Zebrafish Models of Diamond-Blackfan Anemia: A Tool for Understanding the Disease Pathogenesis and Drug Discovery


Abstract
1. Introduction
2. Clinical Features and Associated Genes
3. Mouse and Zebrafish DBA Models
3.1. Mouse Models
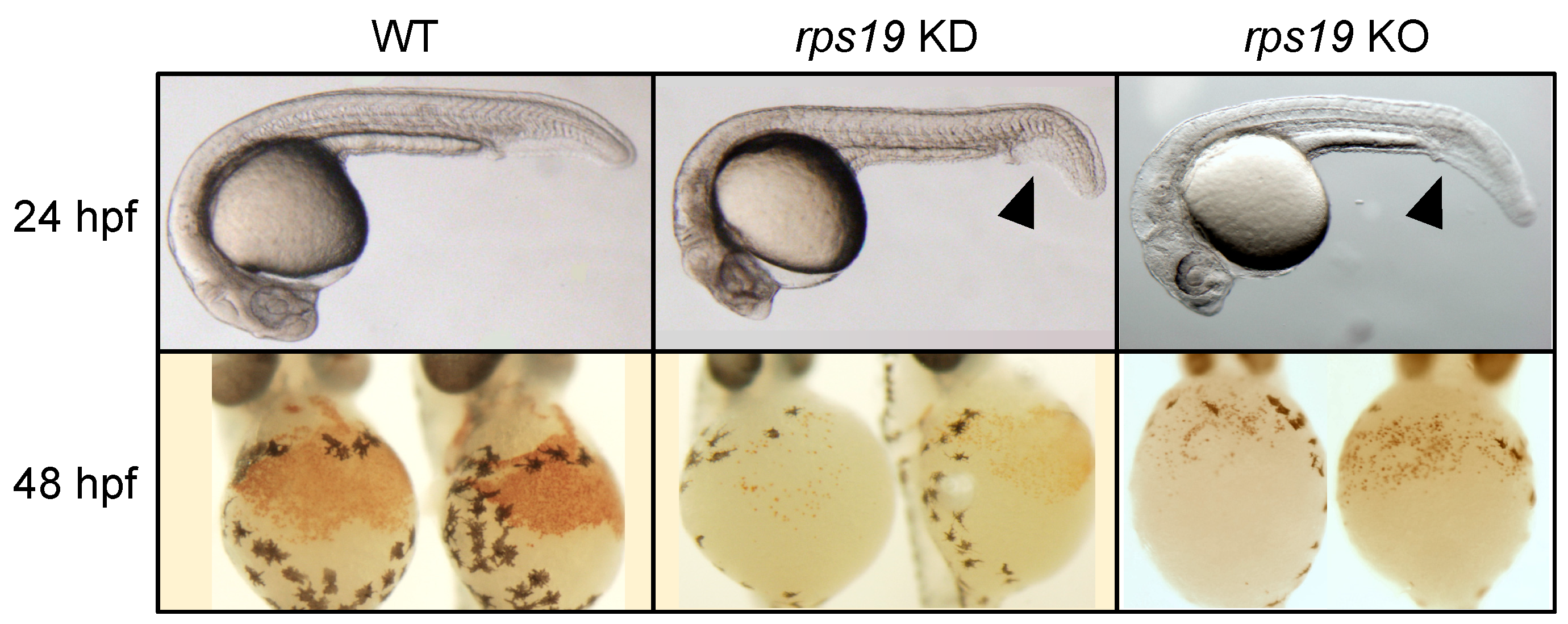
3.2. Zebrafish Models
4. Drug Development
4.1. l-leucine
4.2. Sotatercept
4.3. Trifluoperazine
4.4. SMER28
5. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Narla, A.; Ebert, B.L. Ribosomopathies: Human disorders of ribosome dysfunction. Blood 2010, 115, 3196–3205. [Google Scholar] [CrossRef] [PubMed]
- Aspesi, A.; Ellis, S.R. Rare ribosomopathies: Insights into mechanisms of cancer. Nat. Rev. Cancer 2019, 19, 228–238. [Google Scholar] [CrossRef] [PubMed]
- Diamond, L.K.; Blackfan, K.D. Hypoplastic anemia. Am. J. Dis. Child. 1938, 56, 464–467. [Google Scholar]
- Boria, I.; Garelli, E.; Gazda, H.T.; Aspesi, A.; Quarello, P.; Pavesi, E.; Ferrante, D.; Meerpohl, J.J.; Kartal, M.; Da Costa, L.; et al. The ribosomal basis of Diamond-Blackfan anemia: Mutation and database update. Hum. Mutat. 2010, 31, 1269–1279. [Google Scholar] [CrossRef] [PubMed]
- Lipton, J.M.; Kudisch, M.; Gross, R.; Nathan, D.G. Defective erythroid progenitor differentiation system in congenital hypoplastic (Diamond-Blackfan) anemia. Blood 1986, 67, 962–968. [Google Scholar] [PubMed]
- Garçon, L.; Ge, J.; Manjunath, S.H.; Mills, J.A.; Apicella, M.; Parikh, S.; Sullivan, L.M.; Podsakoff, G.M.; Gadue, P.; French, D.L.; et al. Ribosomal and hematopoietic defects in induced pluripotent stem cells derived from Diamond Blackfan anemia patients. Blood 2013, 122, 912–921. [Google Scholar] [CrossRef] [PubMed]
- Matsson, H.; Davey, E.J.; Draptchinskaia, N.; Hamaguchi, I.; Ooka, A.; Levéen, P.; Forsberg, E.; Karlsson, S.; Dahl, N. Targeted disruption of the ribosomal protein S19 gene is lethal prior to implantation. Mol. Cell. Biol. 2004, 24, 4032–4037. [Google Scholar] [CrossRef]
- Jaako, P.; Flygare, J.; Olsson, K.; Quere, R.; Ehinger, M.; Henson, A.; Ellis, S.; Schambach, A.; Baum, C.; Richter, J.; et al. Mice with ribosomal protein S19 deficiency develop bone marrow failure and symptoms like patients with Diamond-Blackfan anemia. Blood 2011, 118, 6087–6096. [Google Scholar] [CrossRef] [PubMed]
- Ablain, J.; Zon, L.I. Of fish and men: Using zebrafish to fight human diseases. Trends Cell Biol. 2013, 23, 584–586. [Google Scholar] [CrossRef]
- Oyarbide, U.; Topczewski, J.; Corey, S.J. Peering through zebrafish to understand inherited bone marrow failure syndromes. Haematologica 2019, 104, 13–24. [Google Scholar] [CrossRef]
- Amsterdam, A.; Sadler, K.C.; Lai, K.; Farrington, S.; Bronson, R.T.; Lees, J.A.; Hopkins, N. Many ribosomal protein genes are cancer genes in zebrafish. PLoS Biol. 2004, 2, E139. [Google Scholar] [CrossRef] [PubMed]
- Uechi, T.; Nakajima, Y.; Nakao, A.; Torihara, H.; Chakraborty, A.; Inoue, K.; Kenmochi, N. Ribosomal protein gene knockdown causes developmental defects in zebrafish. PLoS ONE 2006, 1, e37. [Google Scholar] [CrossRef] [PubMed]
- Hwang, W.Y.; Fu, Y.; Reyon, D.; Maeder, M.L.; Tsai, S.Q.; Sander, J.D.; Peterson, R.T.; Yeh, J.R.; Joung, J.K. Efficient genome editing in zebrafish using a CRISPR-Cas system. Nat. Biotechnol. 2013, 31, 227–229. [Google Scholar] [CrossRef] [PubMed]
- Uechi, T.; Nakajima, Y.; Chakraborty, A.; Torihara, H.; Higa, S.; Kenmochi, N. Deficiency of ribosomal protein S19 during early embryogenesis leads to reduction of erythrocytes in a zebrafish model of Diamond-Blackfan anemia. Hum. Mol. Genet. 2008, 17, 3204–3211. [Google Scholar] [CrossRef] [PubMed]
- Vlachos, A.; Ball, S.; Dahl, N.; Alter, B.P.; Sheth, S.; Ramenghi, U.; Meerpohl, J.; Karlsson, S.; Liu, J.M.; Leblanc, T.; et al. Diagnosing and treating Diamond Blackfan anaemia: Results of an international clinical consensus conference. Br. J. Haematol. 2008, 142, 859–876. [Google Scholar] [CrossRef] [PubMed]
- Lipton, J.M.; Atsidaftos, E.; Zyskind, I.; Vlachos, A. Improving clinical care and elucidating the pathophysiology of Diamond Blackfan anemia: An update from the Diamond Blackfan Anemia Registry. Pediatr. Blood Cancer 2006, 46, 558–564. [Google Scholar] [CrossRef] [PubMed]
- Draptchinskaia, N.; Gustavsson, P.; Andersson, B.; Pettersson, M.; Willig, T.N.; Dianzani, I.; Ball, S.; Tchernia, G.; Klar, J.; Matsson, H.; et al. The gene encoding ribosomal protein S19 is mutated in Diamond-Blackfan anaemia. Nat. Genet. 1999, 21, 169–175. [Google Scholar] [CrossRef] [PubMed]
- Ulirsch, J.C.; Verboon, J.M.; Kazerounian, S.; Guo, M.H.; Yuan, D.; Ludwig, L.S.; Handsaker, R.E.; Abdulhay, N.J.; Fiorini, C.; Genovese, G.; et al. The genetic landscape of Diamond-Blackfan anemia. Am. J. Hum. Genet. 2018, 103, 930–947. [Google Scholar] [CrossRef] [PubMed]
- Sankaran, V.G.; Ghazvinian, R.; Do, R.; Thiru, P.; Vergilio, J.A.; Beggs, A.H.; Sieff, C.A.; Orkin, S.H.; Nathan, D.G.; Lander, E.S.; et al. Exome sequencing identifies GATA1 mutations resulting in Diamond-Blackfan anemia. J. Clin. Investig. 2012, 122, 2439–2443. [Google Scholar] [CrossRef]
- Gripp, K.W.; Curry, C.; Olney, A.H.; Sandoval, C.; Fisher, J.; Chong, J.X.; Pilchman, L.; Sahraoui, R.; Stabley, D.L.; Sol-Church, K. Diamond-Blackfan anemia with mandibulofacial dystostosis is heterogeneous, including the novel DBA genes TSR2 and RPS28. Am. J. Med. Genet. A. 2014, 164, 2240–2249. [Google Scholar] [CrossRef]
- Ebert, B.; Lipton, J.M. Diamond Blackfan anemia and ribosome biogenesis: Introduction. Semin. Hematol. 2011, 48, 73–74. [Google Scholar] [CrossRef] [PubMed]
- Chakraborty, A.; Uechi, T.; Kenmochi, N. Guarding the ‘translation apparatus’: Defective ribosome biogenesis and the p53 signaling pathway. Wiley Interdiscip. Rev. RNA 2011, 2, 507–522. [Google Scholar] [CrossRef] [PubMed]
- Danilova, N.; Sakamoto, K.M.; Lin, S. Ribosomal protein S19 deficiency in zebrafish leads to developmental abnormalities and defective erythropoiesis through activation of p53 protein family. Blood 2008, 112, 5228–5237. [Google Scholar] [CrossRef] [PubMed]
- McGowan, K.A.; Li, J.Z.; Park, C.Y.; Beaudry, V.; Tabor, H.K.; Sabnis, A.J.; Zhang, W.; Fuchs, H.; de Angelis, M.H.; Myers, R.M.; et al. Ribosomal mutations cause p53-mediated dark skin and pleiotropic effects. Nat. Genet. 2008, 40, 963–970. [Google Scholar] [CrossRef] [PubMed]
- Torihara, H.; Uechi, T.; Chakraborty, A.; Shinya, M.; Sakai, N.; Kenmochi, N. Erythropoiesis failure due to RPS19 deficiency is independent of an activated Tp53 response in a zebrafish model of Diamond-Blackfan anaemia. Br. J. Haematol. 2011, 152, 648–654. [Google Scholar] [CrossRef] [PubMed]
- Chakraborty, A.; Uechi, T.; Nakajima, Y.; Gazda, H.T.; O’Donohue, M.F.; Gleizes, P.E.; Kenmochi, N. Cross talk between TP53 and c-Myc in the pathophysiology of Diamond-Blackfan anemia: Evidence from RPL11-deficient in vivo and in vitro models. Biochem. Biophys. Res. Commun. 2018, 495, 1839–1845. [Google Scholar] [CrossRef] [PubMed]
- Ludwig, L.S.; Gazda, H.T.; Eng, J.C.; Eichhorn, S.W.; Thiru, P.; Ghazvinian, R.; George, T.I.; Gotlib, J.R.; Beggs, A.H.; Sieff, C.A.; et al. Altered translation of GATA1 in Diamond-Blackfan anemia. Nat. Med. 2014, 20, 748–753. [Google Scholar] [CrossRef] [PubMed]
- Khajuria, R.K.; Munschauer, M.; Ulirsch, J.C.; Fiorini, C.; Ludwig, L.S.; McFarland, S.K.; Abdulhay, N.J.; Specht, H.; Keshishian, H.; Mani, D.R.; et al. Ribosome levels selectively regulate translation and lineage commitment in human hematopoiesis. Cell 2018, 173, 90–103. [Google Scholar] [CrossRef] [PubMed]
- Devlin, E.E.; Dacosta, L.; Mohandas, N.; Elliott, G.; Bodine, D.M. A transgenic mouse model demonstrates a dominant negative effect of a point mutation in the RPS19 gene associated with Diamond-Blackfan anemia. Blood 2010, 116, 2826–2835. [Google Scholar] [CrossRef]
- Morgado-Palacin, L.; Varetti, G.; Llanos, S.; Gómez-López, G.; Martinez, D.; Serrano, M. Partial loss of Rpl11 in adult mice recapitulates Diamond-Blackfan anemia and promotes lymphomagenesis. Cell Rep. 2015, 13, 712–722. [Google Scholar] [CrossRef]
- Yadav, G.V.; Chakraborty, A.; Uechi, T.; Kenmochi, N. Ribosomal protein deficiency causes Tp53-independent erythropoiesis failure in zebrafish. Int. J. Biochem. Cell Biol. 2014, 49, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Duan, J.; Ba, Q.; Wang, Z.; Hao, M.; Li, X.; Hu, P.; Zhang, D.; Zhang, R.; Wang, H. Knockdown of ribosomal protein S7 causes developmental abnormalities via p53 dependent and independent pathways in zebrafish. Int. J. Biochem. Cell Biol. 2011, 43, 1218–1227. [Google Scholar] [CrossRef] [PubMed]
- Taylor, A.M.; Humphries, J.M.; White, R.M.; Murphey, R.D.; Burns, C.E.; Zon, L.I. Hematopoietic defects in rps29 mutant zebrafish depend upon p53 activation. Exp. Hematol. 2012, 40, 228–237. [Google Scholar] [CrossRef] [PubMed]
- Danilova, N.; Sakamoto, K.M.; Lin, S. Ribosomal protein L11 mutation in zebrafish leads to haematopoietic and metabolic defects. Br. J. Haematol. 2011, 152, 217–228. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Ear, J.; Yang, Z.; Morimoto, K.; Zhang, B.; Lin, S. Defects of protein production in erythroid cells revealed in a zebrafish Diamond-Blackfan anemia model for mutation in RPS19. Cell Death Dis. 2014, 5, e1352. [Google Scholar] [CrossRef] [PubMed]
- Payne, E.M.; Virgilio, M.; Narla, A.; Sun, H.; Levine, M.; Paw, B.H.; Berliner, N.; Look, A.T.; Ebert, B.L.; Khanna-Gupta, A. l-leucine improves the anemia and developmental defects associated with Diamond-Blackfan anemia and del(5q) MDS by activating the mTOR pathway. Blood 2012, 120, 2214–2224. [Google Scholar] [CrossRef] [PubMed]
- Jaako, P.; Debnath, S.; Olsson, K.; Bryder, D.; Flygare, J.; Karlsson, S. Dietary l-leucine improves the anemia in a mouse model for Diamond-Blackfan anemia. Blood 2012, 120, 2225–2228. [Google Scholar] [CrossRef] [PubMed]
- Stipanuk, M.H. Leucine and protein synthesis: mTOR and beyond. Nutr. Rev. 2007, 65, 122–129. [Google Scholar] [CrossRef] [PubMed]
- Carrancio, S.; Markovics, J.; Wong, P.; Leisten, J.; Castiglioni, P.; Groza, M.C.; Raymon, H.K.; Heise, C.; Daniel, T.; Chopra, R.; et al. An activin receptor IIA ligand trap promotes erythropoiesis resulting in a rapid induction of red blood cells and haemoglobin. Br. J. Haematol. 2014, 165, 870–882. [Google Scholar] [CrossRef] [PubMed]
- Dussiot, M.; Maciel, T.T.; Fricot, A.; Chartier, C.; Negre, O.; Veiga, J.; Grapton, D.; Paubelle, E.; Payen, E.; Beuzard, Y.; et al. An activin receptor IIA ligand trap corrects ineffective erythropoiesis in beta-thalassemia. Nat. Med. 2014, 20, 398–407. [Google Scholar] [CrossRef]
- Ear, J.; Huang, H.; Wilson, T.; Tehrani, Z.; Lindgren, A.; Sung, V.; Laadem, A.; Daniel, T.O.; Chopra, R.; Lin, S. RAP-011 improves erythropoiesis in zebrafish model of Diamond-Blackfan anemia through antagonizing lefty1. Blood 2015, 126, 880–890. [Google Scholar] [CrossRef] [PubMed]
- Taylor, A.M.; Raiser, D.M.; Humphries, J.M.; Ebert, B.L.; Zon, L.I. Calmodulin inhibition rescues the effects of ribosomal protein deficiency by modulating p53 activity in models of Diamond Blackfan anemia. Blood 2012, 120, 512. [Google Scholar]
- Macari, E.R.; Taylor, A.M.; Raiser, D.M.; Siva, K.; McGrath, K.; Humphries, J.M.; Flygare, J.; Ebert, B.L.; Zon, L.I. Calmodulin inhibition rescues DBA models with ribosomal protein deficiency through reduction of RSK signaling. Blood 2016, 128, 332. [Google Scholar]
- Doulatov, S.; Vo, L.T.; Macari, E.R.; Wahlster, L.; Kinney, M.A.; Taylor, A.M.; Barragan, J.; Gupta, M.; McGrath, K.; Lee, H.Y.; et al. Drug discovery for Diamond-Blackfan anemia using reprogrammed hematopoietic progenitors. Sci. Transl. Med. 2017, 9, eaah5645. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Siva, K.; Rzymski, T.; Johansson, L.; Lundbäck, T.; Villacis, L.N.; Ek, F.; Wang, B.; George, A.J.; Wan, Y.; et al. Small molecule screens identify CDK8-inhibitors as candidate Diamond-Blackfan anemia drugs. Blood 2018, 132, 753. [Google Scholar]
- Flygare, J.; Estrada, V.R.; Shin, C.; Gupta, S.; Lodish, H.F. HIF1α synergizes with glucocorticoids to promote BFU-E progenitor self-renewal. Blood 2011, 117, 3435–3444. [Google Scholar] [CrossRef]
- Lee, H.Y.; Gao, X.; Barrasa, M.I.; Li, H.; Elmes, R.R.; Peters, L.L.; Lodish, H.F. PPAR-α and glucocorticoid receptor synergize to promote erythroid progenitor self-renewal. Nature 2015, 522, 474–477. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
| Disease | Gene | Function | Abnormal Phenotypes | |||
|---|---|---|---|---|---|---|
| Blood | Tumor | Skin | Skeleton | |||
| Diamond-Blackfan anemia (DBA) | RPS19 and other 18 RP genes | ribosomal protein | ++ | + | ||
| X-linked dyskeratosis congenita (DC) | DKC1 | rRNA modification enzyme | ++ | + | ++ | |
| Cartilage-hair hypoplasia (CHH) | RMRP | cleavage of 5.8S rRNA | ++ | + | ++ | ++ |
| Shwachman-Diamond syndrome | SBDS | promoting of 60S subunit maturation | ++ | + | ++ | |
| T-cell acute lymphoblastic leukemia (T-ALL) | RPL5, RPL10, RPL22 | ribosomal protein | ++ | ++ | ++ | |
| 5q- syndrome | RPS14 | ribosomal protein | ++ | + | ||
| Treacher-Collins syndrome | TCOF1, POLR1D, POLR1C | rDNA transcription | ++ | |||
| Isolated congenital asplenia | RPSA | ribosomal protein | ||||
| Compound | Function | Disease Model | Clinical Trial, Patent |
|---|---|---|---|
| L-Leucine [36,37,38] | activator of protein synthesis | hCD34+, zebrafish, mouse | NCT01362595 |
| Sotatercept [39,40,41] | activin receptor type II ligand trap | zebrafish | NCT01464164 |
| Trifluoperazine [42,43] | calmodulin inhibitor | hCD34+, zebrafish, mouse | NCT03966053 |
| SMER28 [44] | inducer of autophagy | iPSC, zebrafish, mouse | N/A |
| CDK8 inhibitor [45] | inhibitor of cyclin-dependent kinase 8 | mouse fetal liver cell | WO2017076968Al |
| Dimethyloxalylglycine * [46] | prolyl hydroxylase inhibitor | mBFU-E | N/A |
| GW7647 *, fenofibrate * [47] | PPAR-a agonist | mBFU-E, hCD34+, mouse | N/A |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Uechi, T.; Kenmochi, N. Zebrafish Models of Diamond-Blackfan Anemia: A Tool for Understanding the Disease Pathogenesis and Drug Discovery. Pharmaceuticals 2019, 12, 151. https://doi.org/10.3390/ph12040151
Uechi T, Kenmochi N. Zebrafish Models of Diamond-Blackfan Anemia: A Tool for Understanding the Disease Pathogenesis and Drug Discovery. Pharmaceuticals. 2019; 12(4):151. https://doi.org/10.3390/ph12040151
Chicago/Turabian StyleUechi, Tamayo, and Naoya Kenmochi. 2019. "Zebrafish Models of Diamond-Blackfan Anemia: A Tool for Understanding the Disease Pathogenesis and Drug Discovery" Pharmaceuticals 12, no. 4: 151. https://doi.org/10.3390/ph12040151
APA StyleUechi, T., & Kenmochi, N. (2019). Zebrafish Models of Diamond-Blackfan Anemia: A Tool for Understanding the Disease Pathogenesis and Drug Discovery. Pharmaceuticals, 12(4), 151. https://doi.org/10.3390/ph12040151