The Role of Proteomics in Bacterial Response to Antibiotics




Abstract
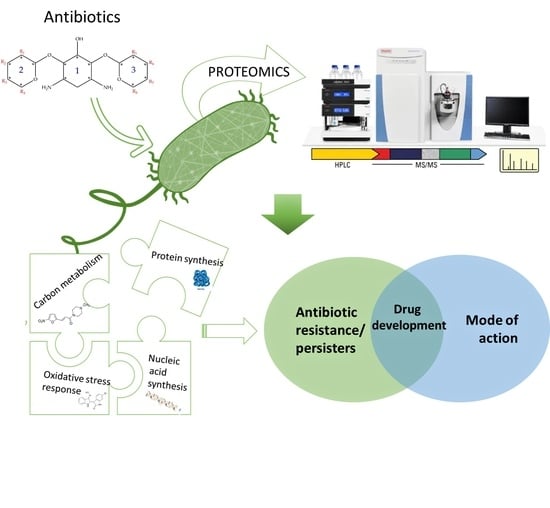
1. Introduction
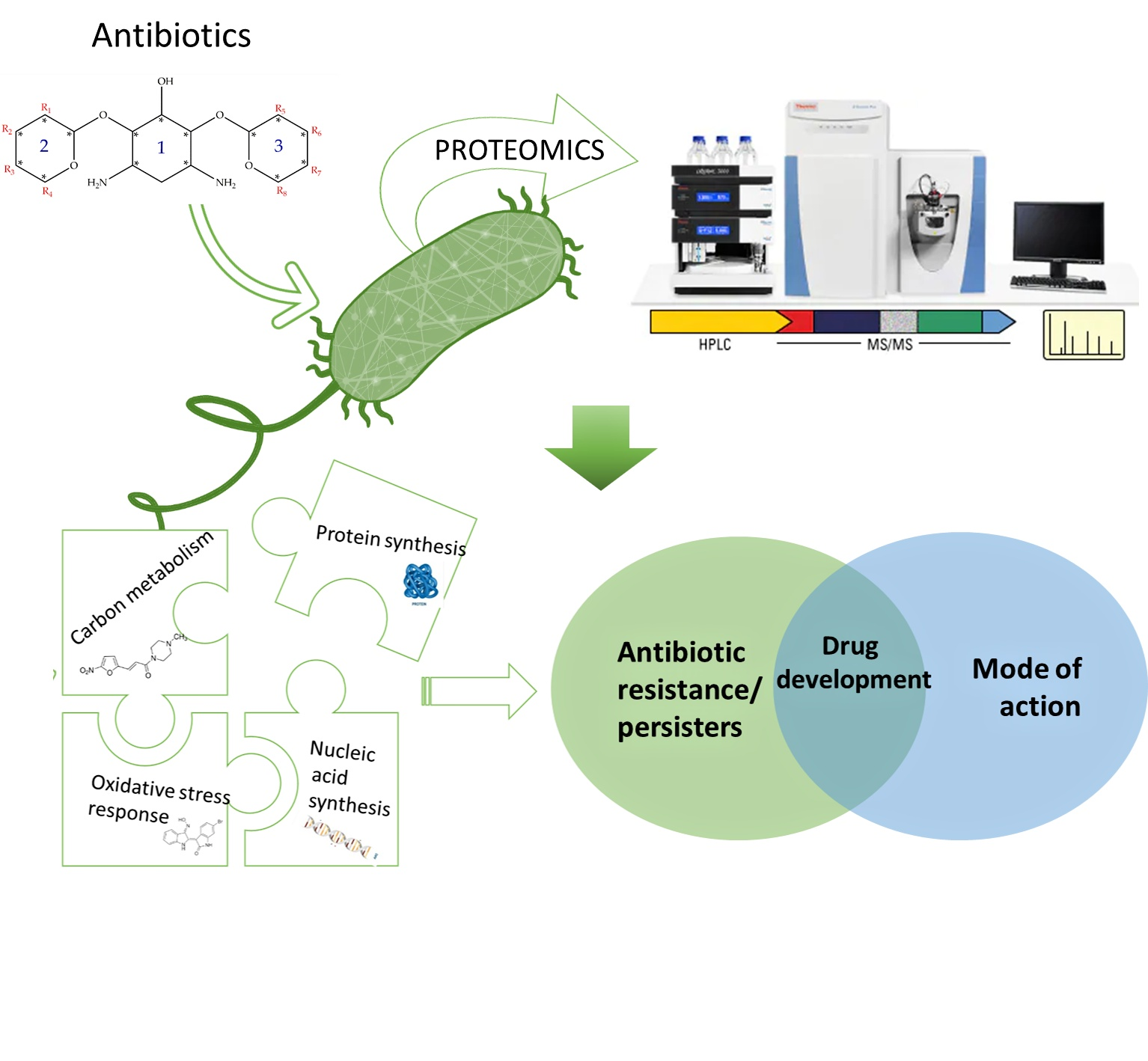
1.1. Multidrug Resistance (MDR), Priority Pathogens and Antibiotic Tolerant Persister Phenotype
1.2. Proteomics by Mass Spectrometry as A Tool and Common Data Acquisition Strategies
1.3. Applications of Proteomic Data Acquisition in Studies of Bacteria
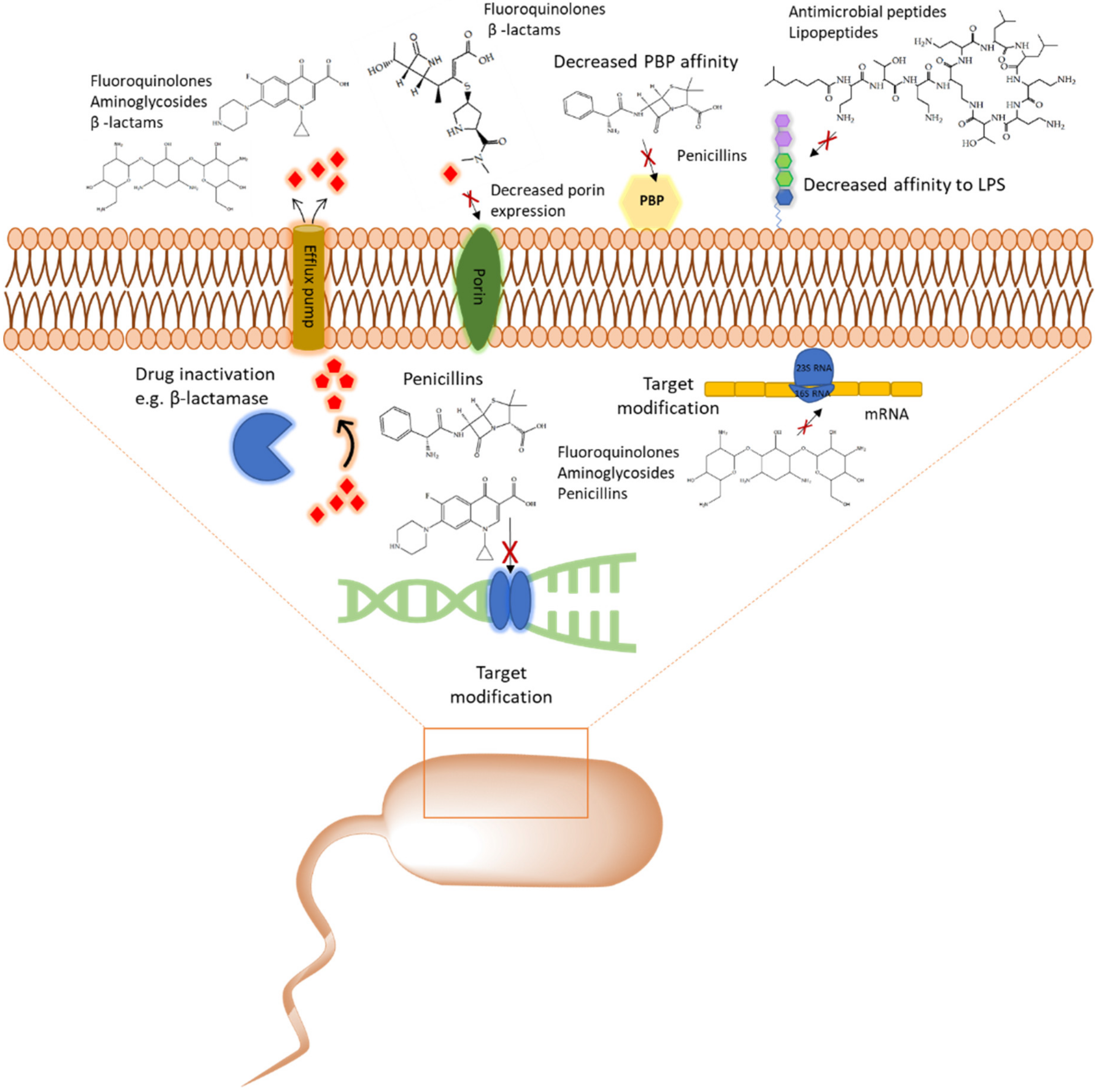
2. The Role of Proteomic Analysis in Generating New Insight about The Mechanism of Action of Antibiotics and Antibiotic Resistance
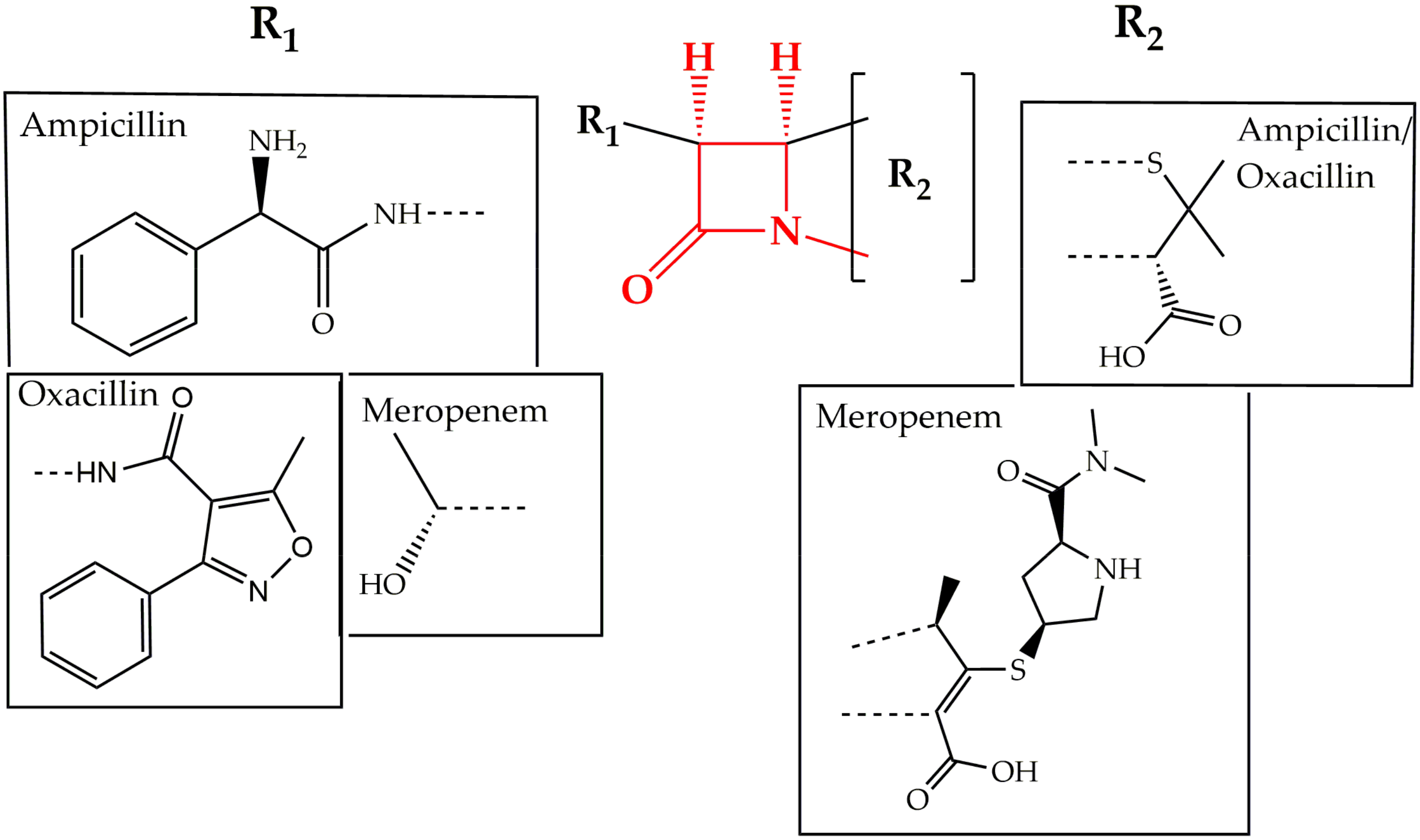
2.1. Cell Wall Synthesis Inhibitors
2.2. Inhibitors of Protein Translation
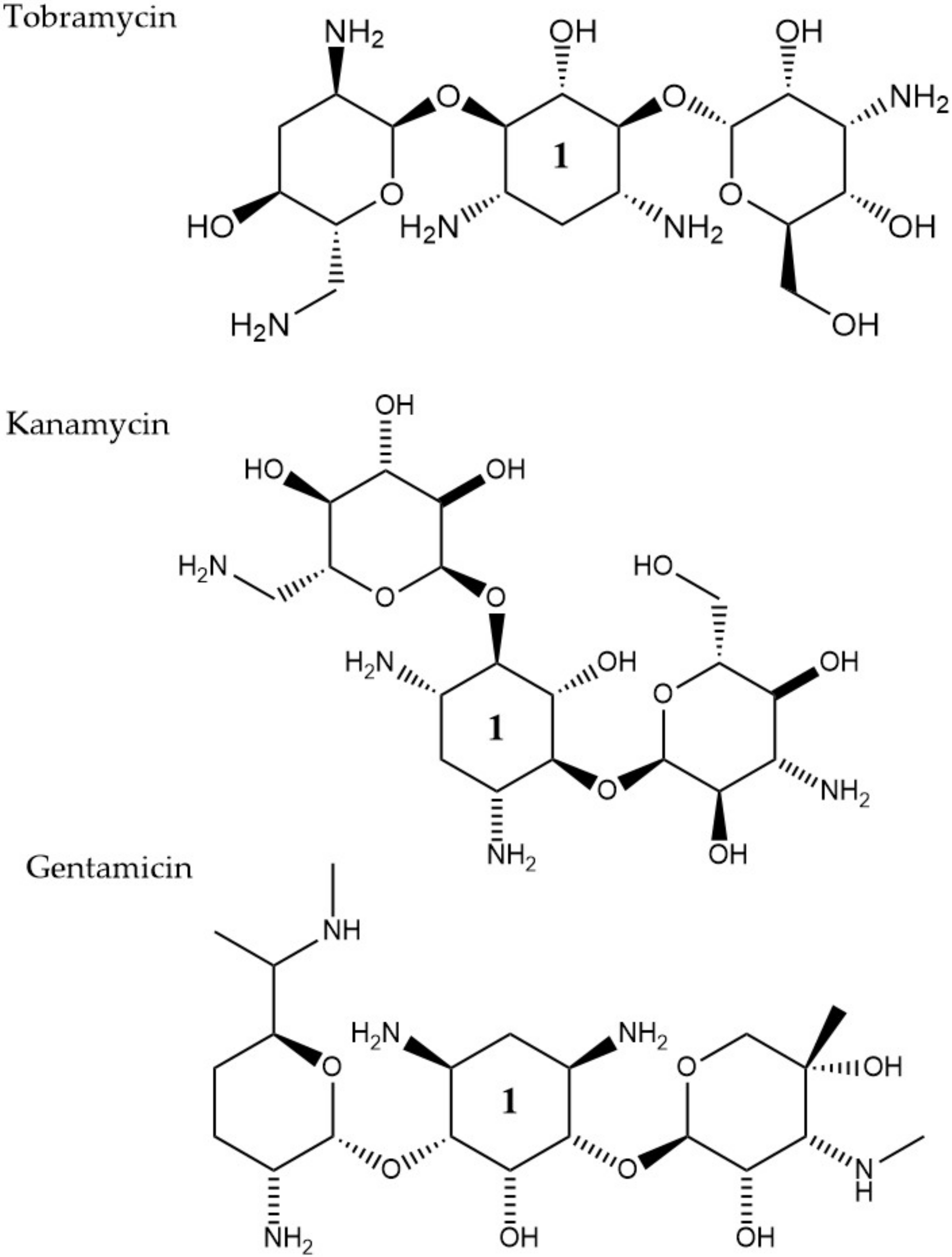
2.2.1. Aminoglycosides
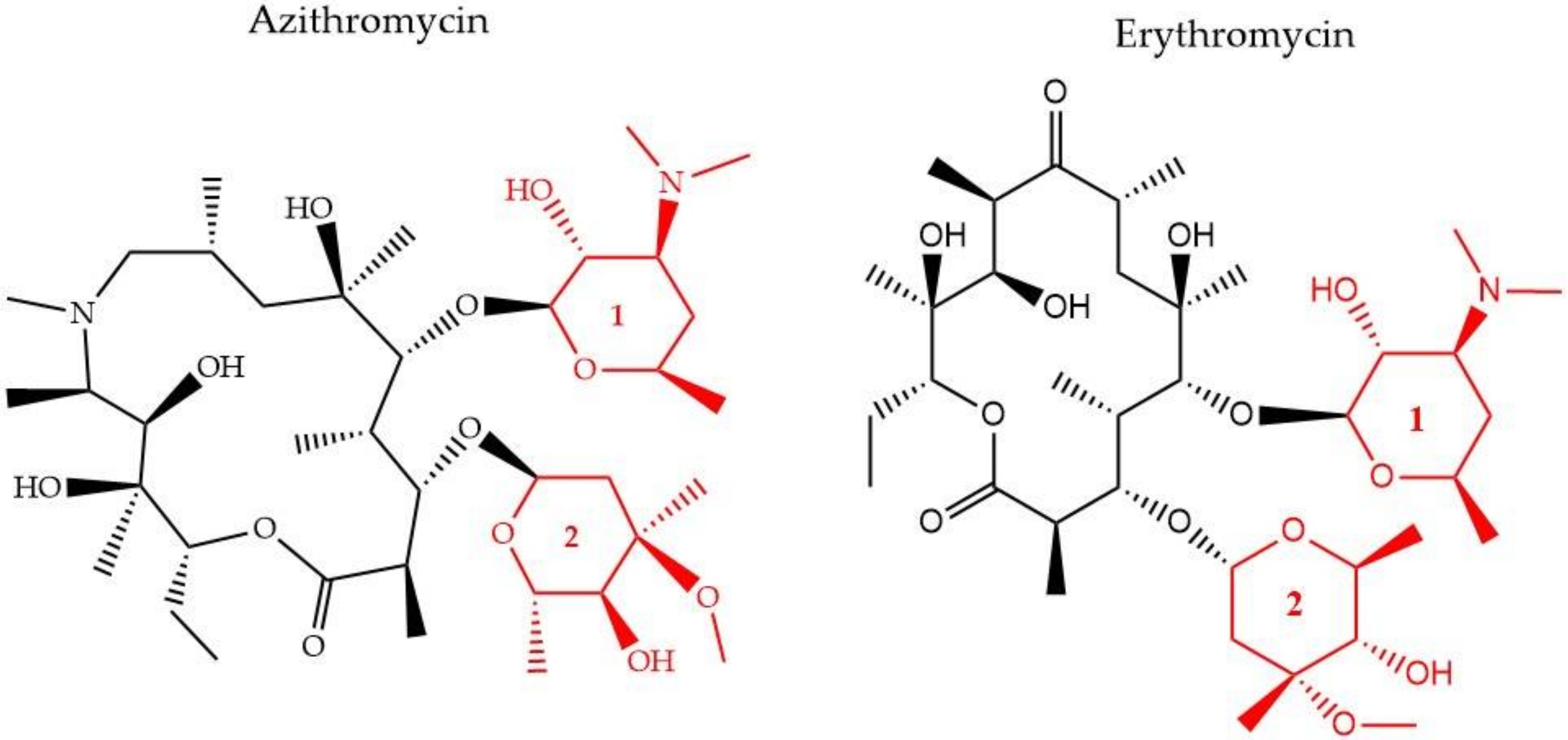
2.2.2. Macrolides
2.3. Inhibitors of DNA Synthesis
2.4. Cyclic Lipopeptides That are Used as Last Resort Drugs in Treatment of Bacterial Infections
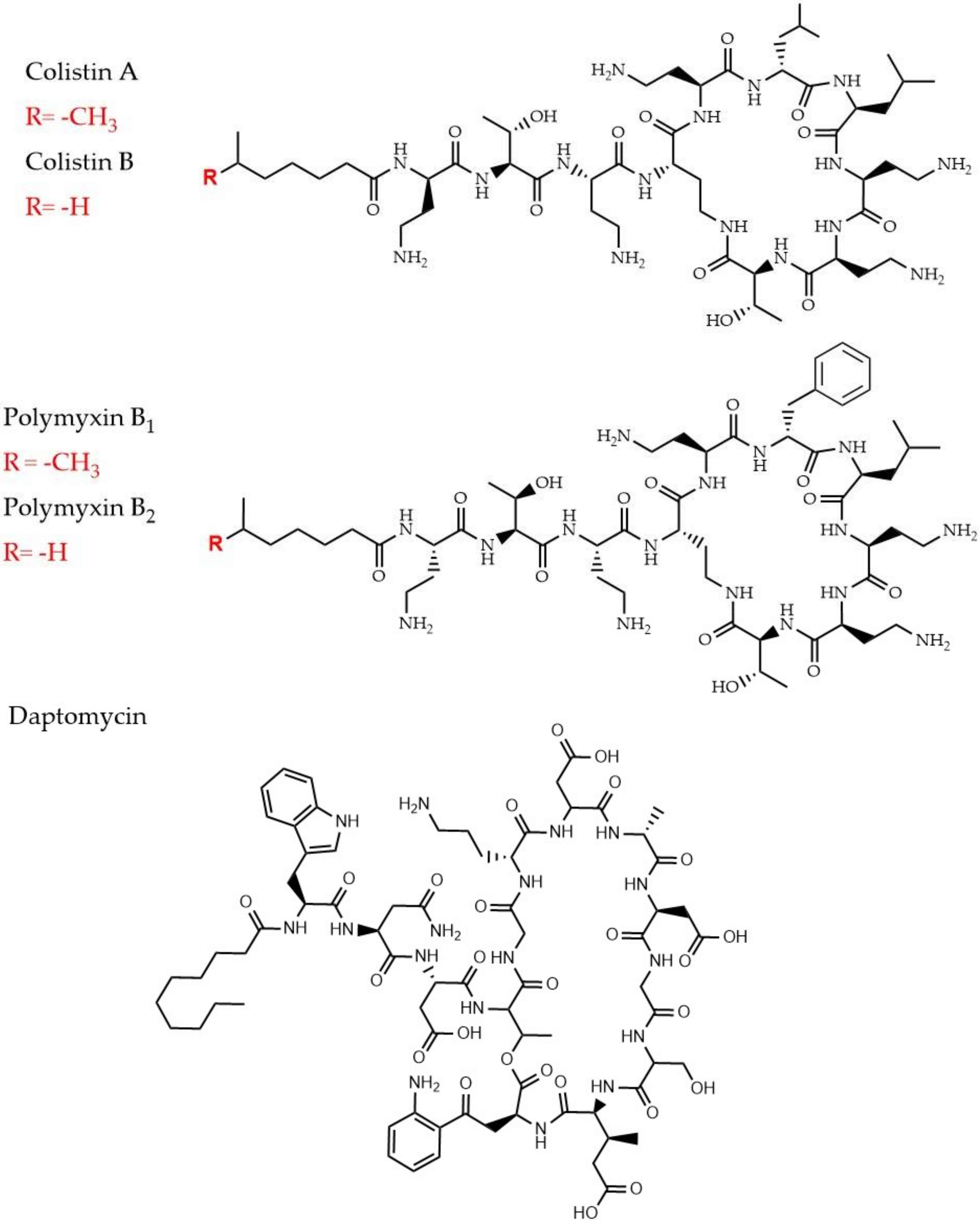
2.4.1. Polymyxins and Colistin
2.4.2. Daptomycin
2.5. Promising Drug Candidates: Antimicrobial Peptides
3. Discussion and Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Prestinaci, F.; Pezzotti, P.; Pantosti, A. Antimicrobial resistance: A global multifaceted phenomenon. Pathog. Glob. Health 2015, 109, 309–318. [Google Scholar] [CrossRef] [PubMed]
- Antibiotic resistance threats in the United States 2019. Available online: https://www.cdc.gov/drugresistance/pdf/threats-report/2019-ar-threats-report-508.pdf (accessed on 10 June 2020).
- Cassini, A.; Högberg, L.D.; Plachouras, D.; Quattrocchi, A.; Hoxha, A.; Simonsen, G.S.; Colomb-Cotinat, M.; Kretzschmar, M.E.; Devleesschauwer, B.; Cecchini, M.; et al. Attributable deaths and disability-adjusted life-years caused by infections with antibiotic-resistant bacteria in the EU and the European Economic Area in 2015: A population-level modelling analysis. Lancet. Infect. Dis. 2019, 19, 56–66. [Google Scholar] [CrossRef]
- The Review on Antimicrobial Resistance. pp. 1–33. Available online: https://Amr-Review.Org/ (accessed on 10 June 2020). [CrossRef]
- Conibear, T.C.R.; Collins, S.L.; Webb, J.S. Role of Mutation in Pseudomonas aeruginosa Biofilm Development. PLoS ONE 2009, 4, e6289. [Google Scholar] [CrossRef] [PubMed]
- Erickson, B.E. Gene Transfer in the Environment. Environ. Sci. Technol. 2001, 35, 20A–22A. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Kapoor, G.; Saigal, S.; Elongavan, A. Action and resistance mechanisms of antibiotics: A guide for clinicians. J. Anaesthesiol. Clin. Pharmacol. 2017, 33, 300. [Google Scholar] [CrossRef]
- Rodrigue, A.; Quentin, Y.; Lazdunski, A.; Méjean, V.; Foglino, M. Cell signalling by oligosaccharides. Two-component systems in Pseudomonas aeruginosa: Why so many? Trends Microbiol. 2000, 8, 498–504. [Google Scholar] [CrossRef]
- Fernández, L.; Jenssen, H.; Bains, M.; Wiegand, I.; Gooderham, W.J.; Hancock, R.E.W. The Two-Component System CprRS Senses Cationic Peptides and Triggers Adaptive Resistance in Pseudomonas aeruginosa Independently of ParRS. Antimicrob. Agents Chemother. 2012, 56, 6212–6222. [Google Scholar] [CrossRef]
- Lewenza, S. Extracellular DNA-induced antimicrobial peptide resistance mechanisms in Pseudomonas aeruginosa. Front. Microbiol. 2013, 4, 21. [Google Scholar] [CrossRef]
- Yen, M.; Peabody, C.R.; Partovi, S.M.; Zhai, Y.; Tseng, Y.; Saier, M.H. Protein-translocating outer membrane porins of Gram-negative bacteria. Biochim. Biophys. Acta (BBA) Biomembr. 2002, 1562, 6–31. [Google Scholar] [CrossRef]
- Kulkarni, A.P.; Nagvekar, V.C.; Veeraraghavan, B.; Warrier, A.R.; TS, D.; Ahdal, J.; Jain, R. Current Perspectives on Treatment of Gram-Positive Infections in India: What Is the Way Forward? Interdiscip. Perspect. Infect. Dis. 2019, 2019, 1–8. [Google Scholar] [CrossRef]
- Santajit, S.; Indrawattana, N. Mechanisms of Antimicrobial Resistance in ESKAPE Pathogens. Biomed Res. Int. 2016, 2016, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Founou, R.C.; Founou, L.L.; Essack, S.Y. Clinical and economic impact of antibiotic resistance in developing countries: A systematic review and meta-analysis. PLoS One 2017, 12, e0189621. [Google Scholar] [CrossRef] [PubMed]
- LaFleur, M.D.; Qi, Q.; Lewis, K. Patients with Long-Term Oral Carriage Harbor High-Persister Mutants of Candida albicans. Antimicrob. Agents Chemother. 2010, 54, 39–44. [Google Scholar] [CrossRef] [PubMed]
- Bartell, J.A.; Cameron, D.R.; Mojsoska, B.; Haagensen, J.A.; Sommer, L.M.; Lewis, K.; Molin, S.; Johansen, H.K. Bacterial persisters in long-term infection: Emergence and fitness in a complex host environment. Under Rev. (PLOS Pathog.) 2020. [Google Scholar] [CrossRef]
- Mulcahy, L.R.; Burns, J.L.; Lory, S.; Lewis, K. Emergence of Pseudomonas aeruginosa strains producing high levels of persister cells in patients with cystic fibrosis. J. Bacteriol. 2010, 192, 6191–6199. [Google Scholar] [CrossRef]
- Balaban, N.Q.; Helaine, S.; Lewis, K.; Ackermann, M.; Aldridge, B.; Andersson, D.I.; Brynildsen, M.P.; Bumann, D.; Camilli, A.; Collins, J.J.; et al. Definitions and guidelines for research on antibiotic persistence. Nat. Rev. Microbiol. 2019, 17, 441–448. [Google Scholar] [CrossRef]
- Pu, Y.; Zhao, Z.; Li, Y.; Zou, J.; Ma, Q.; Zhao, Y.; Ke, Y.; Zhu, Y.; Chen, H.; Baker, M.A.B.; et al. Enhanced Efflux Activity Facilitates Drug Tolerance in Dormant Bacterial Cells. Mol. Cell 2016, 62, 284–294. [Google Scholar] [CrossRef]
- Keren, I.; Shah, D.; Spoering, A.; Kaldalu, N.; Lewis, K. Specialized Persister Cells and the Mechanism of Multidrug Tolerance in Escherichia coli. J. Bacteriol. 2004, 186, 8172–8180. [Google Scholar] [CrossRef]
- Keren, I.; Minami, S.; Rubin, E.; Lewis, K. Characterization and Transcriptome Analysis of Mycobacterium tuberculosis Persisters. MBio 2011, 2, e00100-11. [Google Scholar] [CrossRef]
- Rowe, S.E.; Wagner, N.J.; Li, L.; Beam, J.E.; Wilkinson, A.D.; Radlinski, L.C.; Zhang, Q.; Miao, E.A.; Conlon, B.P. Reactive oxygen species induce antibiotic tolerance during systemic Staphylococcus aureus infection. Nat. Microbiol. 2020, 5, 282–290. [Google Scholar] [CrossRef]
- Kwan, B.W.; Valenta, J.A.; Benedik, M.J.; Wood, T.K. Arrested Protein Synthesis Increases Persister-Like Cell Formation. Antimicrob. Agents Chemother. 2013, 57, 1468–1473. [Google Scholar] [CrossRef] [PubMed]
- Macek, B.; Forchhammer, K.; Hardouin, J.; Weber-Ban, E.; Grangeasse, C.; Mijakovic, I. Protein post-translational modifications in bacteria. Nat. Rev. Microbiol. 2019, 17, 651–664. [Google Scholar] [CrossRef]
- Patel, V.J.; Thalassinos, K.; Slade, S.E.; Connolly, J.B.; Crombie, A.; Murrell, J.C.; Scrivens, J.H. A Comparison of Labeling and Label-Free Mass Spectrometry-Based Proteomics Approaches. J. Proteome Res. 2009, 8, 3752–3759. [Google Scholar] [CrossRef] [PubMed]
- Ong, S.-E.; Blagoev, B.; Kratchmarova, I.; Kristensen, D.B.; Steen, H.; Pandey, A.; Mann, M. Stable isotope labeling by amino acids in cell culture, SILAC, as a simple and accurate approach to expression proteomics. Mol. Cell. Proteomics 2002, 1, 376–386. [Google Scholar] [CrossRef]
- Han, J.; Yi, S.; Zhao, X.; Zheng, Y.; Yang, D.; Du, G.; Yang, X.-Y.; He, Q.-Y.; Sun, X. Improved SILAC method for double labeling of bacterial proteome. J. Proteomics 2019, 194, 89–98. [Google Scholar] [CrossRef] [PubMed]
- Ross, P.L.; Huang, Y.N.; Marchese, J.N.; Williamson, B.; Parker, K.; Hattan, S.; Khainovski, N.; Pillai, S.; Dey, S.; Daniels, S.; et al. Multiplexed Protein Quantitation in Saccharomyces cerevisiae Using Amine-reactive Isobaric Tagging Reagents. Mol. Cell. Proteomics 2004, 3, 1154–1169. [Google Scholar] [CrossRef]
- Thompson, A.; Schäfer, J.; Kuhn, K.; Kienle, S.; Schwarz, J.; Schmidt, G.; Neumann, T.; Hamon, C. Tandem Mass Tags: A Novel Quantification Strategy for Comparative Analysis of Complex Protein Mixtures by MS/MS. Anal. Chem. 2003, 75, 1895–1904. [Google Scholar] [CrossRef]
- Pérez-Llarena, F.J.; Bou, G. Proteomics As a Tool for Studying Bacterial Virulence and Antimicrobial Resistance. Front. Microbiol. 2016, 7, 410. [Google Scholar] [CrossRef]
- Sulaiman, J.E.; Lam, H. Application of proteomics in studying bacterial persistence. Expert Rev. Proteomics 2019, 16, 227–239. [Google Scholar] [CrossRef]
- Yung, Y.P.; McGill, S.L.; Chen, H.; Park, H.; Carlson, R.P.; Hanley, L. Reverse diauxie phenotype in Pseudomonas aeruginosa biofilm revealed by exometabolomics and label-free proteomics. NPJ Biofilms Microbiomes 2019, 5, 31. [Google Scholar] [CrossRef]
- Fagerquist, C.K.; Zaragoza, W.J.; Sultan, O.; Woo, N.; Quinones, B.; Cooley, M.B.; Mandrell, R.E. Top-Down Proteomic Identification of Shiga Toxin 2 Subtypes from Shiga Toxin-Producing Escherichia coli by Matrix-Assisted Laser Desorption Ionization-Tandem Time of Flight Mass Spectrometry. Appl. Environ. Microbiol. 2014, 80, 2928–2940. [Google Scholar] [CrossRef]
- Liu, X.; Gao, B.; Novik, V.; Galán, J.E. Quantitative Proteomics of Intracellular Campylobacter jejuni Reveals Metabolic Reprogramming. PLoS Pathog. 2012, 8, e1002562. [Google Scholar] [CrossRef] [PubMed]
- Vorwerk, S.; Krieger, V.; Deiwick, J.; Hensel, M.; Hansmeier, N. Proteomes of host cell membranes modified by intracellular activities of Salmonella enterica. Mol. Cell. Proteomics 2015, 14, 81–92. [Google Scholar] [CrossRef] [PubMed]
- Zhou, J.; Hao, D.; Wang, X.; Liu, T.; He, C.; Xie, F.; Sun, Y.; Zhang, J. An important role of a “probable ATP-binding component of ABC transporter” during the process of Pseudomonas aeruginosa resistance to fluoroquinolone. Proteomics 2006, 6, 2495–2503. [Google Scholar] [CrossRef] [PubMed]
- Su, H.-C.; Ramkissoon, K.; Doolittle, J.; Clark, M.; Khatun, J.; Secrest, A.; Wolfgang, M.C.; Giddings, M.C. The Development of Ciprofloxacin Resistance in Pseudomonas aeruginosa Involves Multiple Response Stages and Multiple Proteins. Antimicrob. Agents Chemother. 2010, 54, 4626–4635. [Google Scholar] [CrossRef]
- Machado, I.; Coquet, L. Proteomic Changes in Pseudomonas aeruginosa Biofilm Cells after Adaptive Resistance Development. J. Proteomics Bioinform. 2016, 09. [Google Scholar] [CrossRef]
- Babin, B.M.; Atangcho, L.; van Eldijk, M.B.; Sweredoski, M.J.; Moradian, A.; Hess, S.; Tolker-Nielsen, T.; Newman, D.K.; Tirrell, D.A. Selective Proteomic Analysis of Antibiotic-Tolerant Cellular Subpopulations in Pseudomonas aeruginosa Biofilms. MBio 2017, 8. [Google Scholar] [CrossRef]
- Peng, J.; Cao, J.; Ng, F.M.; Hill, J. Pseudomonas aeruginosa develops Ciprofloxacin resistance from low to high level with distinctive proteome changes. J. Proteomics 2017, 152, 75–87. [Google Scholar] [CrossRef]
- Wu, X.; Held, K.; Zheng, C.; Staudinger, B.J.; Chavez, J.D.; Weisbrod, C.R.; Eng, J.K.; Singh, P.K.; Manoil, C.; Bruce, J.E. Dynamic Proteome Response of Pseudomonas aeruginosa to Tobramycin Antibiotic Treatment. Mol. Cell. Proteomics 2015, 14, 2126–2137. [Google Scholar] [CrossRef]
- Koeppen, K.; Barnaby, R.; Jackson, A.A.; Gerber, S.A.; Hogan, D.A.; Stanton, B.A. Tobramycin reduces key virulence determinants in the proteome of Pseudomonas aeruginosa outer membrane vesicles. PLoS One 2019, 14, e0211290. [Google Scholar] [CrossRef]
- LeBel, M. Ciprofloxacin: Chemistry, mechanism of action, resistance, antimicrobial spectrum, pharmacokinetics, clinical trials, and adverse reactions. Pharmacotherapy 1988, 8, 3–33. [Google Scholar] [CrossRef] [PubMed]
- Moffatt, J.H.; Harper, M.; Harrison, P.; Hale, J.D.F.; Vinogradov, E.; Seemann, T.; Henry, R.; Crane, B.; St. Michael, F.; Cox, A.D.; et al. Colistin Resistance in Acinetobacter baumannii Is Mediated by Complete Loss of Lipopolysaccharide Production. Antimicrob. Agents Chemother. 2010, 54, 4971–4977. [Google Scholar] [CrossRef] [PubMed]
- Fernández-Reyes, M.; Rodríguez-Falcón, M.; Chiva, C.; Pachón, J.; Andreu, D.; Rivas, L. The cost of resistance to colistin in Acinetobacter baumannii: A proteomic perspective. Proteomics 2009, 9, 1632–1645. [Google Scholar] [CrossRef]
- Henken, S.; Bohling, J.; Martens-Lobenhoffer, J.; Paton, J.C.; Ogunniyi, A.D.; Briles, D.E.; Salisbury, V.C.; Wedekind, D.; Bode-Böger, S.M.; Welsh, T.; et al. Efficacy profiles of daptomycin for treatment of invasive and noninvasive pulmonary infections with Streptococcus pneumoniae. Antimicrob. Agents Chemother. 2010, 54, 707–717. [Google Scholar] [CrossRef] [PubMed]
- Müller, A.; Grein, F.; Otto, A.; Gries, K.; Orlov, D.; Zarubaev, V.; Girard, M.; Sher, X.; Shamova, O.; Roemer, T.; et al. Differential daptomycin resistance development in Staphylococcus aureus strains with active and mutated gra regulatory systems. Int. J. Med. Microbiol. 2018, 308, 335–348. [Google Scholar] [CrossRef] [PubMed]
- Ma, W.; Zhang, D.; Li, G.; Liu, J.; He, G.; Zhang, P.; Yang, L.; Zhu, H.; Xu, N.; Liang, S. Antibacterial mechanism of daptomycin antibiotic against Staphylococcus aureus based on a quantitative bacterial proteome analysis. J. Proteomics 2017, 150, 242–251. [Google Scholar] [CrossRef]
- Yan, X.; He, B.; Liu, L.; Qu, G.; Shi, J.; Hu, L.; Jiang, G. Antibacterial mechanism of silver nanoparticles in Pseudomonas aeruginosa: Proteomics approach. Metallomics 2018, 10, 557–564. [Google Scholar] [CrossRef]
- Bateman, A.; Martin, M.J.; O’Donovan, C.; Magrane, M.; Apweiler, R.; Alpi, E.; Antunes, R.; Arganiska, J.; Bely, B.; Bingley, M.; et al. UniProt: A hub for protein information. Nucleic Acids Res. 2015. [Google Scholar] [CrossRef]
- Gaviard, C.; Jouenne, T.; Hardouin, J. Proteomics of Pseudomonas aeruginosa: The increasing role of post-translational modifications. Expert Rev. Proteomics 2018, 15, 757–772. [Google Scholar] [CrossRef]
- Boucher, R.C. Cystic fibrosis: A disease of vulnerability to airway surface dehydration. Trends Mol. Med. 2007, 13, 231–240. [Google Scholar] [CrossRef]
- Fujitani, S.; Sun, H.-Y.; Yu, V.L.; Weingarten, J.A. Pneumonia Due to Pseudomonas aeruginosa. Chest 2011, 139, 909–919. [Google Scholar] [CrossRef] [PubMed]
- Rakhimova, E.; Wiehlmann, L.; Brauer, A.L.; Sethi, S.; Murphy, T.F.; Tümmler, B. Pseudomonas aeruginosa Population Biology in Chronic Obstructive Pulmonary Disease. J. Infect. Dis. 2009, 200, 1928–1935. [Google Scholar] [CrossRef] [PubMed]
- Ouidir, T.; Jouenne, T.; Hardouin, J. Post-translational modifications in Pseudomonas aeruginosa revolutionized by proteomic analysis. Biochimie 2016, 125, 66–74. [Google Scholar] [CrossRef] [PubMed]
- Wu, X.; Siehnel, R.J.; Garudathri, J.; Staudinger, B.J.; Hisert, K.B.; Ozer, E.A.; Hauser, A.R.; Eng, J.K.; Manoil, C.; Singh, P.K.; et al. In Vivo Proteome of Pseudomonas aeruginosa in Airways of Cystic Fibrosis Patients. J. Proteome Res. 2019, 18, 2601–2612. [Google Scholar] [CrossRef] [PubMed]
- Ouidir, T.; Cosette, P.; Jouenne, T.; Hardouin, J. Proteomic profiling of lysine acetylation in Pseudomonas aeruginosa reveals the diversity of acetylated proteins. Proteomics 2015, 15, 2152–2157. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, A.; Kochanowski, K.; Vedelaar, S.; Ahrné, E.; Volkmer, B.; Callipo, L.; Knoops, K.; Bauer, M.; Aebersold, R.; Heinemann, M. The quantitative and condition-dependent Escherichia coli proteome. Nat. Biotechnol. 2016, 34, 104–110. [Google Scholar] [CrossRef]
- Sulaiman, J.E.; Hao, C.; Lam, H. Specific Enrichment and Proteomics Analysis of Escherichia coli Persisters from Rifampin Pretreatment. J. Proteome Res. 2018, 17, 3984–3996. [Google Scholar] [CrossRef]
- Sulaiman, J.E.; Lam, H. Proteomic Investigation of Tolerant Escherichia coli Populations from Cyclic Antibiotic Treatment. J. Proteome Res. 2020, 19, 900–913. [Google Scholar] [CrossRef]
- Mücke, P.-A.; Maaß, S.; Kohler, T.P.; Hammerschmidt, S.; Becher, D. Proteomic Adaptation of Streptococcus pneumoniae to the Human Antimicrobial Peptide LL-37. Microorganisms 2020, 8, 413. [Google Scholar] [CrossRef]
- Nakamura, A.; Komatsu, M.; Ohno, Y.; Noguchi, N.; Kondo, A.; Hatano, N. Identification of specific protein amino acid substitutions of extended-spectrum β-lactamase (ESBL)-producing Escherichia coli ST131: A proteomics approach using mass spectrometry. Sci. Rep. 2019, 9, 8555. [Google Scholar] [CrossRef]
- Kim, S.W.; Park, S.B.; Im, S.P.; Lee, J.S.; Jung, J.W.; Gong, T.W.; Lazarte, J.M.S.; Kim, J.; Seo, J.-S.; Kim, J.-H.; et al. Outer membrane vesicles from β-lactam-resistant Escherichia coli enable the survival of β-lactam-susceptible E. coli in the presence of β-lactam antibiotics. Sci. Rep. 2018, 8, 5402. [Google Scholar] [CrossRef] [PubMed]
- Sidjabat, H.E.; Gien, J.; Kvaskoff, D.; Ashman, K.; Vaswani, K.; Reed, S.; McGeary, R.P.; Paterson, D.L.; Bordin, A.; Schenk, G. The use of SWATH to analyse the dynamic changes of bacterial proteome of carbapanemase-producing Escherichia coli under antibiotic pressure. Sci. Rep. 2018, 8, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Hua, X.; Liu, L.; Fang, Y.; Shi, Q.; Li, X.; Chen, Q.; Shi, K.; Jiang, Y.; Zhou, H.; Yu, Y. Colistin Resistance in Acinetobacter baumannii MDR-ZJ06 Revealed by a Multiomics Approach. Front. Cell. Infect. Microbiol. 2017, 7. [Google Scholar] [CrossRef] [PubMed]
- Chua, S.L.; Yam, J.K.H.; Hao, P.; Adav, S.S.; Salido, M.M.; Liu, Y.; Givskov, M.; Sze, S.K.; Tolker-Nielsen, T.; Yang, L. Selective labelling and eradication of antibiotic-tolerant bacterial populations in Pseudomonas aeruginosa biofilms. Nat. Commun. 2016, 7, 10750. [Google Scholar] [CrossRef]
- Lata, M.; Sharma, D.; Deo, N.; Tiwari, P.K.; Bisht, D.; Venkatesan, K. Proteomic analysis of ofloxacin-mono resistant Mycobacterium tuberculosis isolates. J. Proteomics 2015, 127, 114–121. [Google Scholar] [CrossRef]
- Zhang, D.; Li, H.; Lin, X.; Peng, X. Outer membrane proteomics of kanamycin-resistant Escherichia coli identified MipA as a novel antibiotic resistance-related protein. FEMS Microbiol. Lett. 2015, 362, 1–8. [Google Scholar] [CrossRef]
- Liu, X.; Hu, Y.; Pai, P.-J.; Chen, D.; Lam, H. Label-Free Quantitative Proteomics Analysis of Antibiotic Response in Staphylococcus aureus to Oxacillin. J. Proteome Res. 2014, 13, 1223–1233. [Google Scholar] [CrossRef]
- Ma, Y.; Guo, C.; Li, H.; Peng, X.X. Low abundance of respiratory nitrate reductase is essential for Escherichia coli in resistance to aminoglycoside and cephalosporin. J. Proteomics 2013, 87, 78–88. [Google Scholar] [CrossRef]
- Petrackova, D.; Janecek, J.; Bezouskova, S.; Kalachova, L.; Technikova, Z.; Buriankova, K.; Halada, P.; Haladova, K.; Weiser, J. Fitness and proteome changes accompanying the development of erythromycin resistance in a population of Escherichia coli grown in continuous culture. Microbiologyopen 2013, 2, 841–852. [Google Scholar] [CrossRef]
- Pinto, L.; Poeta, P.; Radhouani, H.; Coelho, C.; Carvalho, C.; Rodrigues, J.; Torres, C.; Vitorino, R.; Domingues, P.; Igrejas, G. Proteomic evaluation of escherichia coli isolates from human clinical strains. J. Integr. OMICS 2011, 1, 42–48. [Google Scholar] [CrossRef]
- Bush, K.; Bradford, P.A. β-Lactams and β-Lactamase Inhibitors: An Overview. Cold Spring Harb. Perspect. Med. 2016, 6, a025247. [Google Scholar] [CrossRef] [PubMed]
- Wivagg, C.N.; Bhattacharyya, R.P.; Hung, D.T. Mechanisms of β-lactam killing and resistance in the context of Mycobacterium tuberculosis. J. Antibiot. (Tokyo) 2014, 67, 645–654. [Google Scholar] [CrossRef] [PubMed]
- Tomasz, A. The Mechanism of the Irreversible Antimicrobial Effects of Penicillins: How the Beta-Lactam Antibiotics Kill and Lyse Bacteria. Annu. Rev. Microbiol. 1979, 33, 113–137. [Google Scholar] [CrossRef]
- Tipper, D.J. Mode of action of β-lactam antibiotics. Rev. Infect. Dis. 1979, 1, 39–53. [Google Scholar] [CrossRef]
- Cho, H.; Uehara, T.; Bernhardt, T.G. Beta-lactam antibiotics induce a lethal malfustioning of the bacterial cell wall sunthesis machinery. Cell 2014, 159, 1300–1311. [Google Scholar] [CrossRef]
- Xu, C.; Lin, X.; Ren, H.; Zhang, Y.; Wang, S.; Peng, X. Analysis of outer membrane proteome of Escherichia coli related to resistance to ampicillin and tetracycline. Proteomics 2006, 6, 462–473. [Google Scholar] [CrossRef]
- Krause, K.M.; Serio, A.W.; Kane, T.R.; Connolly, L.E. Aminoglycosides: An overview. Cold Spring Harb. Perspect. Med. 2016, 6, 1–18. [Google Scholar] [CrossRef]
- Hancock, R.E.W.; Farmer, S.W.; Li, Z.; Poolet, K. Interaction of Aminoglycosides with the Outer Membranes and Purified Lipopolysaccharide and OmpF Porin of Escherichia coli. Antimicrob. Agents Chemother. 1991, 35, 1309–1314. [Google Scholar] [CrossRef]
- Taber, H.W.; Mueller, J.P.; Miller, P.F.; Arrow, A.M.Y.S. Bacterial Uptake of Aminoglycoside Antibiotics. J. Antimicrob. Chemother. 1987, 51, 439–457. [Google Scholar] [CrossRef]
- Jana, S.; Deb, J.K. Molecular understanding of aminoglycoside action and resistance. Appl. Microbiol. Biotechnol. 2006, 70, 140–150. [Google Scholar] [CrossRef]
- Ramirez, M.S.; Tolmasky, M.E. Aminoglycoside Modifying Enzymes. Drug Resist. Updat. 2010, 13, 151–171. [Google Scholar] [CrossRef] [PubMed]
- Tsai, A.; Uemura, S.; Johansson, M.; Puglisi, E.V.; Marshall, R.A.; Aitken, C.E.; Korlach, J.; Ehrenberg, M.; Puglisi, J.D. The Impact of Aminoglycosides on the Dynamics of Translation Elongation. Cell Rep. 2013, 3, 497–508. [Google Scholar] [CrossRef] [PubMed]
- Davis, B.D.; Chen, L.; Tai, P.C. Misread protein creates membrane channels: An essential step in the bactericidal action of aminoglycosides. Proc. Natl. Acad. Sci. USA 1986, 83, 6164–6168. [Google Scholar] [CrossRef] [PubMed]
- Serio, A.W.; Keepers, T.; Andrews, L.; Krause, K.M. Aminoglycoside Revival: Review of a Historically Important Class of Antimicrobials Undergoing Rejuvenation. EcoSal Plus 2018, 8. [Google Scholar] [CrossRef] [PubMed]
- Shteinberg, M.; Elborn, J.S. Use of Inhaled Tobramycin in Cystic Fibrosis. Adv. Ther. 2015, 32, 1–9. [Google Scholar] [CrossRef]
- Bulitta, J.B.; Ly, N.S.; Landersdorfer, C.B.; Wanigaratne, N.A.; Velkov, T.; Yadav, R.; Oliver, A.; Martin, L.; Shin, B.S.; Forrest, A.; et al. Two mechanisms of killing of pseudomonas aeruginosa by tobramycin assessed at multiple inocula via mechanism-based modeling. Antimicrob. Agents Chemother. 2015, 59, 2315–2327. [Google Scholar] [CrossRef]
- Yadav, R.; Bulitta, J.B.; Schneider, E.K.; Shin, B.S.; Velkov, T.; Nation, R.L.; Landersdorfer, C.B. Aminoglycoside Concentrations Required for Synergy with Carbapenems against Pseudomonas aeruginosa Determined via Mechanistic Studies and Modeling. Antimicrob. Agents Chemother. 2017, 61, 1–16. [Google Scholar] [CrossRef]
- Park, A.J.; Murphy, K.; Surette, M.D.; Bandoro, C.; Krieger, J.R.; Taylor, P.; Khursigara, C.M. Tracking the Dynamic Relationship between Cellular Systems and Extracellular Subproteomes in Pseudomonas aeruginosa Biofilms. J. Proteome Res. 2015, 14, 4524–4537. [Google Scholar] [CrossRef]
- Reales-Calderón, J.A.; Corona, F.; Monteoliva, L.; Gil, C.; Martínez, J.L. Quantitative proteomics unravels that the post-transcriptional regulator Crc modulates the generation of vesicles and secreted virulence determinants of Pseudomonas aeruginosa. J. Proteomics 2015, 127, 352–364. [Google Scholar] [CrossRef]
- Toyofuku, M.; Roschitzki, B.; Riedel, K.; Eberl, L. Identification of Proteins Associated with the Pseudomonas aeruginosa Biofilm Extracellular Matrix. J. Proteome Res. 2012, 11, 4906–4915. [Google Scholar] [CrossRef]
- Choi, D.-S.; Kim, D.-K.; Choi, S.J.; Lee, J.; Choi, J.-P.; Rho, S.; Park, S.-H.; Kim, Y.-K.; Hwang, D.; Gho, Y.S. Proteomic analysis of outer membrane vesicles derived from Pseudomonas aeruginosa. Proteomics 2011, 11, 3424–3429. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Wang, B.C.; Xu, W.J.; Lin, X.M.; Peng, X.X. Identification and network of outer membrane proteins regulating streptomysin resistance in escherichia coll. J. Proteome Res. 2008, 7, 4040–4049. [Google Scholar] [CrossRef] [PubMed]
- Ćudić, E.; Surmann, K.; Panasia, G.; Hammer, E.; Hunke, S. The role of the two-component systems Cpx and Arc in protein alterations upon gentamicin treatment in Escherichia coli. BMC Microbiol. 2017, 17, 197. [Google Scholar] [CrossRef] [PubMed]
- Dinos, G.P. The macrolide antibiotic renaissance. Br. J. Pharmacol. 2017, 174, 2967–2983. [Google Scholar] [CrossRef]
- Janas, A.; Przybylski, P. 14- and 15-membered lactone macrolides and their analogues and hybrids: Structure, molecular mechanism of action and biological activity. Eur. J. Med. Chem. 2019, 182, 111662. [Google Scholar] [CrossRef]
- Vázquez-Laslop, N.; Mankin, A.S. How Macrolide Antibiotics Work. Trends Biochem. Sci. 2018, 43, 668–684. [Google Scholar] [CrossRef]
- Svetlov, M.S.; Vázquez-Laslop, N.; Mankin, A.S. Kinetics of drug-ribosome interactions defines the cidality of macrolide antibiotics. Proc. Natl. Acad. Sci. USA 2017, 114, 13673–13678. [Google Scholar] [CrossRef]
- Sothiselvam, S.; Neuner, S.; Rigger, L.; Klepacki, D.; Micura, R.; Vázquez-Laslop, N.; Mankin, A.S. Binding of Macrolide Antibiotics Leads to Ribosomal Selection against Specific Substrates Based on Their Charge and Size. Cell Rep. 2016, 16, 1789–1799. [Google Scholar] [CrossRef]
- Davis, A.R.; Gohara, D.W.; Yap, M.-N.F. Sequence selectivity of macrolide-induced translational attenuation. Proc. Natl. Acad. Sci. USA 2014, 111, 15379–15384. [Google Scholar] [CrossRef]
- Yao, W.; Xu, G.; Li, D.; Bai, B.; Wang, H.; Cheng, H.; Zheng, J.; Sun, X.; Lin, Z.; Deng, Q.; et al. Staphylococcus aureus with an erm-mediated constitutive macrolide-lincosamide-streptogramin B resistance phenotype has reduced susceptibility to the new ketolide, solithromycin. BMC Infect. Dis. 2019, 19, 175. [Google Scholar] [CrossRef]
- Schroeder, M.R.; Stephens, D.S. Macrolide Resistance in Streptococcus pneumoniae. Front. Cell. Infect. Microbiol. 2016, 6, 98. [Google Scholar] [CrossRef]
- Gomes, C.; Ruiz-Roldán, L.; Mateu, J.; Ochoa, T.J.; Ruiz, J. Azithromycin resistance levels and mechanisms in Escherichia coli. Sci. Rep. 2019, 9, 6089. [Google Scholar] [CrossRef]
- Fyfe, C.; Grossman, T.H.; Kerstein, K.; Sutcliffe, J. Resistance to Macrolide Antibiotics in Public Health Pathogens. Cold Spring Harb. Perspect. Med. 2016, 6, a025395. [Google Scholar] [CrossRef]
- Chancey, S.T.; Zhou, X.; Zähner, D.; Stephens, D.S. Induction of efflux-mediated macrolide resistance in Streptococcus pneumoniae. Antimicrob. Agents Chemother. 2011, 55, 3413–3422. [Google Scholar] [CrossRef]
- Iannelli, F.; Santoro, F.; Santagati, M.; Docquier, J.-D.; Lazzeri, E.; Pastore, G.; Cassone, M.; Oggioni, M.R.; Rossolini, G.M.; Stefani, S.; et al. Type M Resistance to Macrolides Is Due to a Two-Gene Efflux Transport System of the ATP-Binding Cassette (ABC) Superfamily. Front. Microbiol. 2018, 9. [Google Scholar] [CrossRef]
- Chollet, R.; Chevalier, J.; Bryskier, A.; Pagès, J.-M. The AcrAB-TolC pump is involved in macrolide resistance but not in telithromycin efflux in Enterobacter aerogenes and Escherichia coli. Antimicrob. Agents Chemother. 2004, 48, 3621–3624. [Google Scholar] [CrossRef]
- Andersson, M.I. Development of the quinolones. J. Antimicrob. Chemother. 2003, 51, 1–11. [Google Scholar] [CrossRef]
- Rehman, A.; Patrick, W.M.; Lamont, I.L. Mechanisms of ciprofloxacin resistance in pseudomonas aeruginosa: New approaches to an old problem. J. Med. Microbiol. 2019, 68, 1–10. [Google Scholar] [CrossRef]
- Naqvi, S.A.R.; Roohi, S.; Iqbal, A.; Sherazi, T.A.; Zahoor, A.F.; Imran, M. Ciprofloxacin: From infection therapy to molecular imaging. Mol. Biol. Rep. 2018, 45, 1457–1468. [Google Scholar] [CrossRef]
- Zhang, G.F.; Liu, X.; Zhang, S.; Pan, B.; Liu, M.L. Ciprofloxacin derivatives and their antibacterial activities. Eur. J. Med. Chem. 2018, 146, 599–612. [Google Scholar] [CrossRef]
- Palumbo, M.; Gatto, B.; Zagotto, G.; Palu, G. On the mechanism of action of quinolone drugs. Bull. Johns Hopkins Hosp. 1993, 1, 232–235. [Google Scholar] [CrossRef]
- Drlica, K.; Zhao, X. DNA gyrase, topoisomerase IV, and the 4-quinolones. Microbiol. Mol. Biol. Rev. 1997, 61, 377–392. [Google Scholar] [CrossRef]
- Jensen, P.Ø.; Briales, A.; Brochmann, R.P.; Wang, H.; Kragh, K.N.; Kolpen, M.; Hempel, C.; Bjarnsholt, T.; Høiby, N.; Ciofu, O. Formation of hydroxyl radicals contributes to the bactericidal activity of ciprofloxacin against Pseudomonas aeruginosa biofilms. Pathog. Dis. 2014, 70, 440–443. [Google Scholar] [CrossRef]
- Namvar, A.E.; Bastarahang, S.; Abbasi, N.; Ghehi, G.S.; Farhadbakhtiarian, S.; Arezi, P.; Hosseini, M.; Baravati, S.Z.; Jokar, Z.; Chermahin, S.G. Clinical characteristics of Staphylococcus epidermidis: A systematic review. GMS Hyg. Infect. Control 2014, 9, Doc23. [Google Scholar] [CrossRef]
- Poirel, L.; Jayol, A.; Nordmann, P. Polymyxins: Antibacterial Activity, Susceptibility Testing, and Resistance Mechanisms Encoded by Plasmids or Chromosomes. Clin. Microbiol. Rev. 2017, 30, 557–596. [Google Scholar] [CrossRef]
- Miller, W.R.; Bayer, A.S.; Arias, C.A. Mechanism of Action and Resistance to Daptomycin in Staphylococcus aureus and Enterococci. Cold Spring Harb. Perspect. Med. 2016, 6, a026997. [Google Scholar] [CrossRef]
- Li, J.; Nation, R.L.; Turnidge, J.D.; Milne, R.W.; Coulthard, K.; Rayner, C.R.; Paterson, D.L. Colistin: The re-emerging antibiotic for multidrug-resistant Gram-negative bacterial infections. Lancet Infect. Dis. 2006, 6, 589–601. [Google Scholar] [CrossRef]
- KOCH-WESER, J. Adverse Effects of Sodium Colistimethate. Ann. Intern. Med. 1970, 72, 857. [Google Scholar] [CrossRef]
- Moore, R.A.; Bates, N.C.; Hancock, R.E.W. Interaction of polycationic antibiotics with Pseudomonas aeruginosa lipopolysaccharide and lipid A studied by using dansyl-polymyxin. Antimicrob. Agents Chemother. 1986, 29, 496–500. [Google Scholar] [CrossRef]
- Hancock, R.E.W. Alterations in Outer Membrane Permeability. Annu. Rev. Microbiol. 1984, 38, 237–264. [Google Scholar] [CrossRef]
- Klemperer, R.M.; Gilbert, P.; Meier, A.M.; Cozens, R.M.; Brown, M.R.W. Influence of Suspending Media upon the Susceptibility of Pseudomonas aeruginosa NCTC 6750 and Its Spheroplasts to Polymyxin B. Antimicrob. Agents Chemother. 1979, 15, 147–151. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Dhillon, P.; Yan, H.; Farmer, S.; Hancock, R.E.W. Interactions of Bacterial Cationic Peptide Antibiotics with Outer and Cytoplasmic Membranes of Pseudomonas aeruginosa. Antimicrob. Agents Chemother. 2000, 44, 3317–3321. [Google Scholar] [CrossRef] [PubMed]
- Aquilini, E.; Merino, S.; Knirel, Y.; Regué, M.; Tomás, J. Functional Identification of Proteus mirabilis eptC Gene Encoding a Core Lipopolysaccharide Phosphoethanolamine Transferase. Int. J. Mol. Sci. 2014, 15, 6689–6702. [Google Scholar] [CrossRef] [PubMed]
- Anandan, A.; Evans, G.L.; Condic-Jurkic, K.; O’Mara, M.L.; John, C.M.; Phillips, N.J.; Jarvis, G.A.; Wills, S.S.; Stubbs, K.A.; Moraes, I.; et al. Structure of a lipid A phosphoethanolamine transferase suggests how conformational changes govern substrate binding. Proc. Natl. Acad. Sci. USA 2017, 114, 2218–2223. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Ma, T.; Liu, Q.; Huang, Y.; Hu, C.; Liao, G. Improvement of Daptomycin Production in Streptomyces roseosporus through the Acquisition of Pleuromutilin Resistance. Biomed Res. Int. 2013, 2013, 1–6. [Google Scholar] [CrossRef][Green Version]
- Tally, F.P.; DeBruin, M.F. Development of daptomycin for Gram-positive infections. J. Antimicrob. Chemother. 2000, 46, 523–526. [Google Scholar] [CrossRef]
- Eisenstein, B.I.; Oleson, F.B., Jr.; Baltz, R.H. Daptomycin: From the Mountain to the Clinic, with Essential Help from Francis Tally, MD. Clin. Infect. Dis. 2010, 50, S10–S15. [Google Scholar] [CrossRef]
- Jung, D.; Rozek, A.; Okon, M.; Hancock, R.E.W. Structural Transitions as Determinants of the Action of the Calcium-Dependent Antibiotic Daptomycin. Chem. Biol. 2004, 11, 949–957. [Google Scholar] [CrossRef]
- Chen, Y.F.; Sun, T.L.; Sun, Y.; Huang, H.W. Interaction of daptomycin with lipid bilayers: A lipid extracting effect. Biochemistry 2014. [Google Scholar] [CrossRef]
- Seyfi, R.; Kahaki, F.A.; Ebrahimi, T.; Montazersaheb, S.; Eyvazi, S.; Babaeipour, V.; Tarhriz, V. Antimicrobial Peptides (AMPs): Roles, Functions and Mechanism of Action. Int. J. Pept. Res. Ther. 2019. [Google Scholar] [CrossRef]
- Raheem, N.; Straus, S.K. Mechanisms of Action for Antimicrobial Peptides With Antibacterial and Antibiofilm Functions. Front. Microbiol. 2019, 10, 1–14. [Google Scholar] [CrossRef]
- Mojsoska, B.; Jenssen, H. Peptides and peptidomimetics for antimicrobial drug design. Pharmaceuticals 2015, 8, 366–415. [Google Scholar] [CrossRef] [PubMed]
- Lee, T.-H.N.; Hall, K.; Aguilar, M.-I. Antimicrobial Peptide Structure and Mechanism of Action: A Focus on the Role of Membrane Structure. Curr. Top. Med. Chem. 2015, 16, 25–39. [Google Scholar] [CrossRef] [PubMed]
- Lei, J.; Sun, L.C.; Huang, S.; Zhu, C.; Li, P.; He, J.; Mackey, V.; Coy, D.H.; He, Q.Y. The antimicrobial peptides and their potential clinical applications. Am. J. Transl. Res. 2019, 11, 3919–3931. [Google Scholar] [PubMed]
- Brogden, K.A. Antimicrobial peptides: Pore formers or metabolic inhibitors in bacteria? Nat. Rev. Microbiol. 2005, 3, 238–250. [Google Scholar] [CrossRef] [PubMed]
- Nicolas, P. Multifunctional host defense peptides: Intracellular-targeting antimicrobial peptides. FEBS J. 2009, 276, 6483–6496. [Google Scholar] [CrossRef] [PubMed]
- Le, C.-F.; Fang, C.-M.; Sekaran, D. Intracellular Targeting Mechanisms by Antimicrobial Peptides. Antimicrob. Agents Chemother. 2017, 61, 1–16. [Google Scholar] [CrossRef]
- Zhu, Y.; Mohapatra, S.; Weisshaar, J.C. Rigidification of the Escherichia coli cytoplasm by the human antimicrobial peptide LL-37 revealed by superresolution fluorescence microscopy. Proc. Natl. Acad. Sci. USA 2019, 116, 1017–1026. [Google Scholar] [CrossRef] [PubMed]
- Liu, W.; Dong, S.L.; Xu, F.; Wang, X.Q.; Withers, T.R.; Yu, H.D.; Wang, X. Effect of intracellular expression of antimicrobial peptide LL-37 on growth of escherichia coli strain TOP10 under aerobic and anaerobic conditions. Antimicrob. Agents Chemother. 2013, 57, 4707–4716. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Pathogen | Resistance | Priority | Gram +/− |
|---|---|---|---|
| Acinetobacter baumannii | carbapenem-resistant | Critical | − |
| Pseudomonas aeruginosa | carbapenem-resistant | Critical | − |
| * Enterobacteriaceae | carbapenem-resistant, extended-spectrum β lactamase (ESBL)-producing | Critical | − |
| Enterococcus faecium | vancomycin-resistant | High | + |
| Staphylococcus aureus | methicillin-resistant,vancomycin-intermediate and resistant | High | + |
| Helicobacter pylori | clarithromycin-resistant | High | − |
| Campylobacter spp. | fluoroquinolone-resistant | High | − |
| Salmonellae | fluoroquinolone-resistant | High | − |
| Neisseria gonorrhoeae | cephalosporin-resistant, fluoroquinolone-resistant | High | − |
| Streptococcus pneumoniae | penicillin-non-susceptible | Medium | + |
| Haemophilus influenzae | ampicillin-resistant | Medium | − |
| Shigella spp. | fluoroquinolone-resistant | Medium | − |
| Drug | Bacterial Strain | Primary MOA | Phenotypic Investigation, Target | Time of Exposure | Concentration | Proteomic Approach and Method | Proteome Coverage (%) * | Results, Selected DEPs | Reference |
|---|---|---|---|---|---|---|---|---|---|
| PMB | E. coli | Outer membrane | Planktonic, Adaptive responses, (tolerance) | 30 growth cycles 31 × 16 h | 1 µg/mL 5 µg/mL 10 µg/ml | iTRAQ labeling (TMT) LC-MS/MS | 4,558 (pr. sample) | 1 µg/mL: (66 ↑, 22 ↓) 10 µg/mL: (232 ↑, 82 ↓) | [46] |
| LL-37 | Strep. pneumoniae | Bacterial membrane, DNA | Planktonic, resistance | 1–2 h | 2.5 µg/ml | Label-free, DDA | 68%, 1,293/195 | 23-↑ 29-↓ | [61] |
| CIP | E. coli | DNA gyrase | Persisters | 3 h, cyclic antibiotic exposure | 5 µg/mL (100 × MIC) | Label-free, DDA | 739 | 23 ↑ 12 ↓ | [62] |
| RIF AMP | E. coli | RNA synthesis, cell wall | Persisters | 30 min RIF 3 h AMP | 100 µg/mL RIF, 100 µg/mL AMP | Label-free, DDA | 1,160 | 70 ↓ 35 ↑(persisters) | [59] |
| CST PMB | E. coli | Outer membrane | mcr-1 colistin-resistance | NS | 0, 0.5–1 μg/mL | Label-free, DDA (TOF-MS/MS) DIA | 64.3%, 2,784 | No drug: 26 ↑, 31 ↓ CST 0.5: 75 ↑, 311 ↓ CST 1: 69 ↑, 237 ↓ PMB 0.5: 35 ↑, 219 ↓ PMB 1: 16 ↑, 147 ↓ | [43] |
| TOB | P. aeruginosa | Protein synthesis | Outer membrane vesicles | 24 h | 1 µg/mL (sub-MIC) | Label-free, iBAQ, | 757 | 165 ↓,17 ↑ | [42] |
| - | E. coli (ESBL-ST131) | Cell wall biosynthesis | Characterization of clinical isolates | No exposure | - | MALDI-TOF MS, LC-MS/MS | 10 | - | [62] |
| AgNPs | P. aeruginosa | Cell membrane, ROS generation | Planktonic | 24 h | 0.1–50 µg/mL | Labeling, iTRAQ | ND | 3-↑,5-supressed | [49] |
| AMP CTX CFP | E. coli | Cell wall biosynthesis | Outer membrane vesicles, β-lactam resistance | 12–84 h | AMP: 30 µg/mL CTX: 4 µg/mL CFP: 1.25 µg/mL | SDS-PAGE, MALDI-TOF-MS | 1,639 (273 mapped) 260 (OMVs resistant) 270 (OMVs susceptible) | 83 ↑, 49 ↓ (resistant) | [63] |
| MEM CIP | E. coli(NDM, KPC or IMP) | Cell wall biosynthesis, DNA gyrase | Planktonic, β-lactam resistance | 4 h | sub-MIC 0.3–24 µg/mL MEM 32 µg/mL CIP | DIA | 457 | OmpA: (all strains-CIP) ↓ and (IMP-MEM)↑ HU DNA-bp: (NDM and KPC-MEM) ↑ GroEL/GroES and GrpE: (all strains-MEM) ↑ | [64] |
| DAP | S. aureus | Lipoteichoic acid biosynthesis | Planktonic, biofilm, resistant | 4 months (Daily passages) | 0–31 µg/mL | Labeling | 60%, 1,709 | 349 DEPs, 105 ↑ 80 ↓ | [47] |
| CIP | P. aeruginosa | DNA gyrase | Biofilms- antibiotic tolerant subpopulation | 1.5, 5.5, 14.5 h | 60 µg/Ml (Supra-MIC) | Label (BONCAT enrichment) | > 1,200 | 1.5h: 73 (41 unique) 5.5 h: 187 (80 unique) 14.5: 204 (90 unique) | [39] |
| CIP | P. aeruginosa | DNA gyrase | Planktonic, adaptive resistance | 48 h | 0.125–8 µg/mL | Label-free, DDA | 57.6% 3,251 | mu0125_l: 57 ↑,76 ↓ mu0125_h:62 ↑,92 ↓ mu05_l: 43 ↑, 26 ↓ mu05_h: 48 ↑, 68 ↓ | [40] |
| DAP | S. aureus | Lipoteichoic acidbiosynthesis | Planktonic, resistant and sensitive population | 18 h | 0.25–2 × MIC | Labeling, iTRAQ | 872 | 34 ↑ 17 ↓ | [48] |
| CST | A. baumannii MDR-ZJ06 | Outer membrane | Planktonic, resistance in MDR strains | ON cultures | 8 × MIC 64 × MIC 200 × MIC | Labeling, iTRAQ | 1,582 | 31 ↑ 51 ↓ | [65] |
| CIP BC | P. aeruginosa | DNA gyrase, outer membrane | Outer membrane proteins, Biofilms, adaptive resistance | 12 days | CIP: 6 µg/mL BC: 324 µg/mL | 2-DE SDS-PAGE | 10 proteins/600 spots | 9 ↓ 1 ↑ | [38] |
| CST | P. aeruginosa | Outer membrane | Biofilms, Tolerance | 2-32 h, 8 h | 10 µg/mL-(10 × MIC) | Label - Pulsed-SILAC | 4,250 | 256 ↑ 140 ↓ | [66] |
| OFX | M. tuberculosis | DNA gyrase | Planktonic, Mono-resistance | 36 h | 2 µg/mL (sub-MIC) | 2-DE, MALDI-TOF MS | 14 | 14 ↑ | [67] |
| KAN | E. coli | Protein synthesis | Outer membrane Resistance | 10 sequential subcultures | 6.25 µg/mL (1/2 MIC) | 2-DE, MALDI-TOF MS | 11 | 6 ↑ 5 ↓ | [68] |
| TOB | P. aeruginosa | Protein synthesis | Planktonic, adaptive resistance | DDE: 60 min TCE: 15, 60, 120, 360 min | DDE: 0.1, 0.5, 1µg/mL TCE: 1 µg/mL | DDA | > 1000 (TOB) | 96 ↑ | [41] |
| OXA | MRSA, MSSA | Cell wall biosynthesis | Planktonic | NS | sub-MIC 1/8 × MIC 8 µg/mL 0.125 µg/mL | Label-free, DDA | MRSA: 1,071, (41%) MSSA: 1,034 (40%) | MRSA: 65 ↑,16 ↓ MSSA: 162 ↑, 63 ↓ | [69] |
| SM GEN CEF TET NA | E. coli | Protein synthesis, cell wall synthesis, DNA gyrase | Effect of low abundance of NarG and NarH on resistance | 10 sequential subcultures | 1/2 MIC | 2-DE, MALDI-TOF MS | 94 | CAZ-R: 7↓ 6↑ SM-R: 5 ↓ 1↑ TET-R:7 ↓ 1↑ GEN-R: 9 ↓ 1↑ NA-R: 10 ↓ | [70] |
| ERY | E. coli | Protein synthesis | Planktonic, resistance | 0, 43, 68, 103 h | sub-MIC 10 µg/mL (sub-MIC) | 2-DE, MALDI-TOF/TOF | 35/91 sp. | 43 h: 14 ↑, 3 ↓ 68 h: 6 ↓ 103h: 14 ↑, 1 ↓ | [71] |
| CIP | P. aeruginosa | DNA gyrase | Planktonic, adaptive resistance | 0–48 h | PAO1: 0–8 × MIC (0–4 µg/mL) PAK: 1/2 MIC (0.125 µg/mL) | 2-DE, MALDI-TOF/TOF | 3/650 sp. | 2 proteins with higher phosphorylated/total protein ratio 1 ↑ | [37] |
| β-lactams | E. coli (TEM-52 and CMY-2) | Cell wall biosynthesis | β-lactam resistance mediated | No exposure | - | 2-DE, IEF, MALDI-TOF/TOF | C583 strain: 64 sp. C580 strain: 91 sp. | - | [72] |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tsakou, F.; Jersie-Christensen, R.; Jenssen, H.; Mojsoska, B. The Role of Proteomics in Bacterial Response to Antibiotics. Pharmaceuticals 2020, 13, 214. https://doi.org/10.3390/ph13090214
Tsakou F, Jersie-Christensen R, Jenssen H, Mojsoska B. The Role of Proteomics in Bacterial Response to Antibiotics. Pharmaceuticals. 2020; 13(9):214. https://doi.org/10.3390/ph13090214
Chicago/Turabian StyleTsakou, Foteini, Rosa Jersie-Christensen, Håvard Jenssen, and Biljana Mojsoska. 2020. "The Role of Proteomics in Bacterial Response to Antibiotics" Pharmaceuticals 13, no. 9: 214. https://doi.org/10.3390/ph13090214
APA StyleTsakou, F., Jersie-Christensen, R., Jenssen, H., & Mojsoska, B. (2020). The Role of Proteomics in Bacterial Response to Antibiotics. Pharmaceuticals, 13(9), 214. https://doi.org/10.3390/ph13090214