2.1. Development of the Cycle Network Model of PGHS-1
The derivation of the model of PGHS-1 catalysis represents a special task, as both POX and COX activities are known to be located in different enzyme domains within the same protein complex, and these activities can proceed independently of each other [
18]. Another important feature of PGHS-1 catalysis is that it involves intramolecular electron transfer and formation of multiple redox and binding states of the catalytic domain. The start point of our modelling was the BC mechanism [
5,
9] schematically represented in
Figure 1. The full catalytic cycle of PGHS-1 includes two distinct catalytic activities: a cyclooxygenase (COX) activity converting arachidonic acid (AA) to an intermediate product (prostaglandin G
2 (PGG
2)); and a peroxidase (POX) activity, which further converts PGG
2 into final product PGH
2. Following the approach to the PGHS-1 modelling [
9], we assumed that PGHS-1 catalysis could be presented as a network of microscopic reactions, each of them resulting in a corresponding change in the enzyme catalytic state. Under a catalytic state, we understand the state of the PGHS-1 catalytic domain, which in its turn is defined by a unique combination of the states of its key elementary catalytic constituents, located in the COX and POX active sites. In our model, we considered the following key catalytically active components: fatty acid binding site (COX site), heme prosthetic group (POX site), and Tyr385 residue in the COX site.
We defined the COX site microstates through the presence/absence of AA in the hydrophobic channel of the COX region. The POX state was determined by the redox states of heme iron and protoporphyrin group. Tyr385 residue was assumed to exist in either a ground or radical state. In addition, we considered states of fully inactive enzyme (FIE) [
19]. The detailed description of all-possible states for each catalytic component, considered in our model, is given in
Table 1.
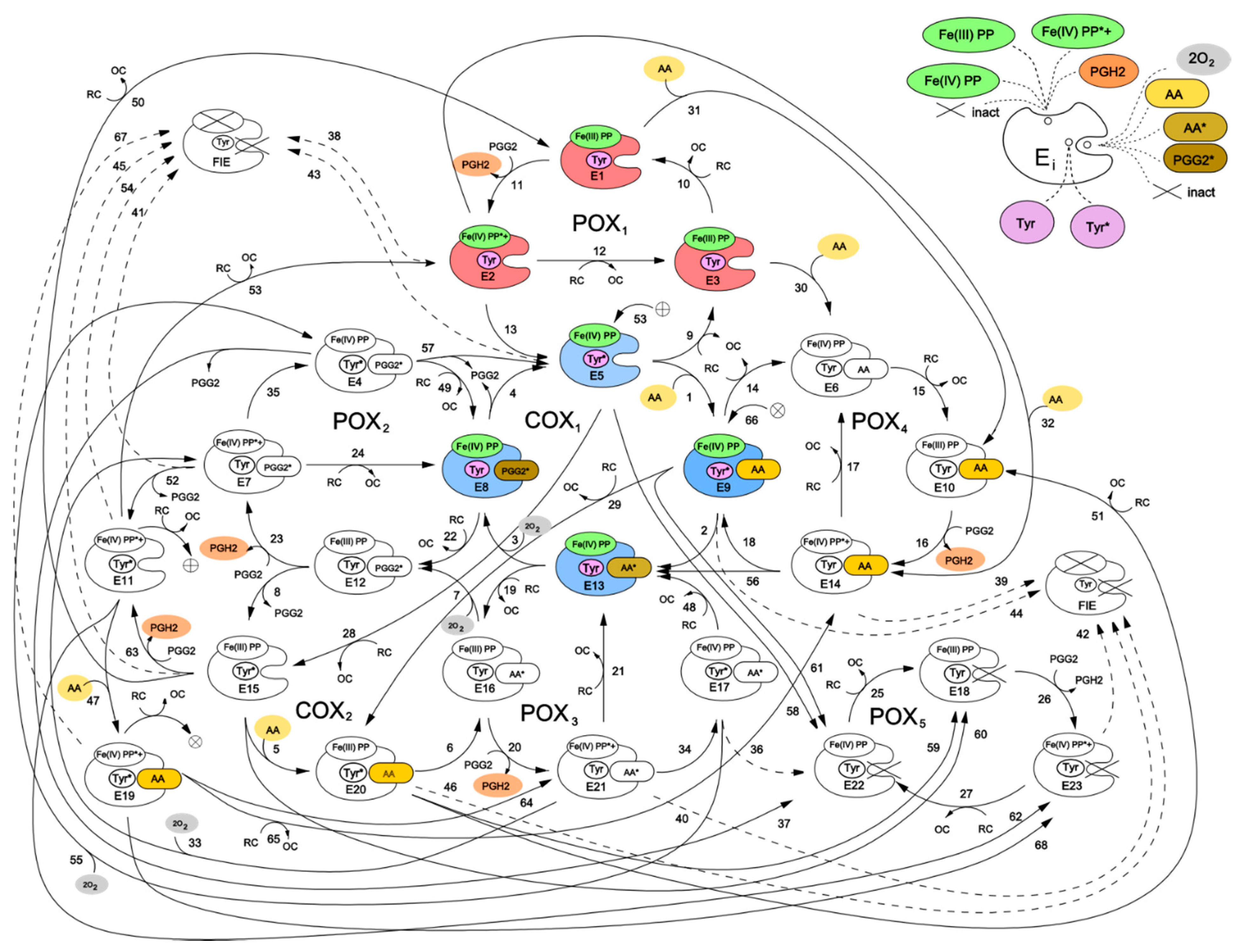
Figure 2 shows a kinetic scheme for the developed catalytic cycle of PGHS-1. The scheme is mainly based on the assumptions of the BC mechanism [
5] and its further extension [
9]. Additionally, we also included in the model some reactions corresponding to an early version of the TC mechanism of PGHS-1 catalysis [
6]. Thus, our scheme represents a generalisation of two previously discussed catalytic mechanisms, and the results obtained in the framework of the model allowed us to draw the conclusions on the role of each mechanism in PGHS-1 catalysis.
We have presented the full catalytic cycle of PGHS-1 as a network of eight interlinked cycles—five of them (POXi) corresponding to the POX activity, and the other three (COXj) to the COX activity of the enzyme. In general, the full catalytic cycle of PGHS-1 can be considered as a complex dynamic network of elementary reactions which describes the totality of possible transitions between multiple states of the PGHS-1 catalytic domain.
Two interconnected cycles—COX
1 (reactions 1–4) and POX
1 (reactions 9–13)—correspond to the BC mechanism. According to this mechanism, PGHS-1 catalysis is initiated in the POX site, and its first step is the reaction of the resting enzyme state [Fe(III), PP] (E
1) with PGG
2 producing PGH
2 and leaving the heme prosthetic group in the state [Fe(IV), PP*+] (E
2). The enzyme state containing heme with two oxidizing equivalents—one on the iron and another one on the porphyrin radical cation—is referred to as Intermediate 1 [
5]. In our model, we described this stage of POX catalysis by bimolecular irreversible reaction (reaction 11) of the resting state with PGG
2, resulting in production of PGH
2 and leaving the enzyme in the E
2 state with heme in [Fe(IV), PP*+] state. The E
2 state can be converted back to the resting state E
1 by 2 consecutive reactions with RC by followed release of oxidized cosubstrate, OC (reactions 12 and 10). Alternatively, the E
2 state can undergo one-electron internal reduction via intramolecular electron transfer from Tyr385 residue in the COX site to protoporphyrin (reaction 13) resulting in the formation of the E
5 state [Fe(IV), PP, Tyr*] containing Tyr385 radical (Tyr385*) and a ferryl heme. This state is referred to as Intermediate II [
5]. The E
5 state can also complete POX cycle and return to the resting state E
1 via reactions 9 and 10 with RC through the formation of [Fe(IV), PP] referred to as Intermediate II [
5]. According to the BC mechanism, the presence of Tyr385 radical is ultimately required for AA oxidation in the COX site. Therefore, the intermediate state E
5 can potentially serve as a starting point for the further cascade of COX reactions.
The majority of previously known models included COX activity as a single Michaelis–Menten type reaction without considering its mechanism in any detail [
10,
13]. In contrast, in our model we describe COX activity by four consecutive reactions (reactions 1–4 in COX
1 cycle) corresponding to the transitions of the enzyme through three intermediate enzyme-substrate complexes (E
9, E
13, and E
8). Note that using Michaelis–Menten approximation to describe the individual reactions involved in a reaction network is not strongly valid.
Reaction 1 describes reversible binding of AA to the enzyme state E
5, resulting in the formation of the state [Fe(IV), PP, Tyr*, AA] (E
9), containing AA in the COX site. Reaction 2 corresponds to the abstraction of a hydrogen atom from AA by Tyr385 radical and formation of the E
13 state containing AA radical in the COX site, [Fe(IV), PP, Tyr, AA*]. Further, AA
* reacts with two oxygen molecules, and the intermediate product PGG
2 radical (PGG
2*) is produced (reaction 3). We assumed that binding of the first and second oxygen molecules is a rapid process, during which the redox state of the heme remains unchanged [
8]. The final step of the COX
1 cycle in our model is irreversible reaction 4, which describes the process of regeneration of Intermediate II with Tyr385 radical (E
5), accompanied by the release of the intermediate product PGG
2 from the COX site.
Taking into account that POX activity of PGHS-1 is observed to proceed independently from COX catalysis [
18], we considered the possibility of several additional POX and COX catalytic cycles, originating from intermediate states of the POX
1 and COX
1 cycles. For example, in the COX
1 cycle, the heme prosthetic group remains in the state [Fe(IV), PP] with one oxidising equivalent on iron, and therefore, each of the enzyme states E
5, E
8, E
9, and E
13 can potentially participate in redox reactions with RC (reactions 19, 22, 28, and 29), producing catalytic states E
12, E
15, E
16, and E
20. The later states can be considered as intermediates of an additional COX
2 cycle. Note that similar COX cycle was included in the model developed by Kulmacz et al. [
18]. The COX
2 cycle differs from the COX
1 cycle only by the state of the heme prosthetic group in the enzyme intermediates. Similarly, each intermediate of the POX
1 cycle has an empty COX binding site which allows for the binding of AA (reactions 30, 31, and 32) and production of the states E
6, E
10, and E
14, respectively. These states can participate in the POX
2 cycle, which differs from the POX
1 cycle by the presence of AA in the COX binding site of its enzyme intermediates. The binding of AA to the resting state (E
1), containing Tyr385 in the ground state, was considered in the PGHS-1 model for the first time, as all previously known models assumed, that AA binds solely to the enzyme containing Tyr385 radical state (Intermediate II, E
5). This allowed us to analyse how the redox state of key catalytic components can influence the enzyme binding to AA.
The third COX
3 cycle, though not explicitly shown in the scheme in
Figure 2, can be defined as a cycle formed by the enzyme states E
11, E
19, E
21, and E
7, involving reactions 47, 64, 33, and 53, respectively. The reactions in the COX
3 cycle are initiated in the result of AA binding to the enzyme being in a two-radical state [Fe(IV), PP*+, Tyr*] (state E
11). Note, that similar COX cycle was considered in the extended BC mechanism model [
10,
18], but its contribution to PGHS-1 functioning was not indicated.
Using similar approach, we introduced into our model the POX3 and POX4 cycles, intermediates of which differ from POX1 intermediates by the occupancy of the COX binding site—it contains AA radical in the POX3 cycle and PGG2 radical in the POX4 cycle. The POX5 cycle was included to account for remaining POX activity, when the COX site is inactivated (see more detailed description below).
In our model, we also included an alternative route for initiation of COX reactions, which was proposed in an early version of the TC mechanism of PGHS-1 function [
5]—a direct hydrogen atom abstraction from AA by protoporphyrin radical cation PP*+ and generation of the AA radical. In our scheme, this mechanism is described by the reaction of AA binding to the initial state E
1 resulting in the production of enzyme-substrate complex E
14, [Fe(III), PP, Tyr, AA], which further converts into intermediate, [Fe(IV), PP, Tyr, AA*] (E
13), containing the arachidonyl radical AA* (reaction 56). Consideration of two alternative mechanisms of AA radical formation in our model allowed us to analyse their possible contributions to overall COX activity.
2.2. Modelling of PGHS-1 Self-Inactivation
PGHS-1 activity undergoes irreversible self-inactivation (SI) during catalysis [
19]. The exact mechanism of this process is still not completely understood [
3]. However, there is a consensus that SI most likely originates from the enzyme damage caused by oxidative reactions, coming from certain radical intermediates generated in PGHS-1 [
20]. Potentially, any enzyme intermediate containing oxidising equivalents or radicals can cause the enzyme damage. One of the key questions discussed in the literature is which intermediate states can serve as the origin for POX and COX inactivation.
Based on the experimental data, we considered several possible mechanisms for enzyme SI in our model. These mechanisms included separate inactivation of the COX and POX activities, originating from enzyme intermediates with different redox state of heme group and containing one or two radicals in the catalytic domains [
20]. All the SI processes accounted for in our model can be conditionally classified into the two following groups.
First, inactivation of POX activity resulting in fully inactivation enzyme (FIE state in
Figure 2) was taken into consideration. According to the BC mechanism, SI of POX activity leads to the subsequent loss of COX activity, as POX reactions are required to regenerate Tyr385 radical state. In our model, this SI mechanism is described by reactions 43–46 and 54. Due to simplicity reasons, we did not distinguish two stages in this process and approximated it by the following one-stage irreversible first order reactions:
(a) Reactions 38–42, 53, and 67 originate from Intermediate I and lead to POX and further COX inactivation due to radical state of heme prosthetic group ([Fe(IV), PP*+]) [
21,
22]. In our model, this SI process can be initiated from seven catalytic states containing radicals such as E
2, E
7, E
11, E
14, E
19, E
21, and E
23;
(b) Reactions 43–46 originate from Intermediate II (states E5, E9, E15, and E20) and lead to POX and further COX inactivation due to the presence of oxidising equivalent Fe(IV) in heme.
Second, we considered the direct inactivation of COX activity with remaining POX activity which can be caused by the presence of radical states in the COX site in the model [
19]. In our model, the remaining POX activity is described by the POX
5 cycle (reactions 25–27). A similar POX cycle was considered in the model developed by Kulmacz et al. [
18]. According to this hypothesis, we assumed that COX inactivation can originate from the enzyme intermediates containing either one or two radicals in the COX catalytic site. Reactions 58–62 and 68 originate from the states containing one Tyr385 radical in the COX site (E
5, E
9, E
11, E
15, E
19, and E
20). Reactions 36 and 37 originate from the states with two radicals in the COX site, [Fe(IV),PP, Tyr*, AA*] (E
17) and [Fe(IV),PP, Tyr*, PGG
2*] (E
4). It should be noted that the remaining POX activity can also further be inactivated due to damage caused by the presence of the radical state in heme (reaction 42).
2.3. Identification of a Set of Kinetic Parameters of PGHS-1
As a result of the joint fitting of three series of the experimental data (see Methods), a unified set of kinetic parameters was determined. The results of fitting are shown in
Figure 3 and presented in
Table 2. For the comparison of the obtained kinetic constants of PGHS-1 with the results of other authors, we also included experimental and theoretical estimations of the corresponding rate constants available from literature in
Table 2.
In the calibration procedure, we used two experimental datasets on PGG
2 and PGH
2 production by PGHS-1 treated by AA and phenol as RC [
11] (
Figure 3a–c). As a result of model fitting against the experimental data, the values of the dissociation constants of AA binding to the COX-site,
Kd,1 (reaction 1 in the COX
1 cycle) and
Kd,12 (reaction 12 in the COX
2 cycle) were estimated (
Table 2). The only experimental estimate available for the stage of AA binding to PGHS-1 is Michaelis constant
Km ranged from 3 μM to 5 μM [
1]. However, the Michaelis constant is an apparent value and generally depends on the concentration of the second PGHS-1 substrate RC (see discussion below) and therefore cannot be directly compared with
Kd,1.
One of the key kinetic parameters of the PGHS-1 catalysis is the reaction rate of intramolecular electron transport [
23] from Tyr385 to the heme group, leading to the formation of the Tyr385 radical. The obtained value for constant
k9 equal to 310 s
−1 is in close agreement with theoretical estimates 300 s
−1 [
22] and 310 s
−1 [
4].
Satisfactory quality of the fitting was achieved at the same values of kinetic parameters of COX activity for similar stages in all the three COX
i cycles considered in the model. Note that the COX
1, COX
2, and COX
3 cycles differ only by the state of the heme prosthetic group in the POX site (see
Section 2.1). The independence of kinetic constants of the COX reactions on the state of the heme group can indicate the absence of cooperativity between the POX and COX sites.
As a result of fitting, we estimated the rate constants for the different mechanisms of PGHS-1 self-inactivation (
Table 2). As discussed in
Section 2.2, we considered three possible mechanisms of SI in the model. For SI originating from Intermediate I ([Fe(IV),PP*+]), we obtained the value of the rate constant
kin1 = 0.6 s
-1, which is in good agreement with experimental data [
4] (
Table 2). For the reactions of COX inactivation (reactions 36, 37, 58–62, and 68) with the remaining POX activity (POX
5 cycle), we obtained the rate constant
kin = 0.72 s
−1 (
Table 2).
A small value of reaction rate k8 of the reduction of protoporphyrin group radical state (Intermediate I, [Fe(IV), PP*+], E2) by RC was obtained to be about zero k8 (0.001 μM−1 s−1); that means that the Intermediate I is rather reduced by intramolecular process of electron abstraction from Tyr385 with radical formation than by exogenous RC.
To estimate uncertainty of the parameters obtained in the fitting procedure of the model against experimental data, we carried out global sensitivity analysis (GSA) of the model (see Methods). GSA revealed that the calibrated model showed uniform sensitivity to the most kinetic parameters (
Figure 4). The model is weakly sensitive to reaction rate
k12 of AA binding to the states in the POX
2 cycle (E
1, E
2 and E
3), while it is sensitive to the dissociation constant
Kd,12 of the same reactions. The model also showed weak sensitivity to parameter
kin2, which corresponds to one of the SI reactions of the enzyme, but it is highly sensitive to parameters
kin1 and
kin characterising the other SI reactions considered in the model.
The model also satisfactory described the third experimental dataset on a pure POX activity of PGHS-1 treated by H
2O
2 and adrenalin as RC [
12] (
Figure 3d). As the result of fitting, another set of the kinetic parameters (
k5,
k6, and
k8) of the POX cycle was obtained, which differs from that identified for the POX reactions in PGHS-1 treated by AA and phenol as RC (
Table 2). This result is consistent with experimental data on the pure POX activity of PGHS-1 in the presence of different peroxides and cosubstrates [
5,
6].
As seen from obtained agreement between theoretical and experimental data (
Figure 3), the model with the unified set of kinetic parameters satisfactorily described the various experimental data such as kinetics of intermediate PGG
2 and final PGH
2 products, as well as consumption of substrate AA and cosubstrate RC. The model with the obtained set of parameters given in
Table 2 was further validated and applied to analysis of regulatory mechanisms of PGHS-1 catalysis.
2.4. Contributions of the Different Enzyme Intermediates and Intra-Enzymatic Reactions to PGHS-1 Catalysis
The catalytic cycle of PGHS-1 (
Figure 2) represents a complex cycle network of intermolecular states with an intricate distribution of internal fluxes between them. To realise their contributions to the PGHS-1 catalysis, we carried out a comparative analysis of the population distribution of intermediate enzyme states and fluxes. This allowed us to estimate the relative impact of different elementary reactions and enzyme intermediates to the overall PGHS-1 function. We determined preferred ways of AA binding to the enzyme, identified key reactions producing intermediate PGG
2 and final product PGH
2, estimated the contribution of the different POX and COX cycles, and compared the role of different mechanisms of enzyme SI.
As mentioned above, in the model, we considered two possible ways of initiation of the COX reaction—namely, two alternative ways of formation of the enzyme–substrate complex containing both Tyr385 radical and AA. The first corresponds to a generally accepted mechanism of the COX reaction initiation in the BC mechanism, when formation of Intermediate II state [Fe(IV), PP, Tyr*] (E
5) precedes the binding of AA (reaction 1). The second, alternative mechanism is the binding of AA to the COX site (reactions 30–32), which can be followed by the formation of the Tyr385 radical state E
9 through reaction 18 (
Figure 2).
The performed flux analysis showed that AA binds to the Intermediate II (E
5) state much more effectively than to the resting state (E
1), Intermediate I (E
2), and E
3 state. Maximum flux
V1(t) exceeds maximum values of
V30(t),
V31(t), and
V32(t) by an order of magnitude (see comparison of
V1(t) and
V31(t) in
Figure 5a). The reaction rate of AA binding to the resting enzyme (
V31) has a high peak in an initial stage of the COX reaction, but its contribution to overall catalysis is negligible because of the short duration of the peak (1 ms).
The results of modelling also showed that the rate constant of electron transfer from Tyr385 residue to heme group depends significantly on the presence of AA in the COX site (reactions 13 and 18). The rate constant
k9 of reaction 13 between enzyme states with the empty COX site equals to 310 s
−1, but the constant of reaction 18 between states with an occupied COX site
k11 is much lower (1.1 s
−1). Thus, reduction of the heme group due to Tyr385 radical formation in the presence of AA in the COX site is weaker than that in the case of the empty COX site. Therefore, substrate–enzyme complex [Fe(IV), PP, Tyr*, AA] (E
9) is formed mainly from Intermediate II according to the experimental data [
5].
To determine what COX
i cycles contribute significantly to the overall COX reaction, we compared the reaction rates of the PGG
2 synthesis in various COX
i cycles (
Table 3) at the different concentrations of RC (10 μM, 100 μM, and 1000 μM). It turned out that the key COX cycle, in which the maximum amount of PGG
2 is produced, is the COX
2 cycle (reaction 8). In our model, the heme group of the COX
2 cycle is in the ground ferric state [Fe(III), PP], as opposed to the kinetic models of PGHS-1 [
5,
9], where the heme group is assumed to remain in the unreduced ferril state [Fe(IV), PP] throughout the COX reaction (the COX
1 cycle in our model).
Calculation of the reaction rates showed that the COX
1 cycle plays a significant role only at low concentrations of RC < 10 μM (
Table 3). This effect results from the increasing connection between COX
1 and COX
2 cycles that causes the reduction of heme group in the COX
1 cycle by RC (reactions 19, 22, 28, and 29) and a decrease of the role of the COX
1 cycle in the overall catalysis. Additionally, the COX
2 cycle significantly contributes to the activation of AA oxygenation reaction in comparison with that in the COX
1 cycle.
We also compared the rates of AA radical formation due to abstraction of a hydrogen atom from AA by the Tyr385 radical in both COX cycles (
Figure 5b). The reaction rate
V6 in the COX
2 cycle was found to be significantly higher than the corresponding rate
V2 in the COX
1 cycle. We also compared the contributions of two different reactions of AA radical formation which can happen either due to the Tyr385 radical according to the BC mechanism of PGHS-1 catalysis or due to another oxidant—oxyferryl heme—according to the early version of the TC mechanism. The calculation showed that the reaction rate V
56 of AA* formation owing to oxyferryl heme electron-transfer was significantly lower than the corresponding reaction V
6 proceeding due to Tyr385 radical (BC mechanism) (
Figure 5b).
To analyse the main reactions contributing considerably to PGH
2 synthesis, we calculated the fluxes in the POX
i cycles in which PGH
2 is produced (
Table 3). The main contribution to PGH
2 production turned out to be made by reaction 23 in the POX
4 cycle. In the PGHS-1 models developed on a basis of the BC mechanism [
8], PGH
2 is produced in reaction 11 in the POX
1 cycle. By contrast, in our model, reaction rate V
11 is negligibly weak in comparison with reaction rate V
23 (
Table 3). To analyse this effect, we calculated the population kinetics of the enzyme states.
Figure 5c shows the time dependence of population of the states, maximum contributing to the PGHS-1 catalysis. These results showed that the POX
1 cycle contributes noticeably at the initial stage of PGH
2 synthesis, when the population of the resting state of the enzyme is maximum. In contrast, state E
12 in the POX
4 cycle works throughout all the catalysis, indicating that the POX
4 cycle is active up to full SI of PGHS-1. Moreover, state E
12 ([Fe(III), PP, Tyr, PGG*], being common for the COX
2 and POX
4 cycles, participates simultaneously in the production of the intermediate product PGG
2 (reaction 23) and the final product PGH
2 (reaction 8). As discussed above, these reactions make maximum contributions to the synthesis of PGG
2 and PGH
2 (
Table 3). Thus, in our model, COX
2 and POX
4 cycles are the main cycles in PGHS-1 catalysis, while the COX
1 and POX
1 cycles contribute only at the initial stage of the PGHS-1 function.
2.5. Model Application to the Investigation of Regulatory Role of AA and RC in PGHS-1 Function
The calibrated model was applied to study the dependence of the PGHS-1 activity on substrate AA and cosubstrate RC concentrations. For comparison of the theoretical results with the experiments, we used the validation experimental data which were not used in the fitting procedure. The comparison of model and experiment results made without any optional fitting was a validation test for the predictive power of the model. This allows the model to be applied to the reliable analysis of in vivo experimental data on the NSAID action.
First, we applied the model to calculate the dependence of the oxygen consumption rate on AA concentration
VO2(
AA). For this, we first calculated the kinetics of the oxygen consumption rate
VO2(
t) and compared the results with the experimental data [
8] (
Figure 6a). As seen, the model satisfactory describes the experimental data on
VO2(
t) and reproduces the time of the enzyme activity (about 20 sec), which is in agreement with the SI time of PGHS-1 [
1]. To plot the dependence of
VO2(
AA), we took the maximum values of the rates
VO2(
t) calculated at different AA concentrations. The theoretical dependence of
VO2(
AA) along with experimental data [
11] are given in
Figure 6b.
We also calculated
VO2(
AA) at different RC concentrations (
Figure 6b). As seen, RC inhibits COX activity of the enzyme, i.e., an increase in RC concentration leads to an increase of apparent Michaelis constant
Km and decrease of the maximum oxygen consumption rate. This means that AA and RC compete each other for the COX site according to reactions 1 and 9 in the catalytic cycle (
Figure 2). Based on these results, we calculated the dependence of the Michaelis constant
Km for AA on RC concentrations,
Km(RC) (
Figure 6c). The linear dependence
Km(RC) looks like the dependence of the apparent Michaelis constant for the substrate on competitive inhibitor concentration, where RC plays a role of an inhibitor. The range of theoretical values of
Km embraces a wide range of the experimental values of Michaelis constant, 3 μM [
1], 5 μM [
9], and 10.2 ± 1.5 μM [
26]. Note that dissociation constants of AA binding with the enzyme obtained in our calculation is significantly lower (
Kd1,AA = 0.1 μM) than
Km (
Table 2).
In the model, RC competes with AA for binding to states E5 and E15 in the COX1 cycle (reactions 1 and 9) and COX2 cycle (reactions 5 and 50), respectively. According with the BC mechanism, redaction of Tyr385 radical by RC leads to the inhibition of COX activity.
Moreover, the discussed mechanism may explain the distinction of the obtained kinetic constants of the reduction rate of Tyr385 radical depending on a state of the COX site. In the parameter set (
Table 2), the rate constant
k10 of reactions 14 and 51 of Tyr385 reduction in states E
9 (COX
1 cycle) and E
20 (COX
2 cycle) with AA in the COX site equals approximately zero in contrast to the corresponding constant
k5 in reaction 9 and 50 of Tyr385* reduction in states E
5 (COX
1 cycle) and E
15 (COX
2 cycle) with the empty COX site (
Table 2). AA in the COX site is proposed not to permit RC to react with Tyr385*. For similar reaction 49 of Tyr385* reduction in states E
4 (POX
2-cycle) with PGG2* in the COX site, the constant equals
k5, which may mean that RC can reduce two radicals state in the COX site.
An increase of
Km with increasing RC concentration is accompanied by a change in the curve shape of
VO2(
AA) from hyperbolic to sigmoidal at high RC concentrations (
Figure 6b). To investigate this positive cooperativity in PGHS-1 function, we plotted the Eadie-Scatchard dependence of ratio
VO2/
AA/PGHS on ratio
VO2/PGHS (
Figure 6d) and provided the corresponding experimental data [
11]. As seen, the theoretical downward-concave curve
VO2/
AA/PGHS vs.
VO2/PGHS significantly differs from the linear Eadie–Scatchard plot corresponding to Michaelis kinetics (
) that confirmed positive cooperativity in COX activity of PGHS-1 [
26].
Positive cooperativity in the COX reactions defines the activation threshold of the PGHS-1 catalysis which depends on both AA and RC concentrations [
11,
13].
Figure 7 shows the activation threshold curve on a plan of AA and RC concentrations which divides the range of AA and RC concentrations into two regions, i.e., PGHS-1 is inactive and active at values of AA and RC concentrations lying below and upper the curve, respectively. The increasing activation threshold at increasing RC concentrations is determined by the inhibition of the autocatalytic COX reaction by RC. The PGHS-1 kinetics at AA and RC concentrations far from the activation threshold are close to the classical Michaelis–Menten kinetics that are typical for in vitro experiments on PGHS-1 kinetics and experiments on its inhibition by NSAIDs. The nonlinear effect in PGHS-1 kinetics becomes more pronounced near the activation threshold, in the range of low AA concentrations (0.05–1 μM), which are likely to relate to cellular conditions [
1]. Below this threshold, the PGHS-1 is in a latent state and can be activated by either increasing AA or decreasing RC concentrations, as shown by the arrows in
Figure 7.











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}