A. FSH-receptor Ala307Thr polymorphism is associated to polycystic ovary syndrome and to a higher responsiveness to exogenous FSH in Italian women. J. Assist. Reprod. Genet. 2011, 28, 925–930. [Google Scholar] [CrossRef]
- Du, T.; Duan, Y.; Li, K.; Zhao, X.; Ni, R.; Li, Y.; Yang, D. Statistical Genomic Approach Identifies Association between FSHR Polymorphisms and Polycystic Ovary Morphology in Women with Polycystic Ovary Syndrome. Biomed. Res. Int. 2015, 2015, 483726. [Google Scholar] [CrossRef]
- Law, N.C.; Hunzicker-Dunn, M.E. Insulin Receptor Substrate 1, the Hub Linking Follicle-stimulating Hormone to Phosphatidylinositol 3-Kinase Activation. J. Biol. Chem. 2016, 291, 4547–4560. [Google Scholar] [CrossRef]
- Chu, Y.L.; Xu, Y.R.; Yang, W.X.; Sun, Y. The role of FSH and TGF-β superfamily in follicle atresia. Aging 2018, 10, 305–321. [Google Scholar] [CrossRef]
- McGee, E.A.; Raj, R.S. Regulators of ovarian preantral follicle development. Semin. Reprod. Med. 2015, 33, 179–184. [Google Scholar] [CrossRef]
- Nagashima, J.B.; Wildt, D.E.; Travis, A.J.; Songsasen, N. Activin promotes growth and antral cavity expansion in the dog ovarian follicle. Theriogenology 2019, 129, 168–177. [Google Scholar] [CrossRef]
- Durlinger, A.L.; Gruijters, M.J.; Kramer, P.; Karels, B.; Kumar, T.R.; Matzuk, M.M.; Rose, U.M.; de Jong, F.H.; Uilenbroek, J.T.; Grootegoed, J.A.; et al. Anti-Müllerian hormone attenuates the effects of FSH on follicle development in the mouse ovary. Endocrinology 2001, 142, 4891–4899. [Google Scholar] [CrossRef]
- Messinis, I.E. Ovarian feedback, mechanism of action and possible clinical implications. Hum. Reprod. Update 2006, 12, 557–571. [Google Scholar] [CrossRef]
- Fang, W.L.; Lai, S.Y.; Lai, W.A.; Lee, M.T.; Liao, C.F.; Ke, F.C.; Hwang, J.J. CRTC2 and Nedd4 ligase involvement in FSH and TGFβ1 upregulation of connexin43 gap junction. J. Mol. Endocrinol. 2015, 55, 263–275. [Google Scholar] [CrossRef]
- Fang, W.L.; Lee, M.T.; Wu, L.S.; Chen, Y.J.; Mason, J.; Ke, F.C.; Hwang, J.J. CREB coactivator CRTC2/TORC2 and its regulator calcineurin crucially mediate follicle-stimulating hormone and transforming growth factor β1 upregulation of steroidogenesis. J. Cell. Physiol. 2012, 227, 2430–2440. [Google Scholar] [CrossRef]
- Lai, W.A.; Yeh, Y.T.; Fang, W.L.; Wu, L.S.; Harada, N.; Wang, P.H.; Ke, F.C.; Lee, W.L.; Hwang, J.J. Calcineurin and CRTC2 mediate FSH and TGFβ1 upregulation of Cyp19a1 and Nr5a in ovary granulosa cells. J. Mol. Endocrinol. 2014, 53, 259–270. [Google Scholar] [CrossRef]
- Chang, H.M.; Qiao, J.; Leung, P.C. Oocyte-somatic cell interactions in the human ovary-novel role of bone morphogenetic proteins and growth differentiation factors. Hum. Reprod. Update 2016, 23, 1–18. [Google Scholar] [CrossRef]
- Piltonen, T.; Morin-Papunen, L.; Koivunen, R.; Perheentupa, A.; Ruokonen, A.; Tapanainen, J.S. Serum anti-Müllerian hormone levels remain high until late reproductive age and decrease during metformin therapy in women with polycystic ovary syndrome. Hum. Reprod. 2005, 20, 1820–1826. [Google Scholar] [CrossRef]
- Foroozanfard, F.; Samimi, M.; Almadani, K.H.; Sehat, M. Effect of metformin on the anti-Müllerian hormone level in infertile women with polycystic ovarian syndrome. Electron. Physician 2017, 9, 5969–5973. [Google Scholar] [CrossRef]
- Fallat, M.E.; Siow, Y.; Marra, M.; Cook, C.; Carrillo, A. Müllerian-inhibiting substance in follicular fluid and serum: A comparison of patients with tubal factor infertility, polycystic ovary syndrome, and endometriosis. Fertil. Steril. 1997, 67, 962–965. [Google Scholar] [CrossRef]
- Pigny, P.; Merlen, E.; Robert, Y.; Cortet-Rudelli, C.; Decanter, C.; Jonard, S.; Dewailly, D. Elevated serum level of anti-mullerian hormone in patients with polycystic ovary syndrome: Relationship to the ovarian follicle excess and to the follicular arrest. J. Clin. Endocrinol. Metab. 2003, 88, 5957–5962. [Google Scholar] [CrossRef] [PubMed]
- Laven, J.S.; Mulders, A.G.; Visser, J.A.; Themmen, A.P.; De Jong, F.H.; Fauser, B.C. Anti-Müllerian hormone serum concentrations in normoovulatory and anovulatory women of reproductive age. J. Clin. Endocrinol. Metab. 2004, 89, 318–323. [Google Scholar] [CrossRef] [PubMed]
- Knight, P.G.; Glister, C. TGF-beta superfamily members and ovarian follicle development. Reproduction 2006, 132, 191–206. [Google Scholar] [CrossRef] [PubMed]
- Pellatt, L.; Rice, S.; Dilaver, N.; Heshri, A.; Galea, R.; Brincat, M.; Brown, K.; Simpson, E.R.; Mason, H.D. Anti-Müllerian hormone reduces follicle sensitivity to follicle-stimulating hormone in human granulosa cells. Fertil. Steril. 2011, 96, 1246–1251. [Google Scholar] [CrossRef]
- Broer, S.L.; Broekmans, F.J.; Laven, J.S.; Fauser, B.C. Anti-Müllerian hormone: Ovarian reserve testing and its potential clinical implications. Hum. Reprod. Update 2014, 20, 688–701. [Google Scholar] [CrossRef]
- Homburg, R.; Ray, A.; Bhide, P.; Gudi, A.; Shah, A.; Timms, P.; Grayson, K. The relationship of serum anti-Mullerian hormone with polycystic ovarian morphology and polycystic ovary syndrome: A prospective cohort study. Hum. Reprod. 2013, 28, 1077–1083. [Google Scholar] [CrossRef]
- Qi, X.; Pang, Y.; Qiao, J. The role of anti-Müllerian hormone in the pathogenesis and pathophysiological characteristics of polycystic ovary syndrome. Eur. J. Obstet. Gynecol. Reprod. Biol. 2016, 199, 82–87. [Google Scholar] [CrossRef]
- Lv, P.P.; Jin, M.; Rao, J.P.; Chen, J.; Wang, L.Q.; Huang, C.C.; Yang, S.Q.; Yao, Q.P.; Feng, L.; Shen, J.M.; et al. Role of anti-Müllerian hormone and testosterone in follicular growth: A cross-sectional study. BMC Endocr. Disord. 2020, 20, 101. [Google Scholar] [CrossRef]
- Pierre, A.; Peigné, M.; Grynberg, M.; Arouche, N.; Taieb, J.; Hesters, L.; Gonzalès, J.; Picard, J.Y.; Dewailly, D.; Fanchin, R.; et al. Loss of LH-induced down-regulation of anti-Müllerian hormone receptor expression may contribute to anovulation in women with polycystic ovary syndrome. Hum. Reprod. 2013, 28, 762–769. [Google Scholar] [CrossRef]
- Garg, D.; Tal, R. The role of AMH in the pathophysiology of polycystic ovarian syndrome. Reprod. Biomed. Online 2016, 33, 15–28. [Google Scholar] [CrossRef] [PubMed]
- Sova, H.; Unkila-Kallio, L.; Tiitinen, A.; Hippeläinen, M.; Perheentupa, A.; Tinkanen, H.; Puukka, K.; Bloigu, R.; Piltonen, T.; Tapanainen, J.S.; et al. Hormone profiling, including anti-Müllerian hormone (AMH), for the diagnosis of polycystic ovary syndrome (PCOS) and characterization of PCOS phenotypes. Gynecol. Endocrinol. 2019, 35, 595–600. [Google Scholar] [CrossRef] [PubMed]
- Fleming, R.; Harborne, L.; MacLaughlin, D.T.; Ling, D.; Norman, J.; Sattar, N.; Seifer, D.B. Metformin reduces serum mullerian-inhibiting substance levels in women with polycystic ovary syndrome after protracted treatment. Fertil. Steril. 2005, 83, 130–136. [Google Scholar] [CrossRef] [PubMed]
- Tomova, A.; Deepinder, F.; Robeva, R.; Kirilov, G.; Mechandjiev, Z.; Kumanov, P. Anti-Müllerian hormone in women with polycystic ovary syndrome before and after therapy with metformin. Horm. Metab. Res. 2011, 43, 723–727. [Google Scholar] [CrossRef]
- Neagu, M.; Cristescu, C. Anti-Műllerian hormone—A prognostic marker for metformin therapy efficiency in the treatment of women with infertility and polycystic ovary syndrome. J. Med. Life. 2012, 5, 462–464. [Google Scholar]
- Wiweko, B.; Susanto, C.A. The Effect of Metformin and Cinnamon on Serum Anti-Mullerian Hormone in Women Having PCOS: A Double-Blind, Randomized, Controlled Trial. J. Hum. Reprod. Sci. 2017, 10, 31–36. [Google Scholar] [CrossRef]
- Chhabra, N.; Malik, S. Effect of Insulin Sensitizers on Raised Serum Anti-mullerian Hormone Levels in Infertile Women with Polycystic Ovarian Syndrome. J. Hum. Reprod. Sci. 2018, 11, 348–352. [Google Scholar] [CrossRef]
- Saleh, B.O.; Ibraheem, W.F.; Ameen, N.S. The role of anti-Mullerian hormone and inhibin B in the assessment of metformin therapy in women with polycystic ovarian syndrome. Saudi Med. J. 2015, 36, 562–567. [Google Scholar] [CrossRef]
- Madsen, H.N.; Lauszus, F.F.; Trolle, B.; Ingerslev, H.J.; Tørring, N. Impact of metformin on anti-Müllerian hormone in women with polycystic ovary syndrome: A secondary analysis of a randomized controlled trial. Acta Obstet. Gynecol. Scand. 2015, 94, 547–551. [Google Scholar] [CrossRef]
- Nascimento, A.D.; Silva Lara, L.A.; Japur de Sá Rosa-e-Silva, A.C.; Ferriani, R.A.; Reis, R.M. Effects of metformin on serum insulin and anti-Mullerian hormone levels and on hyperandrogenism in patients with polycystic ovary syndrome. Gynecol. Endocrinol. 2013, 29, 246–249. [Google Scholar] [CrossRef]
- Grigoryan, O.; Absatarova, J.; Andreeva, E.; Melnichenko, G.; Dedov, I. Effect of metformin on the level of anti-Mullerian hormone in therapy of polycystic ovary syndrome in obese women. Minerva Ginecol. 2014, 66, 85–89. [Google Scholar] [PubMed]
- Dilaver, N.; Pellatt, L.; Jameson, E.; Ogunjimi, M.; Bano, G.; Homburg, R.; Mason, H.; Rice, S. The regulation and signalling of anti-Müllerian hormone in human granulosa cells: Relevance to polycystic ovary syndrome. Hum. Reprod. 2019, 34, 2467–2479. [Google Scholar] [CrossRef] [PubMed]
- Liu, M.; Gao, J.; Zhang, Y.; Li, P.; Wang, H.; Ren, X.; Li, C. Serum levels of TSP-1, NF-κB and TGF-β1 in polycystic ovarian syndrome (PCOS) patients in northern China suggest PCOS is associated with chronic inflammation. Clin. Endocrinol. 2015, 83, 913–922. [Google Scholar] [CrossRef] [PubMed]
- Goldman, S.; Shalev, E. MMPS and TIMPS in ovarian physiology and pathophysiology. Front. Biosci. 2004, 9, 2474–2483. [Google Scholar] [CrossRef] [PubMed]
- Lewandowski, K.C.; Komorowski, J.; O’Callaghan, C.J.; Tan, B.K.; Chen, J.; Prelevic, G.M.; Randeva, H.S. Increased circulating levels of matrix metalloproteinase-2 and -9 in women with the polycystic ovary syndrome. J. Clin. Endocrinol. Metab. 2006, 91, 1173–1177. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.S.; Wang, Q.; Fu, X.H.; Huang, X.H.; Chen, X.L.; Cao, L.Q.; Chen, L.Z.; Tan, H.X.; Li, W.; Bi, J.; et al. Involvement of PI3K/PTEN/AKT/mTOR pathway in invasion and metastasis in hepatocellular carcinoma: Association with MMP-9. Hepatol. Res. 2009, 39, 177–186. [Google Scholar] [CrossRef]
- Li, X.; Yang, Z.; Song, W.; Zhou, L.; Li, Q.; Tao, K.; Zhou, J.; Wang, X.; Zheng, Z.; You, N.; et al. Overexpression of Bmi-1 contributes to the invasion and metastasis of hepatocellular carcinoma by increasing the expression of matrix metalloproteinase (MMP)-2, MMP-9 and vascular endothelial growth factor via the PTEN/PI3K/Akt pathway. Int. J. Oncol. 2013, 43, 793–802. [Google Scholar] [CrossRef]
- Jalving, M.; Gietema, J.A.; Lefrandt, J.D.; de Jong, S.; Reyners, A.K.; Gans, R.O.; de Vries, E.G. Metformin: Taking away the candy for cancer? Eur. J. Cancer 2010, 46, 2369–2380. [Google Scholar] [CrossRef]
- Chen, Z.; Wei, H.; Zhao, X.; Xin, X.; Peng, L.; Ning, Y.; Wang, Y.; Lan, Y.; Zhang, Q. Metformin treatment alleviates polycystic ovary syndrome by decreasing the expression of MMP-2 and MMP-9 via H19/miR-29b-3p and AKT/mTOR/autophagy signaling pathways. J. Cell. Physiol. 2019, 234, 19964–19976. [Google Scholar] [CrossRef]
- Sam, S.; Dunaif, A. Polycystic ovary syndrome: Syndrome XX? Trends Endocrinol. Metab. 2003, 14, 365–370. [Google Scholar] [CrossRef] [PubMed]
- Teede, H.J.; Hutchison, S.K.; Zoungas, S. The management of insulin resistance in polycystic ovary syndrome. Trends Endocrinol. Metab. 2007, 18, 273–279. [Google Scholar] [CrossRef] [PubMed]
- Kai, Y.; Kawano, Y.; Yamamoto, H.; Narahara, H. A possible role for AMP-activated protein kinase activated by metformin and AICAR in human granulosa cells. Reprod. Biol. Endocrinol. 2015, 13, 27. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.; Zheng, J.; Cui, N.; Jiang, L.; Zhou, H.; Zhang, D.; Hao, G. Baicalin ameliorates polycystic ovary syndrome through AMP-activated protein kinase. J. Ovarian Res. 2019, 12, 109. [Google Scholar] [CrossRef]
- Gambineri, A.; Tomassoni, F.; Gasparini, D.I.; Di Rocco, A.; Mantovani, V.; Pagotto, U.; Altieri, P.; Sanna, S.; Fulghesu, A.M.; Pasquali, R. Organic cation transporter 1 polymorphisms predict the metabolic response to metformin in women with the polycystic ovary syndrome. J. Clin. Endocrinol. Metab. 2010, 95, 204–208. [Google Scholar] [CrossRef] [PubMed]
- Schweighofer, N.; Lerchbaum, E.; Trummer, O.; Schwetz, V.; Pieber, T.; Obermayer-Pietsch, B. Metformin resistance alleles in polycystic ovary syndrome: Pattern and association with glucose metabolism. Pharmacogenomics 2014, 15, 305–317. [Google Scholar] [CrossRef]
- Mofo Mato, E.P.; Guewo-Fokeng, M.; Essop, M.F.; Owira, P.M.O. Genetic polymorphisms of organic cation transporter 1 (OCT1) and responses to metformin therapy in individuals with type 2 diabetes: A systematic review. Medicine 2018, 97, e11349. [Google Scholar] [CrossRef]
- Di Pietro, M.; Velazquez, C.; Matzkin, M.E.; Frungieri, M.B.; Peña, M.G.; de Zúñiga, I.; Pascuali, N.; Irusta, G.; Bianchi, M.S.; Parborell, F.; et al. Metformin has a direct effect on ovarian cells that is dependent on organic cation transporters. Mol. Cell. Endocrinol. 2020, 499, 110591. [Google Scholar] [CrossRef]
- Brownfoot, F.C.; Hastie, R.; Hannan, N.J.; Cannon, P.; Tuohey, L.; Parry, L.J.; Senadheera, S.; Illanes, S.E.; Kaitu’u-Lino, T.J.; Tong, S. Metformin as a prevention and treatment for preeclampsia: Effects on soluble fms-like tyrosine kinase 1 and soluble endoglin secretion and endothelial dysfunction. Am. J. Obstet. Gynecol. 2016, 214, 356.e1–356.e15. [Google Scholar] [CrossRef]
- Wouldes, T.A.; Battin, M.; Coat, S.; Rush, E.C.; Hague, W.M.; Rowan, J.A. Neurodevelopmental outcome at 2 years in offspring of women randomised to metformin or insulin treatment for gestational diabetes. Arch. Dis. Child. Fetal Neonatal Ed. 2016, 101, 488–493. [Google Scholar] [CrossRef]
- Gray, S.G.; McGuire, T.M.; Cohen, N.; Little, P.J. The emerging role of metformin in gestational diabetes mellitus. Diabetes Obes. Metab. 2017, 19, 765–772. [Google Scholar] [CrossRef] [PubMed]
- Mirghani Dirar, A.; Doupis, J. Gestational diabetes from A to Z. World J. Diabetes 2017, 8, 489–511. [Google Scholar] [CrossRef] [PubMed]
- Romero, R.; Erez, O.; Hüttemann, M.; Maymon, E.; Panaitescu, B.; Conde-Agudelo, A.; Pacora, P.; Yoon, B.H.; Grossman, L.I. Metformin, the aspirin of the 21st century: Its role in gestational diabetes mellitus, prevention of preeclampsia and cancer, and the promotion of longevity. Am. J. Obstet. Gynecol. 2017, 217, 282–302. [Google Scholar] [CrossRef] [PubMed]
- Kalafat, E.; Sukur, Y.E.; Abdi, A.; Thilaganathan, B.; Khalil, A. Metformin for prevention of hypertensive disorders of pregnancy in women with gestational diabetes or obesity: Systematic review and meta-analysis of randomized trials. Ultrasound Obstet. Gynecol. 2018, 52, 706–714. [Google Scholar] [CrossRef]
- Priya, G.; Kalra, S. A Review of Insulin Resistance in Type 1 Diabetes: Is There a Place for Adjunctive Metformin? Diabetes Ther. 2018, 9, 349–361. [Google Scholar] [CrossRef]
- Farahvar, S.; Walfisch, A.; Sheiner, E. Gestational diabetes risk factors and long-term consequences for both mother and offspring: A literature review. Expert Rev. Endocrinol. Metab. 2019, 14, 63–74. [Google Scholar] [CrossRef]
- McIntyre, H.D.; Catalano, P.; Zhang, C.; Desoye, G.; Mathiesen, E.R.; Damm, P. Gestational diabetes mellitus. Nat. Rev. Dis. Primers 2019, 5, 47. [Google Scholar] [CrossRef]
- Jorquera, G.; Echiburú, B.; Crisosto, N.; Sotomayor-Zárate, R.; Maliqueo, M.; Cruz, G. Metformin during Pregnancy: Effects on Offspring Development and Metabolic Function. Front. Pharmacol. 2020, 11, 653. [Google Scholar] [CrossRef]
- Benhalima, K.; Devlieger, R.; Van Assche, A. Screening and management of gestational diabetes. Best Pract. Res. Clin. Obstet. Gynaecol. 2015, 29, 339–349. [Google Scholar] [CrossRef]
- Chiefari, E.; Arcidiacono, B.; Foti, D.; Brunetti, A. Gestational diabetes mellitus: An updated overview. J. Endocrinol. Investig. 2017, 40, 899–909. [Google Scholar] [CrossRef]
- Bashir, M.; Aboulfotouh, M.; Dabbous, Z.; Mokhtar, M.; Siddique, M.; Wahba, R.; Ibrahim, A.; Brich, S.A.; Konje, J.C.; Abou-Samra, A.B. Metformin-treated-GDM has lower risk of macrosomia compared to diet-treated GDM—A retrospective cohort study. J. Matern. Fetal Neonatal Med. 2020, 33, 2366–2371. [Google Scholar] [CrossRef] [PubMed]
- Shepherd, E.; Gomersall, J.C.; Tieu, J.; Han, S.; Crowther, C.A.; Middleton, P. Combined diet and exercise interventions for preventing gestational diabetes mellitus. Cochrane Database Syst. Rev. 2017, 11, CD010443. [Google Scholar] [CrossRef] [PubMed]
- Laredo-Aguilera, J.A.; Gallardo-Bravo, M.; Rabanales-Sotos, J.A.; Cobo-Cuenca, A.I.; Carmona-Torres, J.M. Physical Activity Programs during Pregnancy Are Effective for the Control of Gestational Diabetes Mellitus. Int. J. Environ. Res. Public Health 2020, 17, 6151. [Google Scholar] [CrossRef] [PubMed]
- Griffith, R.J.; Alsweiler, J.; Moore, A.E.; Brown, S.; Middleton, P.; Shepherd, E.; Crowther, C.A. Interventions to prevent women from developing gestational diabetes mellitus: An overview of Cochrane Reviews. Cochrane Database Syst. Rev. 2020, 6, CD012394. [Google Scholar] [CrossRef]
- Crawford, T.J.; Crowther, C.A.; Alsweiler, J.; Brown, J. Antenatal dietary supplementation with myo-inositol in women during pregnancy for preventing gestational diabetes. Cochrane Database Syst. Rev. 2015, 2015, CD011507. [Google Scholar] [CrossRef]
- Bao, W.; Song, Y.; Bertrand, K.A.; Tobias, D.K.; Olsen, S.F.; Chavarro, J.E.; Mills, J.L.; Hu, F.B.; Zhang, C. Prepregnancy habitual intake of vitamin D from diet and supplements in relation to risk of gestational diabetes mellitus: A prospective cohort study. J. Diabetes 2018, 10, 373–379. [Google Scholar] [CrossRef]
- Zhang, Y.; Gong, Y.; Xue, H.; Xiong, J.; Cheng, G. Vitamin D and gestational diabetes mellitus: A systematic review based on data free of Hawthorne effect. BJOG 2018, 125, 784–793. [Google Scholar] [CrossRef]
- Brown, J.; Grzeskowiak, L.; Williamson, K.; Downie, M.R.; Crowther, C.A. Insulin for the treatment of women with gestational diabetes. Cochrane Database Syst. Rev. 2017, 11, CD012037. [Google Scholar] [CrossRef]
- Brown, J.; Martis, R.; Hughes, B.; Rowan, J.; Crowther, C.A. Oral anti-diabetic pharmacological therapies for the treatment of women with gestational diabetes. Cochrane Database Syst. Rev. 2017, 1, CD011967. [Google Scholar] [CrossRef]
- Liang, H.L.; Ma, S.J.; Xiao, Y.N.; Tan, H.Z. Comparative efficacy and safety of oral antidiabetic drugs and insulin in treating gestational diabetes mellitus: An updated PRISMA-compliant network meta-analysis. Medicine 2017, 96, e7939. [Google Scholar] [CrossRef]
- Helal, K.F.; Badr, M.S.; Rafeek, M.E.; Elnagar, W.M.; Lashin, M.E. Can glyburide be advocated over subcutaneous insulin for perinatal outcomes of women with gestational diabetes? A systematic review and meta-analysis. Arch. Gynecol. Obstet. 2020, 301, 19–32. [Google Scholar] [CrossRef] [PubMed]
- Nicolaou, V.; Soepnel, L.; Huddle, K.R.; Levitt, N.; Klipstein-Grobusch, K.; Norris, S.A. Maternal and neonatal outcomes following the introduction of oral hypoglycaemic agents for gestational diabetes mellitus were comparable to insulin monotherapy in two historical cohorts. S. Afr. Med. J. 2020, 110, 154–158. [Google Scholar] [CrossRef] [PubMed]
- Moore, L.E.; Briery, C.M.; Clokey, D.; Martin, R.W.; Williford, N.J.; Bofill, J.A.; Morrison, J.C. Metformin and insulin in the management of gestational diabetes mellitus: Preliminary results of a comparison. J. Reprod. Med. 2007, 52, 1011–1015. [Google Scholar]
- Rowan, J.A.; Hague, W.M.; Gao, W.; Battin, M.R.; Moore, M.P.; MiG Trial Investigators. Metformin versus insulin for the treatment of gestational diabetes. N. Engl. J. Med. 2008, 358, 2003–2015. [Google Scholar] [CrossRef]
- Ijäs, H.; Vääräsmäki, M.; Saarela, T.; Keravuo, R.; Raudaskoski, T. A follow-up of a randomised study of metformin and insulin in gestational diabetes mellitus: Growth and development of the children at the age of 18 months. BJOG 2015, 122, 994–1000. [Google Scholar] [CrossRef] [PubMed]
- Niromanesh, S.; Alavi, A.; Sharbaf, F.R.; Amjadi, N.; Moosavi, S.; Akbari, S. Metformin compared with insulin in the management of gestational diabetes mellitus: A randomized clinical trial. Diabetes Res. Clin. Pract. 2012, 98, 422–429. [Google Scholar] [CrossRef] [PubMed]
- Gui, J.; Liu, Q.; Feng, L. Metformin vs insulin in the management of gestational diabetes: A meta-analysis. PLoS ONE 2013, 8, e64585. [Google Scholar] [CrossRef] [PubMed]
- Tertti, K.; Ekblad, U.; Koskinen, P.; Vahlberg, T.; Rönnemaa, T. Metformin vs. insulin in gestational diabetes. A randomized study characterizing metformin patients needing additional insulin. Diabetes Obes. Metab. 2013, 15, 246–251. [Google Scholar] [CrossRef] [PubMed]
- Butalia, S.; Gutierrez, L.; Lodha, A.; Aitken, E.; Zakariasen, A.; Donovan, L. Short- and long-term outcomes of metformin compared with insulin alone in pregnancy: A systematic review and meta-analysis. Diabet. Med. 2017, 34, 27–36. [Google Scholar] [CrossRef]
- Feng, Y.; Yang, H. Metformin—A potentially effective drug for gestational diabetes mellitus: A systematic review and meta-analysis. J. Matern. Fetal Neonatal Med. 2017, 30, 1874–1881. [Google Scholar] [CrossRef]
- Hickman, M.A.; McBride, R.; Boggess, K.A.; Strauss, R. Metformin compared with insulin in the treatment of pregnant women with overt diabetes: A randomized controlled trial. Am. J. Perinatol. 2013, 30, 483–490. [Google Scholar] [CrossRef] [PubMed]
- Spaulonci, C.P.; Bernardes, L.S.; Trindade, T.C.; Zugaib, M.; Francisco, R.P. Randomized trial of metformin vs insulin in the management of gestational diabetes. Am. J. Obstet. Gynecol. 2013, 209, 34.e1–34.e7. [Google Scholar] [CrossRef] [PubMed]
- Ibrahim, M.I.; Hamdy, A.; Shafik, A.; Taha, S.; Anwar, M.; Faris, M. The role of adding metformin in insulin-resistant diabetic pregnant women: A randomized controlled trial. Arch. Gynecol. Obstet. 2014, 289, 959–965. [Google Scholar] [CrossRef]
- Ruholamin, S.; Eshaghian, S.; Allame, Z. Neonatal outcomes in women with gestational diabetes mellitus treated with metformin in compare with insulin: A randomized clinical trial. J. Res. Med. Sci. 2014, 19, 970–975. [Google Scholar]
- Ainuddin, J.; Karim, N.; Hasan, A.A.; Naqvi, S.A. Metformin versus insulin treatment in gestational diabetes in pregnancy in a developing country: A randomized control trial. Diabetes Res. Clin. Pract. 2015, 107, 290–299. [Google Scholar] [CrossRef] [PubMed]
- Ainuddin, J.A.; Karim, N.; Zaheer, S.; Ali, S.S.; Hasan, A.A. Metformin treatment in type 2 diabetes in pregnancy: An active controlled, parallel-group, randomized, open label study in patients with type 2 diabetes in pregnancy. J. Diabetes Res. 2015, 2015, 325851. [Google Scholar] [CrossRef] [PubMed]
- Balsells, M.; García-Patterson, A.; Solà, I.; Roqué, M.; Gich, I.; Corcoy, R. Glibenclamide, metformin, and insulin for the treatment of gestational diabetes: A systematic review and meta-analysis. BMJ 2015, 350, h102. [Google Scholar] [CrossRef] [PubMed]
- Kitwitee, P.; Limwattananon, S.; Limwattananon, C.; Waleekachonlert, O.; Ratanachotpanich, T.; Phimphilai, M.; Nguyen, T.V.; Pongchaiyakul, C. Metformin for the treatment of gestational diabetes: An updated meta-analysis. Diabetes Res. Clin. Pract. 2015, 109, 521–532. [Google Scholar] [CrossRef]
- Refuerzo, J.S.; Gowen, R.; Pedroza, C.; Hutchinson, M.; Blackwell, S.C.; Ramin, S. A pilot randomized, controlled trial of metformin versus insulin in women with type 2 diabetes mellitus during pregnancy. Am. J. Perinatol. 2015, 30, 163–170. [Google Scholar] [CrossRef]
- Bettencourt-Silva, R.; Neves, J.S.; Ferreira, M.J.; Souteiro, P.; Belo, S.; Oliveira, A.I.; Carvalho, D.; Namora, G.; Montenegro, N.; Queirós, J. Metformin in overweight and obese women with gestational diabetes: A propensity score-matched study. Endocrine 2019, 66, 192–200. [Google Scholar] [CrossRef]
- Su, D.F.; Wang, X.Y. Metformin vs insulin in the management of gestational diabetes: A systematic review and meta-analysis. Diabetes Res. Clin. Pract. 2014, 104, 353–357. [Google Scholar] [CrossRef] [PubMed]
- Zawiejska, A.; Wender-Ozegowska, E.; Grewling-Szmit, K.; Brazert, M.; Brazert, J. Short-term antidiabetic treatment with insulin or metformin has a similar impact on the components of metabolic syndrome in women with gestational diabetes mellitus requiring antidiabetic agents: Results of a prospective, randomised study. J. Physiol. Pharmacol. 2016, 67, 227–233. [Google Scholar] [PubMed]
- Jiang, Y.F.; Chen, X.Y.; Ding, T.; Wang, X.F.; Zhu, Z.N.; Su, S.W. Comparative efficacy and safety of OADs in management of GDM: Network meta-analysis of randomized controlled trials. J. Clin. Endocrinol. Metab. 2015, 100, 2071–2080. [Google Scholar] [CrossRef] [PubMed]
- Farrar, D.; Simmonds, M.; Bryant, M.; Sheldon, T.A.; Tuffnell, D.; Golder, S.; Lawlor, D.A. Treatments for gestational diabetes: A systematic review and meta-analysis. BMJ Open 2017, 7, e015557. [Google Scholar] [CrossRef] [PubMed]
- Guo, L.; Ma, J.; Tang, J.; Hu, D.; Zhang, W.; Zhao, X. Comparative Efficacy and Safety of Metformin, Glyburide, and Insulin in Treating Gestational Diabetes Mellitus: A Meta-Analysis. J. Diabetes Res. 2019, 2019, 9804708. [Google Scholar] [CrossRef] [PubMed]
- Kaitu’u-Lino, T.J.; Brownfoot, F.C.; Beard, S.; Cannon, P.; Hastie, R.; Nguyen, T.V.; Binder, N.K.; Tong, S.; Hannan, N.J. Combining metformin and esomeprazole is additive in reducing sFlt-1 secretion and decreasing endothelial dysfunction—Implications for treating preeclampsia. PLoS ONE. 2018, 13, e0188845. [Google Scholar] [CrossRef] [PubMed]
- Brownfoot, F.C.; Hastie, R.; Hannan, N.J.; Cannon, P.; Nguyen, T.V.; Tuohey, L.; Cluver, C.; Tong, S.; Kaitu’u-Lino, T.J. Combining metformin and sulfasalazine additively reduces the secretion of antiangiogenic factors from the placenta: Implications for the treatment of preeclampsia. Placenta. 2020, 95, 78–83. [Google Scholar] [CrossRef]
- Jena, M.K.; Sharma, N.R.; Petitt, M.; Maulik, D.; Nayak, N.R. Pathogenesis of Preeclampsia and Therapeutic Approaches Targeting the Placenta. Biomolecules 2020, 10, 953. [Google Scholar] [CrossRef]
- Wang, F.; Cao, G.; Yi, W.; Li, L.; Cao, X. Effect of Metformin on a Preeclampsia-Like Mouse Model Induced by High-Fat Diet. Biomed. Res. Int. 2019, 2019, 6547019. [Google Scholar] [CrossRef]
- Roberts, J.M.; Escudero, C. The placenta in preeclampsia. Pregnancy Hypertens. 2012, 2, 72–83. [Google Scholar] [CrossRef]
- Cerdeira, A.S.; Agrawal, S.; Staff, A.C.; Redman, C.W.; Vatish, M. Angiogenic factors: Potential to change clinical practice in pre-eclampsia? BJOG 2018, 125, 1389–1395. [Google Scholar] [CrossRef] [PubMed]
- Duckitt, K.; Harrington, D. Risk factors for pre-eclampsia at antenatal booking: Systematic review of controlled studies. BMJ 2005, 330, 565. [Google Scholar] [CrossRef]
- Masuyama, H.; Segawa, T.; Sumida, Y.; Masumoto, A.; Inoue, S.; Akahori, Y.; Hiramatsu, Y. Different profiles of circulating angiogenic factors and adipocytokines between early- and late-onset pre-eclampsia. BJOG 2010, 117, 314–320. [Google Scholar] [CrossRef]
- Barjaktarovic, M.; Korevaar, T.I.M.; Jaddoe, V.W.V.; de Rijke, Y.B.; Peeters, R.P.; Steegers, E.A.P. Human chorionic gonadotropin and risk of pre-eclampsia: Prospective population-based cohort study. Ultrasound Obstet. Gynecol. 2019, 54, 477–483. [Google Scholar] [CrossRef] [PubMed]
- Pazzagli, L.; Abdi, L.; Kieler, H.; Cesta, C.E. Metformin versus insulin use for treatment of gestational diabetes and delivery by caesarean section: A nationwide Swedish cohort study. Eur. J. Obstet. Gynecol. Reprod. Biol. 2020, 254, 271–276. [Google Scholar] [CrossRef]
- Simeonova-Krstevska, S.; Bogoev, M.; Bogoeva, K.; Zisovska, E.; Samardziski, I.; Velkoska-Nakova, V.; Livrinova, V.; Todorovska, I.; Sima, A.; Blazevska-Siljanoska, V. Maternal and Neonatal Outcomes in Pregnant Women with Gestational Diabetes Mellitus Treated with Diet, Metformin or Insulin. Open Access Maced. J. Med. Sci. 2018, 6, 803–807. [Google Scholar] [CrossRef] [PubMed]
- Cassina, M.; Donà, M.; Di Gianantonio, E.; Litta, P.; Clementi, M. First-trimester exposure to metformin and risk of birth defects: A systematic review and meta-analysis. Hum. Reprod. Update 2014, 20, 656–669. [Google Scholar] [CrossRef]
- Given, J.E.; Loane, M.; Garne, E.; Addor, M.C.; Bakker, M.; Bertaut-Nativel, B.; Gatt, M.; Klungsoyr, K.; Lelong, N.; Morgan, M.; et al. Metformin exposure in first trimester of pregnancy and risk of all or specific congenital anomalies: Exploratory case-control study. BMJ 2018, 361, k2477. [Google Scholar] [CrossRef]
- Panchaud, A.; Rousson, V.; Vial, T.; Bernard, N.; Baud, D.; Amar, E.; De Santis, M.; Pistelli, A.; Dautriche, A.; Beau-Salinas, F.; et al. Pregnancy outcomes in women on metformin for diabetes or other indications among those seeking teratology information services. Br. J. Clin. Pharmacol. 2018, 84, 568–578. [Google Scholar] [CrossRef]
- Polasek, T.M.; Doogue, M.P.; Thynne, T.R.J. Metformin treatment of type 2 diabetes mellitus in pregnancy: Update on safety and efficacy. Ther. Adv. Drug. Saf. 2018, 9, 287–295. [Google Scholar] [CrossRef]
- Rowan, J.A.; Rush, E.C.; Plank, L.D.; Lu, J.; Obolonkin, V.; Coat, S.; Hague, W.M. Metformin in gestational diabetes: The offspring follow-up (MiG TOFU): Body composition and metabolic outcomes at 7–9 years of age. BMJ Open Diabetes Res. Care 2018, 6, e000456. [Google Scholar] [CrossRef] [PubMed]
- Hyer, S.; Balani, J.; Shehata, H. Metformin in Pregnancy: Mechanisms and Clinical Applications. Int. J. Mol. Sci. 2018, 19, 1954. [Google Scholar] [CrossRef] [PubMed]
- Xu, Q.; Xie, Q. Long-term effects of prenatal exposure to metformin on the health of children based on follow-up studies of randomized controlled trials: A systematic review and meta-analysis. Arch. Gynecol. Obstet. 2019, 299, 1295–1303. [Google Scholar] [CrossRef] [PubMed]
- Singh, N.; Madhu, M.; Vanamail, P.; Malik, N.; Kumar, S. Efficacy of metformin in improving glycaemic control & perinatal outcome in gestational diabetes mellitus: A non-randomized study. Indian J. Med. Res. 2017, 145, 623–628. [Google Scholar] [CrossRef] [PubMed]
- Feig, D.S.; Donovan, L.E.; Zinman, B.; Sanchez, J.J.; Asztalos, E.; Ryan, E.A.; Fantus, I.G.; Hutton, E.; Armson, A.B.; Lipscombe, L.L.; et al. Metformin in women with type 2 diabetes in pregnancy (MiTy): A multicentre, international, randomised, placebo-controlled trial. Lancet Diabetes Endocrinol. 2020, 8, 834–844. [Google Scholar] [CrossRef]
- Ashoush, S.; El-Said, M.; Fathi, H.; Abdelnaby, M. Identification of metformin poor responders, requiring supplemental insulin, during randomization of metformin versus insulin for the control of gestational diabetes mellitus. J. Obstet. Gynaecol. Res. 2016, 42, 640–647. [Google Scholar] [CrossRef]
- Blair, R.A.; Rosenberg, E.A.; Palermo, N.E. The Use of Non-insulin Agents in Gestational Diabetes: Clinical Considerations in Tailoring Therapy. Curr. Diab. Rep. 2019, 19, 158. [Google Scholar] [CrossRef]
- Cluver, C.; Walker, S.P.; Mol, B.W.; Hall, D.; Hiscock, R.; Brownfoot, F.C.; Kaitu’u-Lino, T.J.; Tong, S. A double blind, randomised, placebo-controlled trial to evaluate the efficacy of metformin to treat preterm pre-eclampsia (PI2 Trial): Study protocol. BMJ Open 2019, 9, e025809. [Google Scholar] [CrossRef]
- Tarry-Adkins, J.L.; Aiken, C.E.; Ozanne, S.E. Neonatal, infant, and childhood growth following metformin versus insulin treatment for gestational diabetes: A systematic review and meta-analysis. PLoS Med. 2019, 16, e1002848. [Google Scholar] [CrossRef]
- Ekpebegh, C.O.; Coetzee, E.J.; van der Merwe, L.; Levitt, N.S. A 10-year retrospective analysis of pregnancy outcome in pregestational Type 2 diabetes: Comparison of insulin and oral glucose-lowering agents. Diabet. Med. 2007, 24, 253–258. [Google Scholar] [CrossRef]
- Cesta, C.E.; Cohen, J.M.; Pazzagli, L.; Bateman, B.T.; Bröms, G.; Einarsdóttir, K.; Furu, K.; Havard, A.; Heino, A.; Hernandez-Diaz, S.; et al. Antidiabetic medication use during pregnancy: An international utilization study. BMJ Open Diabetes Res. Care 2019, 7, e000759. [Google Scholar] [CrossRef] [PubMed]
- Hawthorne, G. Metformin use and diabetic pregnancy-has its time come? Diabet. Med. 2006, 23, 223–227. [Google Scholar] [CrossRef] [PubMed]
- Rai, L.; Meenakshi, D.; Kamath, A. Metformin—A convenient alternative to insulin for Indian women with diabetes in pregnancy. Indian J. Med. Sci. 2009, 63, 491–497. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Waheed, S.; Malik, F.P.; Mazhar, S.B. Efficacy of metformin versus insulin in the management of pregnancy with diabetes. J. Coll. Physicians Surg. Pak. 2013, 23, 866–869. [Google Scholar] [PubMed]
- Beyuo, T.; Obed, S.A.; Adjepong-Yamoah, K.K.; Bugyei, K.A.; Oppong, S.A.; Marfoh, K. Metformin versus Insulin in the Management of Pre-Gestational Diabetes Mellitus in Pregnancy and Gestational Diabetes Mellitus at the Korle Bu Teaching Hospital: A Randomized Clinical Trial. PLoS ONE 2015, 10, e0125712. [Google Scholar] [CrossRef]
- Feig, D.S.; Murphy, K.; Asztalos, E.; Tomlinson, G.; Sanchez, J.; Zinman, B.; Ohlsson, A.; Ryan, E.A.; Fantus, I.G.; Armson, A.B.; et al. Metformin in women with type 2 diabetes in pregnancy (MiTy): A multi-center randomized controlled trial. BMC Pregnancy Childbirth 2016, 16, 173. [Google Scholar] [CrossRef]
- Bailey, C.J. Metformin in women with type 2 diabetes in pregnancy. Lancet Diabetes Endocrinol. 2020, 8, 802–803. [Google Scholar] [CrossRef]
- Elmaraezy, A.; Abushouk, A.I.; Emara, A.; Elshahat, O.; Ahmed, H.; Mostafa, I.M. Effect of metformin on maternal and neonatal outcomes in pregnant obese non-diabetic women: A meta-analysis. Int. J. Reprod. Biomed. 2017, 15, 461–470. [Google Scholar] [CrossRef]
- Dodd, J.M.; Grivell, R.M.; Deussen, A.R.; Hague, W.M. Metformin for women who are overweight or obese during pregnancy for improving maternal and infant outcomes. Cochrane Database Syst. Rev. 2018, 7, CD010564. [Google Scholar] [CrossRef]
- Dodd, J.M.; Louise, J.; Deussen, A.R.; Grivell, R.M.; Dekker, G.; McPhee, A.J.; Hague, W. Effect of metformin in addition to dietary and lifestyle advice for pregnant women who are overweight or obese: The GRoW randomised, double-blind, placebo-controlled trial. Lancet Diabetes Endocrinol. 2019, 7, 15–24. [Google Scholar] [CrossRef]
- Hillman, N.; Herranz, L.; Vaquero, P.M.; Villarroel, A.; Fernandez, A.; Pallardo, L.F. Is pregnancy outcome worse in type 2 than in type 1 diabetic women? Diabetes Care 2006, 29, 2557–2558. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Persson, M.; Norman, M.; Hanson, U. Obstetric and perinatal outcomes in type 1 diabetic pregnancies: A large, population-based study. Diabetes Care 2009, 32, 2005–2009. [Google Scholar] [CrossRef] [PubMed]
- Starikov, R.; Inman, K.; Chen, K.; Lopes, V.; Coviello, E.; Pinar, H.; He, M. Comparison of placental findings in type 1 and type 2 diabetic pregnancies. Placenta 2014, 35, 1001–1006. [Google Scholar] [CrossRef] [PubMed]
- Owens, L.A.; Sedar, J.; Carmody, L.; Dunne, F. Comparing type 1 and type 2 diabetes in pregnancy- similar conditions or is a separate approach required? BMC Pregnancy Childbirth 2015, 15, 69. [Google Scholar] [CrossRef] [PubMed]
- Stone, R.G.; Scully, P.; Troy, E.; Moloney, Y.; Quinn, A.; Noctor, E.; Neylon, O.; Slevin, J.; Murphy, A.; O’Gorman, C. Pregnancy outcomes in women with onset of type 1 diabetes mellitus less than 18 years of age. BMJ Open Diabetes Res. Care 2020, 8, e001080. [Google Scholar] [CrossRef] [PubMed]
- Morikawa, M.; Kato-Hirayama, E.; Mayama, M.; Saito, Y.; Nakagawa, K.; Umazume, T.; Chiba, K.; Kawaguchi, S.; Okuyama, K.; Watari, H. Glycemic control and fetal growth of women with diabetes mellitus and subsequent hypertensive disorders of pregnancy. PLoS ONE 2020, 15, e0230488. [Google Scholar] [CrossRef]
- García-Patterson, A.; Gich, I.; Amini, S.B.; Catalano, P.M.; de Leiva, A.; Corcoy, R. Insulin requirements throughout pregnancy in women with type 1 diabetes mellitus: Three changes of direction. Diabetologia 2010, 53, 446–451. [Google Scholar] [CrossRef]
- Cyganek, K.; Klupa, T.; Szopa, M.; Katra, B.; Małecki, M.T. Medical care of pregnant women with type 1 diabetes: Current guidelines and clinical practice. Pol. Arch. Med. Wewn. 2013, 123, 59–65. [Google Scholar] [CrossRef]
- Murphy, H.R. Intensive Glycemic Treatment during Type 1 Diabetes Pregnancy: A Story of (Mostly) Sweet Success! Diabetes Care 2018, 41, 1563–1571. [Google Scholar] [CrossRef]
- Steel, J.M.; Johnstone, F.D.; Hume, R.; Mao, J.H. Insulin requirements during pregnancy in women with type I diabetes. Obstet. Gynecol. 1994, 83, 253–258. [Google Scholar]
- Abell, S.K.; Boyle, J.A.; de Courten, B.; Knight, M.; Ranasinha, S.; Regan, J.; Soldatos, G.; Wallace, E.M.; Zoungas, S.; Teede, H.J. Contemporary type 1 diabetes pregnancy outcomes: Impact of obesity and glycaemic control. Med. J. Aust. 2016, 205, 162–167. [Google Scholar] [CrossRef] [PubMed]
- McGrath, R.T.; Glastras, S.J.; Hocking, S.L.; Fulcher, G.R. Large-for-Gestational-Age Neonates in Type 1 Diabetes and Pregnancy: Contribution of Factors Beyond Hyperglycemia. Diabetes Care 2018, 41, 1821–1828. [Google Scholar] [CrossRef] [PubMed]
- Gómez, A.M.; Marín Carrillo, L.F.; Arévalo Correa, C.M.; Muñoz Velandia, O.M.; Rondón Sepúlveda, M.A.; Silva Herrera, J.L.; Henao Carrillo, D.C. Maternal-Fetal Outcomes in 34 Pregnant Women with Type 1 Diabetes in Sensor-Augmented Insulin Pump Therapy. Diabetes Technol. Ther. 2017, 19, 417–422. [Google Scholar] [CrossRef]
- Polsky, S.; Wu, M.; Bode, B.W.; DuBose, S.N.; Goland, R.S.; Maahs, D.M.; Foster, N.C.; Peters, A.L.; Levy, C.J.; Shah, V.N.; et al. Diabetes Technology Use Among Pregnant and Nonpregnant Women with T1D in the T1D Exchange. Diabetes Technol. Ther. 2018, 20, 517–523. [Google Scholar] [CrossRef] [PubMed]
- Moon, R.J.; Bascombe, L.A.; Holt, R.I. The addition of metformin in type 1 diabetes improves insulin sensitivity, diabetic control, body composition and patient well-being. Diabetes Obes. Metab. 2007, 9, 143–145. [Google Scholar] [CrossRef]
- Al Khalifah, R.A.; Alnhdi, A.; Alghar, H.; Alanazi, M.; Florez, I.D. The effect of adding metformin to insulin therapy for type 1 diabetes mellitus children: A systematic review and meta-analysis. Pediatr. Diabetes 2017, 18, 664–673. [Google Scholar] [CrossRef]
- Livingstone, R.; Boyle, J.G.; Petrie, J.R.; REMOVAL Study Team. A new perspective on metformin therapy in type 1 diabetes. Diabetologia 2017, 60, 1594–1600. [Google Scholar] [CrossRef]
- Beysel, S.; Unsal, I.O.; Kizilgul, M.; Caliskan, M.; Ucan, B.; Cakal, E. The effects of metformin in type 1 diabetes mellitus. BMC Endocr. Disord. 2018, 18, 1. [Google Scholar] [CrossRef]
- Priya, G.; Kalra, S. Metformin in the management of diabetes during pregnancy and lactation. Drugs Context 2018, 7, 212523. [Google Scholar] [CrossRef]
- Ping, F.; Deng, M.; Zhai, X.; Song, Y.; Xiao, X. Real-World Experience of Adding Metformin in Pregnant Women with Type 1 Diabetes in a Chinese Population: A Retrospective Cohort. Diabetes Ther. 2019, 10, 1089–1097. [Google Scholar] [CrossRef]
- Chavarro, J.E.; Toth, T.L.; Wright, D.L.; Meeker, J.D.; Hauser, R. Body mass index in relation to semen quality, sperm DNA integrity, and serum reproductive hormone levels among men attending an infertility clinic. Fertil. Steril. 2010, 93, 2222–2231. [Google Scholar] [CrossRef] [PubMed]
- Hofny, E.R.; Ali, M.E.; Abdel-Hafez, H.Z.; Kamal Eel, D.; Mohamed, E.E.; Abd El-Azeem, H.G.; Mostafa, T. Semen parameters and hormonal profile in obese fertile and infertile males. Fertil. Steril. 2010, 94, 581–584. [Google Scholar] [CrossRef] [PubMed]
- Brand, J.S.; van der Tweel, I.; Grobbee, D.E.; Emmelot-Vonk, M.H.; van der Schouw, Y.T. Testosterone, sex hormone-binding globulin and the metabolic syndrome: A systematic review and meta-analysis of observational studies. Int. J. Epidemiol. 2011, 40, 189–207. [Google Scholar] [CrossRef] [PubMed]
- La Vignera, S.; Condorelli, R.; Vicari, E.; D’Agata, R.; Calogero, A.E. Diabetes mellitus and sperm parameters. J. Androl. 2012, 33, 145–153. [Google Scholar] [CrossRef] [PubMed]
- Dupont, C.; Faure, C.; Sermondade, N.; Boubaya, M.; Eustache, F.; Clément, P.; Briot, P.; Berthaut, I.; Levy, V.; Cedrin-Durnerin, I.; et al. Obesity leads to higher risk of sperm DNA damage in infertile patients. Asian J. Androl. 2013, 15, 622–625. [Google Scholar] [CrossRef] [PubMed]
- Corona, G.; Bianchini, S.; Sforza, A.; Vignozzi, L.; Maggi, M. Hypogonadism as a possible link between metabolic diseases and erectile dysfunction in aging men. Hormones 2015, 14, 569–578. [Google Scholar] [CrossRef]
- Pergialiotis, V.; Prodromidou, A.; Frountzas, M.; Korou, L.M.; Vlachos, G.D.; Perrea, D. Diabetes mellitus and functional sperm characteristics: A meta-analysis of observational studies. J. Diabetes Complicat. 2016, 30, 1167–1176. [Google Scholar] [CrossRef]
- Lu, X.; Huang, Y.; Zhang, H.; Zhao, J. Effect of diabetes mellitus on the quality and cytokine content of human semen. J. Reprod. Immunol. 2017, 123, 1–2. [Google Scholar] [CrossRef]
- Condorelli, R.A.; La Vignera, S.; Mongioì, L.M.; Alamo, A.; Calogero, A.E. Diabetes Mellitus and Infertility: Different Pathophysiological Effects in Type 1 and Type 2 on Sperm Function. Front. Endocrinol. 2018, 9, 268. [Google Scholar] [CrossRef] [PubMed]
- Dhindsa, S.; Ghanim, H.; Batra, M.; Dandona, P. Hypogonadotropic Hypogonadism in Men with Diabesity. Diabetes Care 2018, 41, 1516–1525. [Google Scholar] [CrossRef] [PubMed]
- Rastrelli, G.; Filippi, S.; Sforza, A.; Maggi, M.; Corona, G. Metabolic Syndrome in Male Hypogonadism. Front. Horm. Res. 2018, 49, 131–155. [Google Scholar] [CrossRef] [PubMed]
- Martins, A.D.; Majzoub, A.; Agawal, A. Metabolic Syndrome and Male Fertility. World J. Men’s Health 2019, 37, 113–127. [Google Scholar] [CrossRef] [PubMed]
- Agarwal, A.; Said, T.M. Oxidative stress, DNA damage and apoptosis in male infertility: A clinical approach. BJU Int. 2005, 95, 503–507. [Google Scholar] [CrossRef] [PubMed]
- Hagiuda, J.; Ishikawa, H.; Furuuchi, T.; Hanawa, Y.; Marumo, K. Relationship between dyslipidaemia and semen quality and serum sex hormone levels: An infertility study of 167 Japanese patients. Andrologia 2014, 46, 131–135. [Google Scholar] [CrossRef] [PubMed]
- Alahmar, A.T. Role of Oxidative Stress in Male Infertility: An Updated Review. J. Hum. Reprod. Sci. 2019, 12, 4–18. [Google Scholar] [CrossRef] [PubMed]
- Zhao, L.; Pang, A. Effects of Metabolic Syndrome on Semen Quality and Circulating Sex Hormones: A Systematic Review and Meta-Analysis. Front. Endocrinol. 2020, 11, 428. [Google Scholar] [CrossRef]
- Bhattacharya, S.M.; Ghosh, M.; Nandi, N. Diabetes mellitus and abnormalities in semen analysis. J. Obstet. Gynaecol. Res. 2014, 40, 167–171. [Google Scholar] [CrossRef]
- Colli, L.G.; Belardin, L.B.; Echem, C.; Akamine, E.H.; Antoniassi, M.P.; Andretta, R.R.; Mathias, L.S.; Rodrigues, S.F.P.; Bertolla, R.P.; de Carvalho, M.H.C. Systemic arterial hypertension leads to decreased semen quality and alterations in the testicular microcirculation in rats. Sci. Rep. 2019, 9, 11047. [Google Scholar] [CrossRef]
- Stokes, V.J.; Anderson, R.A.; George, J.T. How does obesity affect fertility in men–and what are the treatment options? Clin. Endocrinol. 2015, 82, 633–638. [Google Scholar] [CrossRef]
- Morgante, G.; Tosti, C.; Orvieto, R.; Musacchio, M.C.; Piomboni, P.; De Leo, V. Metformin improves semen characteristics of oligo-terato-asthenozoospermic men with metabolic syndrome. Fertil. Steril. 2011, 95, 2150–2152. [Google Scholar] [CrossRef]
- Liu, C.Y.; Chang, T.C.; Lin, S.H.; Wu, S.T.; Cha, T.L.; Tsao, C.W. Metformin Ameliorates Testicular Function and Spermatogenesis in Male Mice with High-Fat and High-Cholesterol Diet-Induced Obesity. Nutrients 2020, 12, 1932. [Google Scholar] [CrossRef] [PubMed]
- Ozata, M.; Oktenli, C.; Bingol, N.; Ozdemir, I.C. The effects of metformin and diet on plasma testosterone and leptin levels in obese men. Obes. Res. 2001, 9, 662–667. [Google Scholar] [CrossRef] [PubMed]
- Al-Kuraishy, H.M.; Al-Gareeb, A.I. Erectile Dysfunction and Low Sex Drive in Men with Type 2 DM: The Potential Role of Diabetic Pharmacotherapy. J. Clin. Diagn. Res. 2016, 10, FC21–FC26. [Google Scholar] [CrossRef] [PubMed]
- Pelusi, C.; Giagulli, V.A.; Baccini, M.; Fanelli, F.; Mezzullo, M.; Fazzini, A.; Bianchi, N.; Carbone, M.D.; De Pergola, G.; Mastroroberto, M.; et al. Clomiphene citrate effect in obese men with low serum testosterone treated with metformin due to dysmetabolic disorders: A randomized, double-blind, placebo-controlled study. PLoS ONE 2017, 12, e0183369. [Google Scholar] [CrossRef]
- Tosca, L.; Froment, P.; Rame, C.; McNeilly, J.R.; McNeilly, A.S.; Maillard, V.; Dupont, J. Metformin decreases GnRH- and activin-induced gonadotropin secretion in rat pituitary cells: Potential involvement of adenosine 5’ monophosphate-activated protein kinase (PRKA). Biol. Reprod. 2011, 84, 351–362. [Google Scholar] [CrossRef]
- Casulari, L.A.; Caldas, A.D.; Domingues Casulari Motta, L.; Lofrano-Porto, A. Effects of metformin and short-term lifestyle modification on the improvement of male hypogonadism associated with metabolic syndrome. Minerva Endocrinol. 2010, 35, 145–151. [Google Scholar]
- Guay, A.T.; Bansal, S.; Heatley, G.J. Effect of raising endogenous testosterone levels in impotent men with secondary hypogonadism: Double blind placebo-controlled trial with clomiphene citrate. J. Clin. Endocrinol. Metab. 1995, 80, 3546–3552. [Google Scholar] [CrossRef]
- Giagulli, V.A.; Silvestrini, A.; Bruno, C.; Triggiani, V.; Mordente, A.; Mancini, A. Is There Room for SERMs or SARMs as Alternative Therapies for Adult Male Hypogonadism? Int. J. Endocrinol. 2020, 2020, 9649838. [Google Scholar] [CrossRef]
- Aggerholm, A.S.; Thulstrup, A.M.; Toft, G.; Ramlau-Hansen, C.H.; Bonde, J.P. Is overweight a risk factor for reduced semen quality and altered serum sex hormone profile? Fertil. Steril. 2008, 90, 619–626. [Google Scholar] [CrossRef]
- Hammoud, A.O.; Wilde, N.; Gibson, M.; Parks, A.; Carrell, D.T.; Meikle, A.W. Male obesity and alteration in sperm parameters. Fertil. Steril. 2008, 90, 2222–2225. [Google Scholar] [CrossRef]
- Allan, C.A.; McLachlan, R.I. Androgens and obesity. Curr. Opin. Endocrinol. Diabetes Obes. 2010, 17, 224–232. [Google Scholar] [CrossRef] [PubMed]
- Mah, P.M.; Wittert, G.A. Obesity and testicular function. Mol. Cell. Endocrinol. 2010, 316, 180–186. [Google Scholar] [CrossRef] [PubMed]
- Bellastella, G.; Menafra, D.; Puliani, G.; Colao, A.; Savastano, S.; Obesity Programs of nutrition, Education, Research and Assessment (OPERA) Group. How much does obesity affect the male reproductive function? Int. J. Obes. Suppl. 2019, 9, 50–64. [Google Scholar] [CrossRef] [PubMed]
- Lee, Y.; Dang, J.T.; Switzer, N.; Yu, J.; Tian, C.; Birch, D.W.; Karmali, S. Impact of Bariatric Surgery on Male Sex Hormones and Sperm Quality: A Systematic Review and Meta-Analysis. Obes. Surg. 2019, 29, 334–346. [Google Scholar] [CrossRef]
- Hart, R.J.; Doherty, D.A.; Mori, T.A.; Adams, L.A.; Huang, R.C.; Minaee, N.; Handelsman, D.J.; McLachlan, R.; Norman, R.J.; Dickinson, J.E.; et al. Features of the metabolic syndrome in late adolescence are associated with impaired testicular function at 20 years of age. Hum. Reprod. 2019, 34, 389–402. [Google Scholar] [CrossRef]
- Kapoor, D.; Channer, K.S.; Jones, T.H. Rosiglitazone increases bioactive testosterone and reduces waist circumference in hypogonadal men with type 2 diabetes. Diab. Vasc. Dis. Res. 2008, 5, 135–137. [Google Scholar] [CrossRef]
- Wong, L.; Chen, H.M.; Lai, S.Q.; Yang, H.Z.; Kuang, J.; Pei, J.H. Effects of sulfonylurea as initial treatment on testosterone of middle-aged men with type 2 diabetes: A 16-week, pilot study. J. Diabetes Investig. 2015, 6, 454–459. [Google Scholar] [CrossRef]
- Attia, S.M.; Helal, G.K.; Alhaider, A.A. Assessment of genomic instability in normal and diabetic rats treated with metformin. Chem. Biol. Interact. 2009, 180, 296–304. [Google Scholar] [CrossRef]
- Rabbani, S.I.; Devi, K.; Khanam, S. Role of Pioglitazone with Metformin or Glimepiride on Oxidative Stress-induced Nuclear Damage and Reproductive Toxicity in Diabetic Rats. Malays. J. Med. Sci. 2010, 17, 3–11. [Google Scholar]
- Yan, W.J.; Mu, Y.; Yu, N.; Yi, T.L.; Zhang, Y.; Pang, X.L.; Cheng, D.; Yang, J. Protective effects of metformin on reproductive function in obese male rats induced by high-fat diet. J. Assist. Reprod. Genet. 2015, 32, 1097–1104. [Google Scholar] [CrossRef]
- Ghasemnejad-Berenji, M.; Ghazi-Khansari, M.; Yazdani, I.; Nobakht, M.; Abdollahi, A.; Ghasemnejad-Berenji, H.; Mohajer Ansari, J.; Pashapour, S.; Dehpour, A.R. Effect of metformin on germ cell-specific apoptosis, oxidative stress and epididymal sperm quality after testicular torsion/detorsion in rats. Andrologia 2018, 50. [Google Scholar] [CrossRef] [PubMed]
- Nna, V.U.; Bakar, A.B.A.; Ahmad, A.; Mohamed, M. Diabetes-induced testicular oxidative stress, inflammation, and caspase-dependent apoptosis: The protective role of metformin. Arch. Physiol. Biochem. 2018, 4, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Nna, V.U.; Abu Bakar, A.B.; Ahmad, A.; Eleazu, C.O.; Mohamed, M. Oxidative Stress, NF-κB-Mediated Inflammation and Apoptosis in the Testes of Streptozotocin-Induced Diabetic Rats: Combined Protective Effects of Malaysian Propolis and Metformin. Antioxidants 2019, 8, 465. [Google Scholar] [CrossRef]
- Annie, L.; Jeremy, M.; Gurusubramanian, G.; Derkach, K.V.; Shpakov, A.O.; Roy, V.K. Effect of metformin on testicular expression and localization of leptin receptor and levels of leptin in the diabetic mice. Mol. Reprod. Dev. 2020, 87, 620–629. [Google Scholar] [CrossRef]
- Derkach, K.V.; Bakhtyukov, A.A.; Romanova, I.V.; Zorina, I.I.; Bayunova, L.V.; Bondareva, V.M.; Morina, I.Y.; Kumar Roy, V.; Shpakov, A.O. The effect of metformin treatment on the basal and gonadotropin-stimulated steroidogenesis in male rats with type 2 diabetes mellitus. Andrologia 2020, e13816. [Google Scholar] [CrossRef] [PubMed]
- Ayuob, N.N.; Murad, H.A.; Ali, S.S. Impaired expression of sex hormone receptors in male reproductive organs of diabetic rat in response to oral antidiabetic drugs. Folia Histochem. Cytobiol. 2015, 53, 35–48. [Google Scholar] [CrossRef]
- Nna, V.U.; Bakar, A.B.A.; Ahmad, A.; Mohamed, M. Down-regulation of steroidogenesis-related genes and its accompanying fertility decline in streptozotocin-induced diabetic male rats: Ameliorative effect of metformin. Andrology 2019, 7, 110–123. [Google Scholar] [CrossRef] [PubMed]
- Morgan, D.H.; Ghribi, O.; Hui, L.; Geiger, J.D.; Chen, X. Cholesterol-enriched diet disrupts the blood-testis barrier in rabbits. Am. J. Physiol. Endocrinol. Metab. 2014, 307, E1125–E1130. [Google Scholar] [CrossRef]
- Yu, C.; Jiang, F.; Zhang, M.; Luo, D.; Shao, S.; Zhao, J.; Gao, L.; Zuo, C.; Guan, Q. HC diet inhibited testosterone synthesis by activating endoplasmic reticulum stress in testicular Leydig cells. J. Cell Mol. Med. 2019, 23, 3140–3150. [Google Scholar] [CrossRef] [PubMed]
- Fejes, I.; Koloszár, S.; Závaczki, Z.; Daru, J.; Szöllösi, J.; Pál, A. Effect of body weight on testosterone/estradiol ratio in oligozoospermic patients. Arch. Androl. 2006, 52, 97–102. [Google Scholar] [CrossRef]
- Asghari, A.; Akbari, G.; Meghdadi, A.; Mortazavi, P. Protective effect of metformin on testicular ischemia/reperfusion injury in rats. Acta Cir. Bras. 2016, 31, 411–416. [Google Scholar] [CrossRef]
- Asghari, A.; Akbari, G.; Meghdadi, A.; Mortazavi, P. Effects of melatonin and metformin co-administration on testicular ischemia/reperfusion injury in rats. J. Pediatr. Urol. 2016, 12, 410.e1–410.e7. [Google Scholar] [CrossRef] [PubMed]
- Nasrolahi, O.; Khaneshi, F.; Rahmani, F.; Razi, M. Honey and metformin ameliorated diabetes-induced damages in testes of rat; correlation with hormonal changes. Iran. J. Reprod. Med. 2013, 11, 1013–1020. [Google Scholar] [PubMed]
- Nna, V.U.; Bakar, A.B.A.; Ahmad, A.; Umar, U.Z.; Suleiman, J.B.; Zakaria, Z.; Othman, Z.A.; Mohamed, M. Malaysian propolis and metformin mitigate subfertility in streptozotocin-induced diabetic male rats by targeting steroidogenesis, testicular lactate transport, spermatogenesis and mating behaviour. Andrology 2020, 8, 731–746. [Google Scholar] [CrossRef] [PubMed]
- Mardanshahi, T.; Rezaei, N.; Zare, Z.; Malekzadeh Shafaroudi, M.; Mohammadi, H. Effects of L-Carnitine on the sperm parameters disorders, apoptosis of spermatogenic cells and testis histopathology in diabetic Rats. Int. J. Reprod. Biomed. 2018, 17, 325–336. [Google Scholar] [CrossRef] [PubMed]
- Hurtado de Llera, A.; Martin-Hidalgo, D.; Gil, M.C.; Garcia-Marin, L.J.; Bragado, M.J. AMP-activated kinase AMPK is expressed in boar spermatozoa and regulates motility. PLoS ONE 2012, 7, e38840. [Google Scholar] [CrossRef]
- Calle-Guisado, V.; de Llera, A.H.; Martin-Hidalgo, D.; Mijares, J.; Gil, M.C.; Alvarez, I.S.; Bragado, M.J.; Garcia-Marin, L.J. AMP-activated kinase in human spermatozoa: Identification, intracellular localization, and key function in the regulation of sperm motility. Asian J. Androl. 2017, 19, 707–714. [Google Scholar] [CrossRef]
- Nguyen, T.M. Impact of 5’-amp-activated Protein Kinase on Male Gonad and Spermatozoa Functions. Front. Cell Dev. Biol. 2017, 5, 25. [Google Scholar] [CrossRef]
- Nguyen, T.M.D. Role of AMPK in mammals reproduction: Specific controls and whole-body energy sensing. Comptes Rendus Biol. 2019, 342, 1–6. [Google Scholar] [CrossRef]
- Martin-Hidalgo, D.; de Llera, H.A.; Calle-Guisado, V.; Gonzalez-Fernandez, L.; Garcia-Marin, L.; Bragado, M.J. AMPK Function in Mammalian Spermatozoa. Int. J. Mol. Sci. 2018, 19, 3293. [Google Scholar] [CrossRef]
- Kwon, O.; Kim, K.W.; Kim, M.S. Leptin signalling pathways in hypothalamic neurons. Cell. Mol. Life Sci. 2016, 73, 1457–1477. [Google Scholar] [CrossRef] [PubMed]
- Wang, B.; Cheng, K.K. Hypothalamic AMPK as a Mediator of Hormonal Regulation of Energy Balance. Int. J. Mol. Sci. 2018, 19, 3552. [Google Scholar] [CrossRef] [PubMed]
- Tosca, L.; Chabrolle, C.; Dupont, J. L’AMPK: Un lien entre métabolisme et reproduction? [AMPK: A link between metabolism and reproduction?]. Med. Sci. 2008, 24, 297–300. [Google Scholar] [CrossRef]



Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Shpakov, A.O. Improvement Effect of Metformin on Female and Male Reproduction in Endocrine Pathologies and Its Mechanisms. Pharmaceuticals 2021, 14, 42. https://doi.org/10.3390/ph14010042
Shpakov AO. Improvement Effect of Metformin on Female and Male Reproduction in Endocrine Pathologies and Its Mechanisms. Pharmaceuticals. 2021; 14(1):42. https://doi.org/10.3390/ph14010042
Chicago/Turabian StyleShpakov, Alexander O. 2021. "Improvement Effect of Metformin on Female and Male Reproduction in Endocrine Pathologies and Its Mechanisms" Pharmaceuticals 14, no. 1: 42. https://doi.org/10.3390/ph14010042
APA StyleShpakov, A. O. (2021). Improvement Effect of Metformin on Female and Male Reproduction in Endocrine Pathologies and Its Mechanisms. Pharmaceuticals, 14(1), 42. https://doi.org/10.3390/ph14010042