ADP-Mediated Upregulation of Expression of CD62P on Human Platelets Is Critically Dependent on Co-Activation of P2Y1 and P2Y12 Receptors

 , , and
, , and
Abstract
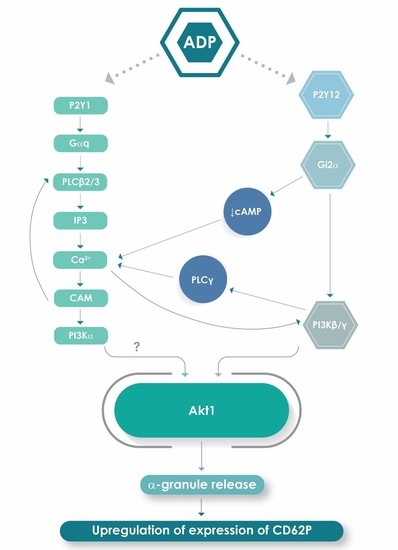
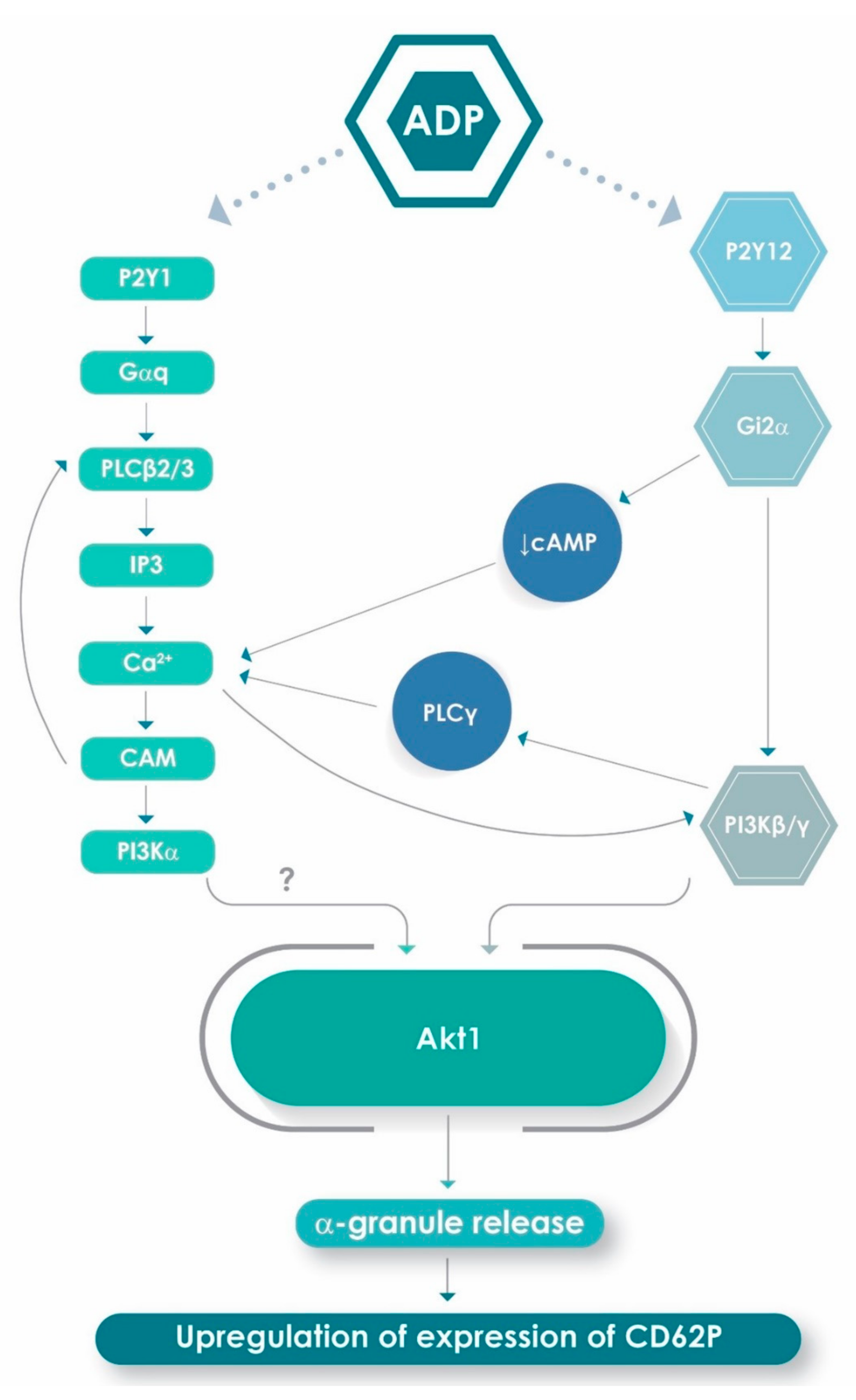
1. Introduction
2. Results
2.1. Purified Platelet Counts
2.2. Effects of MRS2500 and PSB0739 on Expression of CD62P by ADP-Activated Platelets
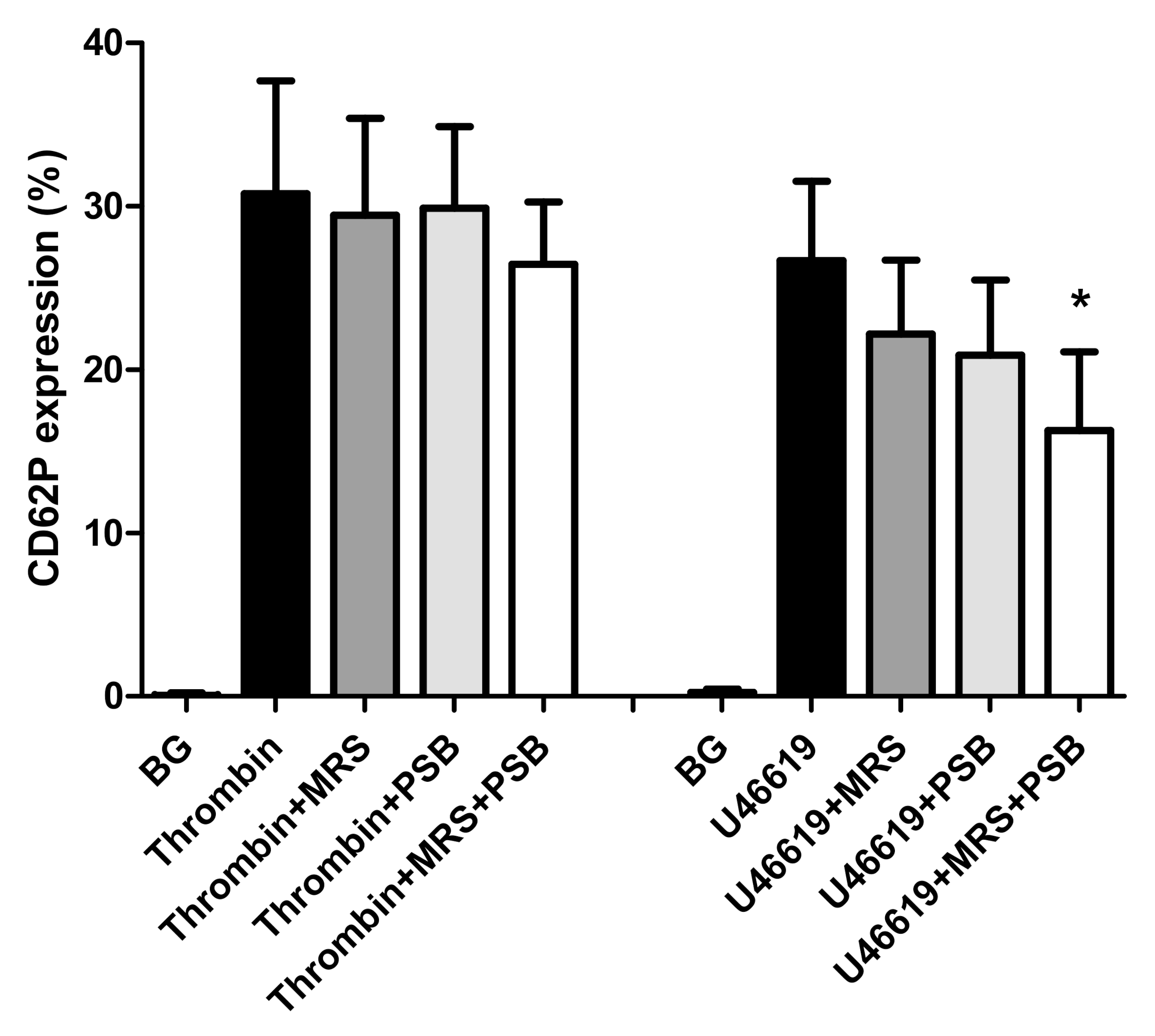
2.3. Effects of MRS2500 and PSB0739 on Expression of CD62P by Thrombin- and U46619-Activated Platelets
2.4. Effects of 2-APB, Calmidazolium Chloride, U73122 and Wortmannin on Expression of CD62P by ADP-Activated Platelets
2.5. Neutrophil:Platelet (NP) Aggregation
2.6. Effects of MRS2500 and PSB0739 on Akt1 Phosphorylation
3. Discussion
4. Materials and Methods
4.1. Ethical Statement
4.2. Chemicals
4.3. Platelet-Rich Plasma and Buffy Coat Preparation
4.4. Purified Platelet Suspensions
4.5. Measurement of Expression of CD62P by Platelets Activated with ADP
4.6. Neutrophil:Platelet (NP) Aggregate Formation
4.7. Measurement of Akt1 Phosphorylation
4.8. Expression and Statistical Analysis of Results
5. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Wagner, D.D. New links between inflammation and thrombosis. Arterioscler. Thromb. Vasc. Biol. 2005, 25, 1321–1324. [Google Scholar] [CrossRef] [PubMed]
- Caudrillier, A.; Kessenbrock, K.; Gilliss, B.M.; Nguyen, J.X.; Marques, M.B.; Monestier, M.; Toy, P.; Werb, Z.; Looney, M.R. Platelets induce neutrophil extracellular traps in transfusion-related acute lung injury. J. Clin. Investig. 2012, 122, 2661–2671. [Google Scholar] [CrossRef] [PubMed]
- Maugeri, N.; Rovere-Querini, P.; Evangelista, V.; Godino, C.; Demetrio, M.; Baldini, M.; Figini, F.; Coppi, G.; Slavich, M.; Camera, M.; et al. An intense and short-lasting burst of neutrophil activation differentiates early acute myocardial infarction from systemic inflammatory syndromes. PLoS ONE 2012, 7, e39484. [Google Scholar] [CrossRef] [PubMed]
- Page, C.; Pitchford, S. Neutrophil and platelet complexes and their relevance to neutrophil recruitment and activation. Int. Immunopharmacol. 2013, 17, 1176–1184. [Google Scholar] [CrossRef]
- Sreeramkumar, V.; Adrover, J.M.; Ballesteros, I.; Cuartero, M.I.; Rossaint, J.; Bilbao, I.; Nácher, M.; Pitaval, C.; Radovanovic, I.; Fukui, Y.; et al. Neutrophils scan for activated platelets to initiate inflammation. Science 2014, 346, 1234–1238. [Google Scholar] [CrossRef]
- Constantinescu-Bercu, A.; Salles-Crawley, I.I.; Woollard, K.J.; Crawley, J.T.B. Novel platelet-neutrophil interaction via activated αIIbβ3 mediates NETosis under flow. bioRxiv 2018, 373670. Available online: https://www.biorxiv.org/content/biorxiv/early/2018/07/21/373670.full.pdf (accessed on 2 September 2019). [CrossRef]
- Lisman, T. Platelet-neutrophil interactions as drivers of inflammatory and thrombotic disease. Cell Tissue Res. 2018, 371, 567–576. [Google Scholar] [CrossRef]
- Hamzeh-Cognasse, H.; Damien, P.; Chabert, A.; Pozzetto, B.; Cognasse, F.; Garraud, O. Platelets and infections—Complex interactions with bacteria. Front. Immunol. 2015, 6, 82. [Google Scholar] [CrossRef]
- Ali, R.A.; Wuescher, L.M.; Dona, K.R.; Worth, R.G. Platelets mediate host defense against Staphylococcus aureus through direct bactericidal activity and by enhancing macrophage activities. J. Immunol. 2017, 198, 344–351. [Google Scholar] [CrossRef]
- Amison, R.T.; Momi, S.; Morris, A.; Manni, G.; Keir, S.; Gresele, P.; Page, C.P.; Pitchford, S.C. RhoA signaling through platelet P2Y1 receptor controls leukocyte recruitment in allergic mice. J. Allergy Clin. Immunol. 2015, 135, 528–538. [Google Scholar] [CrossRef]
- Martinod, K.; Wagner, D.D. Thrombosis: Tangled up in NETs. Blood 2014, 123, 2768–2776. [Google Scholar] [CrossRef] [PubMed]
- Kimball, A.S.; Obi, A.T.; Diaz, J.A.; Henke, P.K. The emerging role of NETs in venous thrombosis and immunothrombosis. Front. Immunol. 2016, 7, 236. [Google Scholar] [CrossRef] [PubMed]
- Pfeiler, S.; Stark, K.; Massberg, S.; Engelmann, B. Propagation of thrombosis by neutrophils and extracellular nucleosome networks. Haematologica 2017, 102, 206–213. [Google Scholar] [CrossRef] [PubMed]
- De Bont, C.M.; Boelens, W.C.; Pruijn, G.J.M. NETosis, complement, and coagulation: A triangular relationship. Cell Mol. Immunol. 2019, 16, 19–27. [Google Scholar] [CrossRef] [PubMed]
- Bigalke, B.; Lindemann, S.; Ehlers, R.; Seizer, P.; Daub, K.; Langer, H.; Schonberger, T.; Kremmer, E.; Siegel-Axel, D.; May, A.E.; et al. Expression of platelet collagen receptor glycoprotein VI is associated with acute coronary syndrome. Eur. Heart J. 2006, 27, 2165–2169. [Google Scholar] [CrossRef]
- Offermanns, S. Activation of platelet function through G protein-coupled receptors. Circ. Res. 2006, 99, 1293–1304. [Google Scholar] [CrossRef]
- Ollivier, V.; Syvannarath, V.; Gros, A.; Butt, A.; Loyau, S.; Jandrot-Perrus, M.; Ho-Tin-Noé, B. Collagen can selectively trigger a platelet secretory phenotype via glycoprotein VI. PLoS ONE 2014, 9, e104712. [Google Scholar] [CrossRef]
- Gachet, C. Identification, characterization, and inhibition of the platelet ADP receptors. Int. J. Hematol. 2001, 74, 375–381. [Google Scholar] [CrossRef]
- Gremmel, T.; Yanachkov, I.B.; Yanachkova, M.; Wright, G.E.; Wider, J.; Undyala, V.V.; Michelson, A.D.; Frelinger, A.L., 3rd; Przyklenk, K. Synergistic inhibition of both P2Y1 and P2Y12 adenosine diphosphate receptors as novel approach to rapidly attenuate platelet-mediated thrombosis. Arterioscler. Thromb. Vasc. Biol. 2016, 36, 501–509. [Google Scholar] [CrossRef]
- Gachet, C. P2Y12 receptors in platelets and other hematopoietic and non-hematopoietic cells. Purinergic Signal. 2012, 8, 609–619. [Google Scholar] [CrossRef]
- Storey, R.F.; Sanderson, H.M.; White, A.E.; May, J.A.; Cameron, K.E.; Heptinstall, S. The central role of the P2T receptor in amplification of human platelet activation, aggregation, secretion and procoagulant activity. Br. J. Haematol. 2000, 110, 925–934. [Google Scholar] [CrossRef] [PubMed]
- Storey, R.F.; Judge, H.M.; Wilcox, R.G.; Heptinstall, S. Inhibition of ADP-induced P-selectin expression and platelet-leukocyte conjugate formation by clopidogrel and the P2Y12 receptor antagonist AR-C69931MX but not aspirin. Thromb. Haemost. 2002, 88, 488–494. [Google Scholar] [CrossRef] [PubMed]
- Leon, C.; Ravanat, C.; Freund, M.; Cazenave, J.P.; Gachet, C. Differential involvement of the P2Y1 and P2Y12 receptors in platelet procoagulant activity. Arterioscler. Thromb. Vasc. Biol. 2003, 23, 1941–1947. [Google Scholar] [CrossRef] [PubMed]
- Oestreich, J.H.; Ferraris, S.P.; Steinhubl, S.R.; Akers, W.S. Pharmacodynamic interplay of the P2Y1, P2Y12, and TxA2 pathways in platelets: The potential of triple antiplatelet therapy with P2Y1 receptor antagonism. Thromb. Res. 2013, 131, e64–e70. [Google Scholar] [CrossRef] [PubMed]
- Amison, R.T.; O’Shaughnessy, B.G.; Arnold, S.; Cleary, S.J.; Nandi, M.; Pitchford, S.C.; Bragonzi, A.; Page, C.P. Platelet depletion impairs host defense to pulmonary infection with Pseudomonas aeruginosa in mice. Am. J. Respir. Cell Mol. Biol. 2018, 58, 331–340. [Google Scholar] [CrossRef]
- Hechler, B.; Nonne, C.; Roh, E.J.; Cattaneo, M.; Cazenave, J.P.; Lanza, F.; Jacobson, K.A.; Gachet, C. MRS2500 [2-iodo-N6-methyl-(N)-methanocarba-2′-deoxyadenosine-3′,5′-bisphosphate], a potent, selective, and stable antagonist of the platelet P2Y1 receptor with strong antithrombotic activity in mice. J. Pharmacol. Exp. Ther. 2006, 316, 556–563. [Google Scholar] [CrossRef]
- Horváth, G.; Gölöncsér, F.; Csölle, C.; Király, K.; Andó, R.D.; Baranyi, M.; Koványi, B.; Máté, Z.; Hoffmann, K.; Algaier, I.; et al. Central P2Y12 receptor blockade alleviates inflammatory and neuropathic pain and cytokine production in rodents. Neurobiol. Dis. 2014, 70, 162–178. [Google Scholar] [CrossRef]
- Woulfe, D.; Jiang, H.; Morgans, A.; Monks, R.; Birnbaum, M.; Brass, L.F. Defects in secretion, aggregation, and thrombus formation in platelets from mice lacking Akt2. J. Clin. Investig. 2004, 113, 441–450. [Google Scholar] [CrossRef]
- Woulfe, D.S. Akt signaling in platelets and thrombosis. Expert Rev. Hematol. 2010, 3, 81–91. [Google Scholar] [CrossRef]
- Jin, J.; Kunapuli, S.P. Coactivation of two different G protein-coupled receptors is essential for ADP-induced platelet aggregation. Proc. Natl. Acad. Sci. USA 1998, 95, 8070–8074. [Google Scholar] [CrossRef]
- Nylander, S.; Mattsson, C.; Ramström, S.; Lindahl, T.L. Synergistic action between inhibition of P2Y12/P2Y1 and P2Y12/thrombin in ADP- and thrombin-induced human platelet activation. Br. J. Pharmacol. 2004, 142, 1325–1331. [Google Scholar] [CrossRef] [PubMed]
- Ohlmann, P.; de Castro, S.; Brown, G.G., Jr.; Gachet, C.; Jacobson, K.A.; Harden, T.K. Quantification of recombinant and platelet P2Y(1) receptors utilizing a [125I]-labeled high-affinity antagonist 2-iodo-N6-methyl-(N)-methanocarba-2′-deoxyadenosine-3′,5′-bisphosphate ([125I]MRS2500). Pharmacol. Res. 2010, 62, 344–351. [Google Scholar] [CrossRef] [PubMed]
- Koupenova, M.; Ravid, K. Biology of platelet purinergic receptors and implications for platelet heterogeneity. Front. Pharmacol. 2018, 9, 37. [Google Scholar] [CrossRef] [PubMed]
- López, J.J.; Redondo, P.C.; Salido, G.M.; Pariente, J.A.; Rosado, J.A. Two distinct Ca2+ compartments show differential sensitivity to thrombin, ADP and vasopressin in human platelets. Cell Signal. 2006, 18, 373–381. [Google Scholar] [CrossRef] [PubMed]
- Hill, T.D.; Campos-Gonzalez, R.; Kindmark, H.; Boynton, A.L. Inhibition of inositol trisphosphate-stimulated calcium mobilization by calmodulin antagonists in rat liver epithelial cells. J. Biol. Chem. 1988, 263, 16479–16484. [Google Scholar]
- Kasri, N.N.; Török, K.; Galione, A.; Garnham, C.; Callewaert, G.; Missiaen, L.; Parys, J.B.; De Smedt, H. Endogenously bound calmodulin is essential for the function of the inositol 1,4,5-trisphosphate receptor. J. Biol. Chem. 2006, 281, 8332–8338. [Google Scholar] [CrossRef]
- Hardy, A.R.; Jones, M.L.; Mundell, S.J.; Poole, A.W. Reciprocal cross-talk between P2Y1 and P2Y12 receptors at the level of calcium signaling in human platelets. Blood 2004, 104, 1745–1752. [Google Scholar] [CrossRef]
- Cattaneo, M. P2Y12 receptors: Structure and function. J. Thromb. Haemost. 2015, 13, S10–S16. [Google Scholar] [CrossRef]
- Rameh, L.E.; Rhee, S.G.; Spokes, K.; Kazlauskas, A.; Cantley, L.C.; Cantley, L.G. Phosphoinositide 3-kinase regulates phospholipase Cγ-mediated calcium signaling. J. Biol. Chem. 1998, 273, 23750–23757. [Google Scholar] [CrossRef]
- Barker, S.A.; Lujan, D.; Wilson, B.S. Multiple roles for PI 3-kinase in the regulation of PLCγ activity and Ca2+ mobilization in antigen-stimulated mast cells. J. Leukoc. Biol. 1999, 65, 321–329. [Google Scholar] [CrossRef]
- Sun, D.S.; Lo, S.J.; Lin, C.H.; Yu, M.S.; Huang, C.Y.; Chen, Y.F.; Chang, H.H. Calcium oscillation and phosphatidylinositol 3-kinase positively regulate integrin αIIbβ3-mediated outside-in signaling. J. Biomed. Sci. 2005, 12, 321–333. [Google Scholar] [CrossRef] [PubMed]
- McCullar, J.S.; Larsen, S.A.; Millimaki, R.A.; Filtz, T.M. Calmodulin is a phospholipase C-β interacting protein. J. Biol. Chem. 2003, 278, 33708–33713. [Google Scholar] [CrossRef] [PubMed]
- Arthur, J.F.; Shen, Y.; Mu, F.T.; Leon, C.; Gachet, C.; Berndt, M.C.; Andrews, R.K. Calmodulin interacts with the platelet ADP receptor P2Y1. Biochem. J. 2006, 398, 339–343. [Google Scholar] [CrossRef] [PubMed]
- Joyal, J.L.; Burks, D.J.; Pons, S.; Matter, W.F.; Vlahos, C.J.; White, M.F.; Sacks, D.B. Calmodulin activates phosphatidylinositol 3-kinase. J. Biol. Chem. 1997, 272, 28183–28186. [Google Scholar] [CrossRef]
- Nussinov, R.; Wang, G.; Tsai, C.J.; Jang, H.; Lu, S.; Banerjee, A.; Zhang, J.; Gaponenko, V. Calmodulin and PI3K signaling in KRAS cancers. Trends Cancer 2017, 3, 214–224. [Google Scholar] [CrossRef]
- Gilio, K.; Munnix, I.C.; Mangin, P.; Cosemans, J.M.; Feijge, M.A.; van der Meijden, P.E.; Olieslagers, S.; Chrzanowska-Wodnicka, M.B.; Lillian, R.; Schoenwaelder, S.; et al. Non-redundant roles of phosphoinositide 3-kinase isoforms α and β in glycoprotein VI-induced platelet signaling and thrombus formation. J. Biol. Chem. 2009, 284, 33750–33762. [Google Scholar] [CrossRef]
- Franke, H.; Sauer, C.; Rudolph, C.; Krügel, U.; Hengstler, J.G.; Illes, P. P2 receptor-mediated stimulation of the PI3-K/Akt-pathway in vivo. Glia 2009, 57, 1031–1045. [Google Scholar] [CrossRef]
- Wypych, D.; Pomorski, P. P2Y1 nucleotide receptor silencing and its effect on glioma C6 calcium signaling. Acta Biochim. Pol. 2012, 59, 711–717. [Google Scholar] [CrossRef]
- Zucoloto, A.Z.; Jenne, C.N. Platelet-neutrophil interplay: Insights into neutrophil extracellular trap (NET)-driven coagulation in infection. Front. Cardiovasc. Med. 2019, 6, 85. [Google Scholar] [CrossRef]
- Briasoulis, A.; Telila, T.; Palla, M.; Siasos, G.; Tousoulis, D. P2Y12 receptor antagonists: Which one to choose? A systematic review and meta-analysis. Curr. Pharm. Des. 2016, 22, 4568–4576. [Google Scholar] [CrossRef]
- Moon, J.Y.; Franchi, F.; Rollini, F.; Angiolillo, D.J. Role for thrombin receptor antagonism with vorapaxar in secondary prevention of atherothrombotic events: From bench to bedside. J. Cardiovasc. Pharmacol. Ther. 2018, 23, 23–37. [Google Scholar] [CrossRef] [PubMed]
- Nel, J.G.; Durandt, C.; Mitchell, T.J.; Feldman, C.; Anderson, R.; Tintinger, G.R. Pneumolysin mediates platelet activation in vitro. Lung 2016, 194, 589–593. [Google Scholar] [CrossRef] [PubMed]
- Nel, J.G.; Durandt, C.; Theron, A.J.; Tintinger, G.R.; Pool, R.; Richards, G.A.; Mitchell, T.J.; Feldman, C.; Anderson, R. Pneumolysin mediates heterotypic aggregation of neutrophils and platelets in vitro. J. Infect. 2017, 74, 599–608. [Google Scholar] [CrossRef] [PubMed]
- Anderson, R.; Theron, A.J.; Nel, J.G.; Durandt, C.; Cholo, M.C.; Feldman, C.; Tintinger, G.R. Clofazimine, but not isoniazid or rifampicin, augments platelet activation in vitro. Front. Pharmacol. 2018, 9, 1335. [Google Scholar] [CrossRef]
- Poeckel, D.; Tausch, L.; Altmann, A.; Feisst, C.; Klinkhardt, U.; Graff, J.; Harder, S.; Werz, O. Induction of central signalling pathways and select functional effects in human platelets by β-boswellic acid. Br. J. Pharmacol. 2005, 146, 514–524. [Google Scholar] [CrossRef]
- O’Brien, K.A.; Stojanovic-Terpo, A.; Hay, N.; Du, X. An important role for Akt3 in platelet activation and thrombosis. Blood 2011, 118, 4215–4223. [Google Scholar] [CrossRef]
- Moroi, A.J.; Watson, S.P. Akt and mitogen-activated protein kinase enhance C-type lectin-like receptor 2-mediated platelet activation by inhibition of glycogen synthase kinase 3α/β. J. Thromb. Haemost. 2015, 13, 1139–1150. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| System | % CD62 Expression | % Inhibition |
|---|---|---|
| BG | 0.61 ± 0.22 | |
| ADP 100 μmol·L−1 | 42.4 ± 8 | |
| MRS 0.155 μmol·L−1 | 30.2 ± 8.7 * | 29 |
| MRS 0.31 μmol·L−1 | 22 ± 8.8 * | 48 |
| PSB 2.5 μmol·L−1 | 26.5 ± 9.3 * | 37 |
| MRS 0.155 μmol·L−1 + PSB 2.5 μmol·L−1 | 17 ± 7.4 * | 60 |
| MRS 0.31 μmol·L−1 + PSB 2.5 μmol·L−1 | 12 ± 6.6 * | 72 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Anderson, R.; Theron, A.J.; Steel, H.C.; Nel, J.G.; Tintinger, G.R. ADP-Mediated Upregulation of Expression of CD62P on Human Platelets Is Critically Dependent on Co-Activation of P2Y1 and P2Y12 Receptors. Pharmaceuticals 2020, 13, 420. https://doi.org/10.3390/ph13120420
Anderson R, Theron AJ, Steel HC, Nel JG, Tintinger GR. ADP-Mediated Upregulation of Expression of CD62P on Human Platelets Is Critically Dependent on Co-Activation of P2Y1 and P2Y12 Receptors. Pharmaceuticals. 2020; 13(12):420. https://doi.org/10.3390/ph13120420
Chicago/Turabian StyleAnderson, Ronald, Annette J. Theron, Helen C. Steel, Jan G. Nel, and Gregory R. Tintinger. 2020. "ADP-Mediated Upregulation of Expression of CD62P on Human Platelets Is Critically Dependent on Co-Activation of P2Y1 and P2Y12 Receptors" Pharmaceuticals 13, no. 12: 420. https://doi.org/10.3390/ph13120420
APA StyleAnderson, R., Theron, A. J., Steel, H. C., Nel, J. G., & Tintinger, G. R. (2020). ADP-Mediated Upregulation of Expression of CD62P on Human Platelets Is Critically Dependent on Co-Activation of P2Y1 and P2Y12 Receptors. Pharmaceuticals, 13(12), 420. https://doi.org/10.3390/ph13120420