3-Vinylazetidin-2-Ones: Synthesis, Antiproliferative and Tubulin Destabilizing Activity in MCF-7 and MDA-MB-231 Breast Cancer Cells

 ,
, 
Abstract
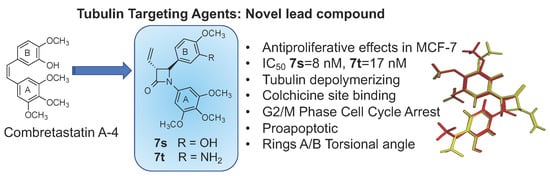
:
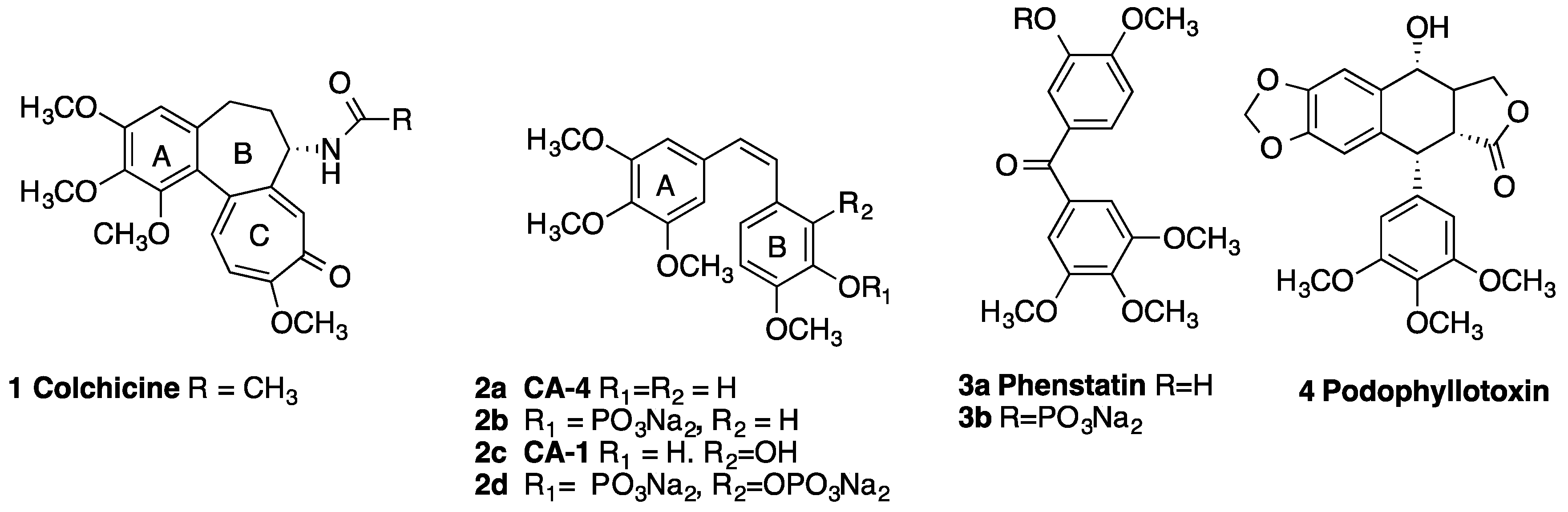
1. Introduction
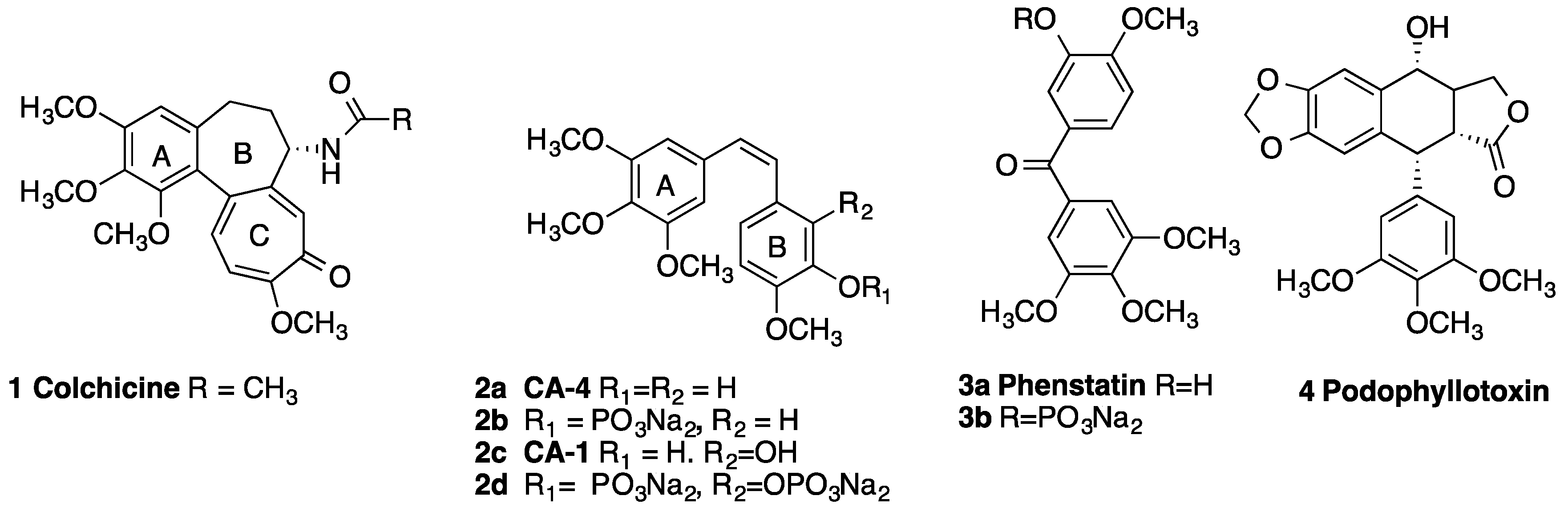
2. Results and Discussion
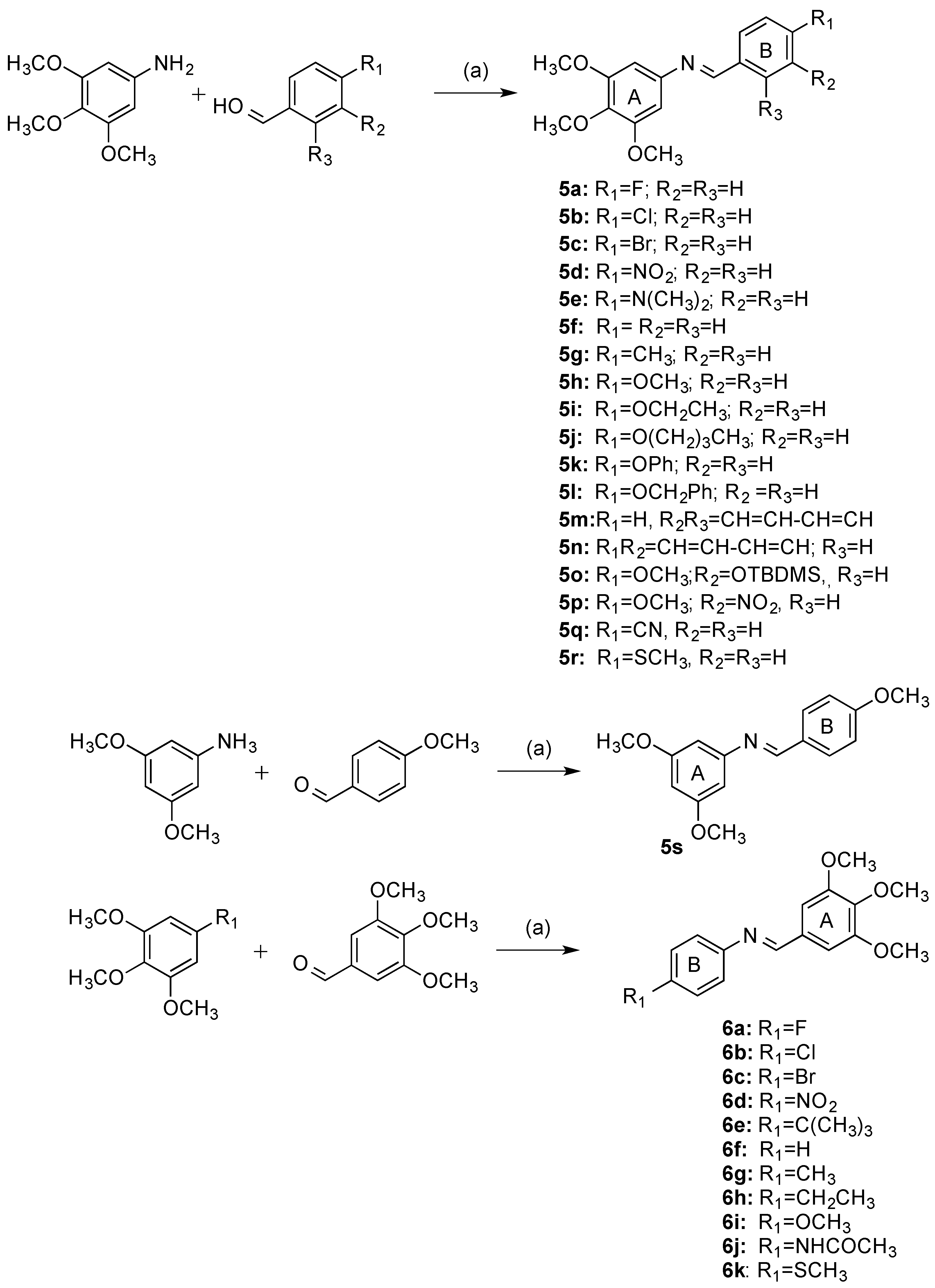
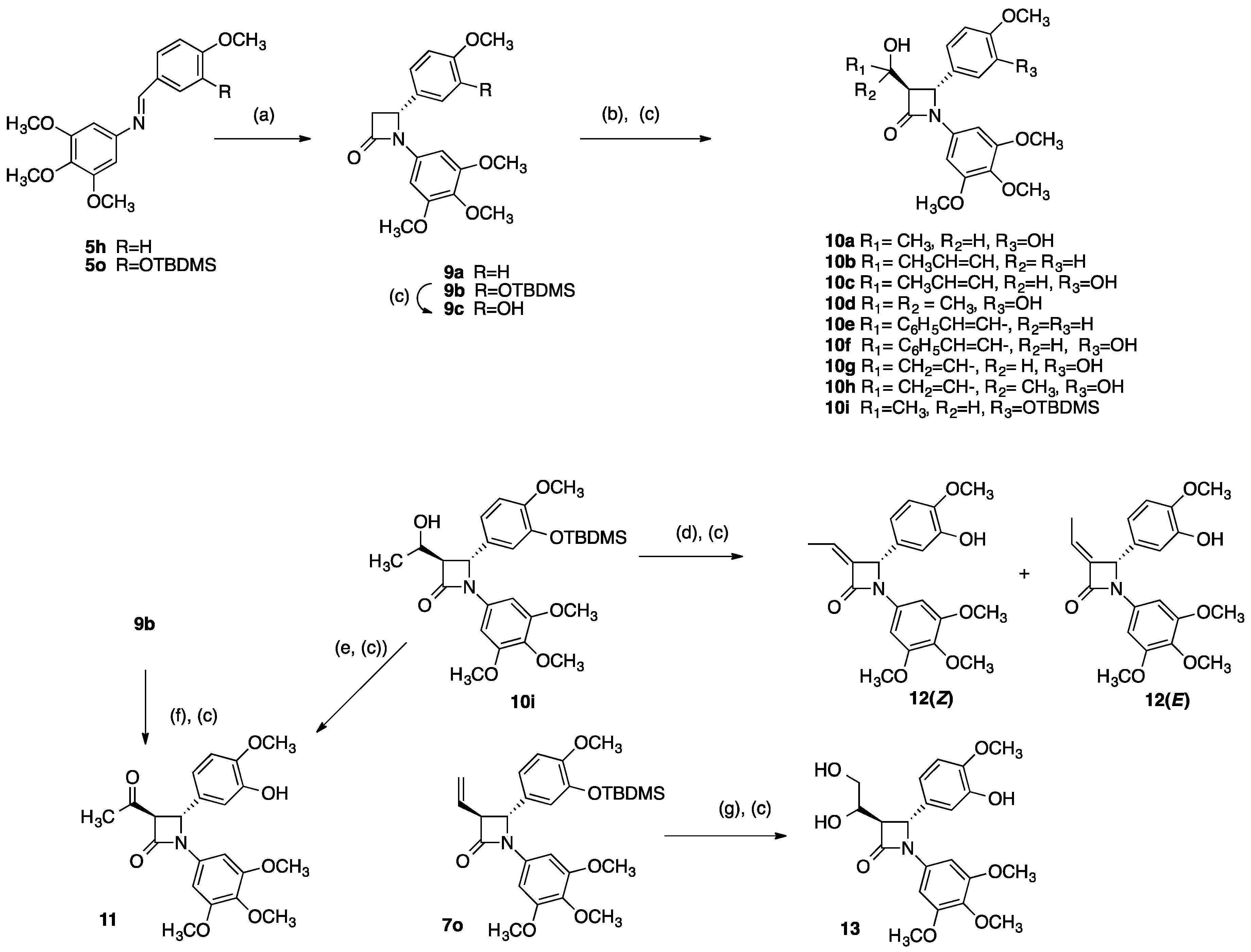
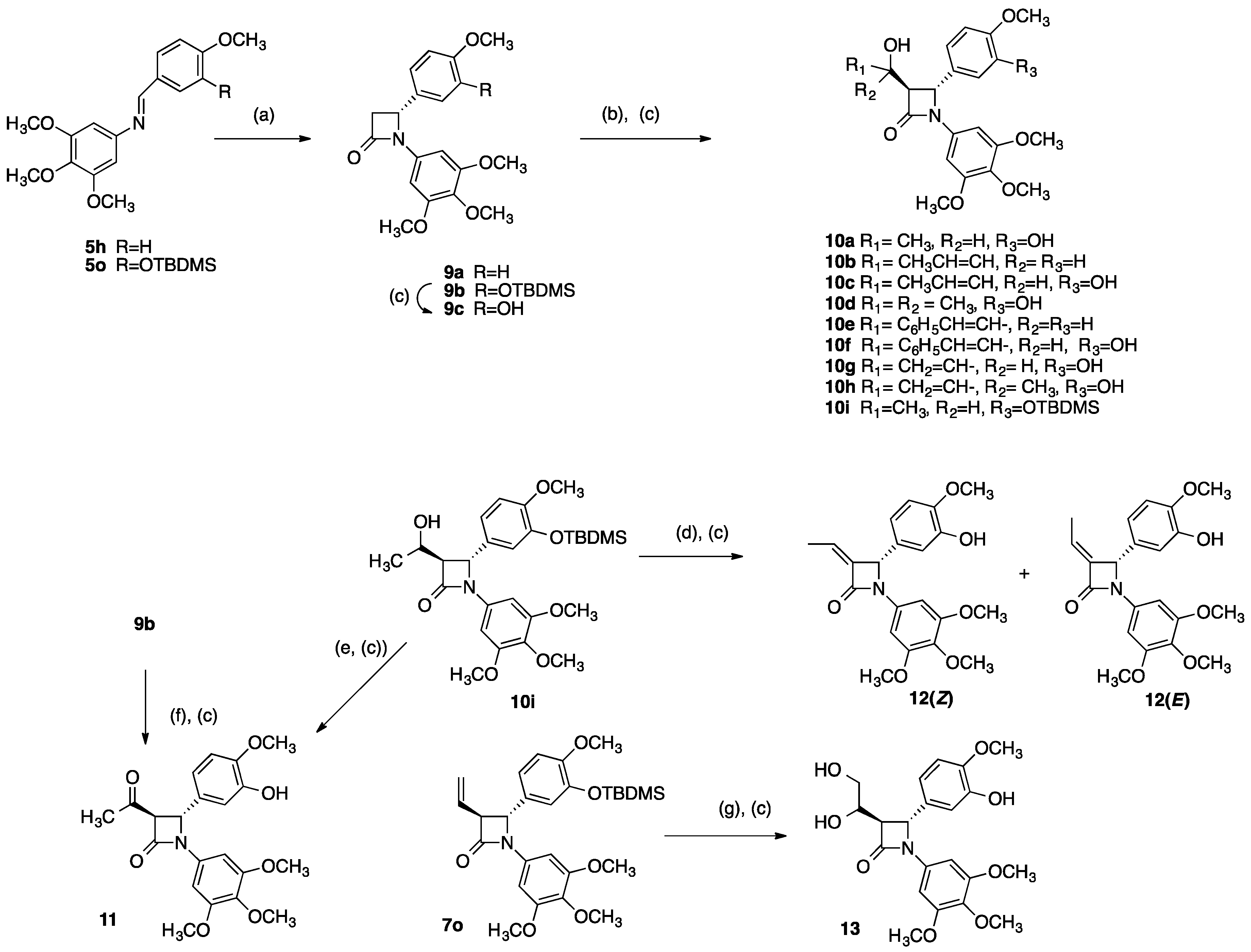
2.1. Chemistry: Synthesis of β-lactams
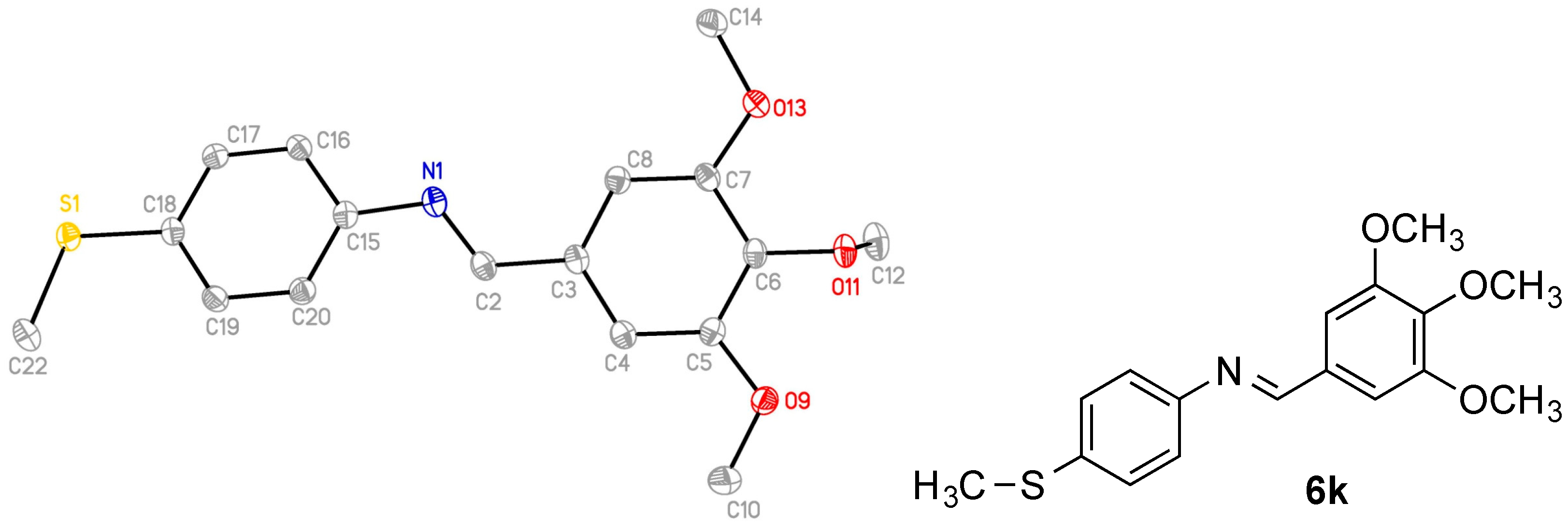
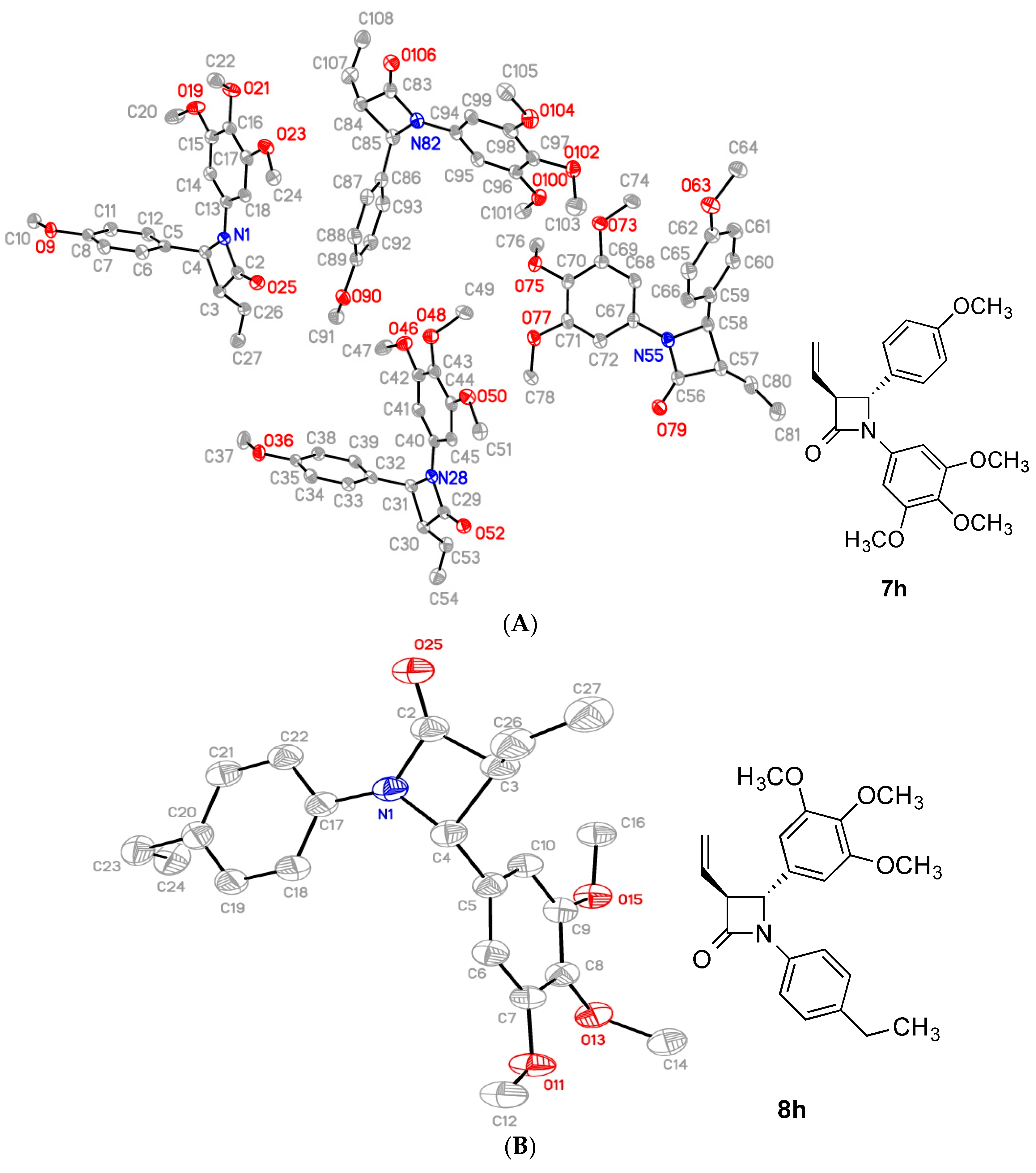
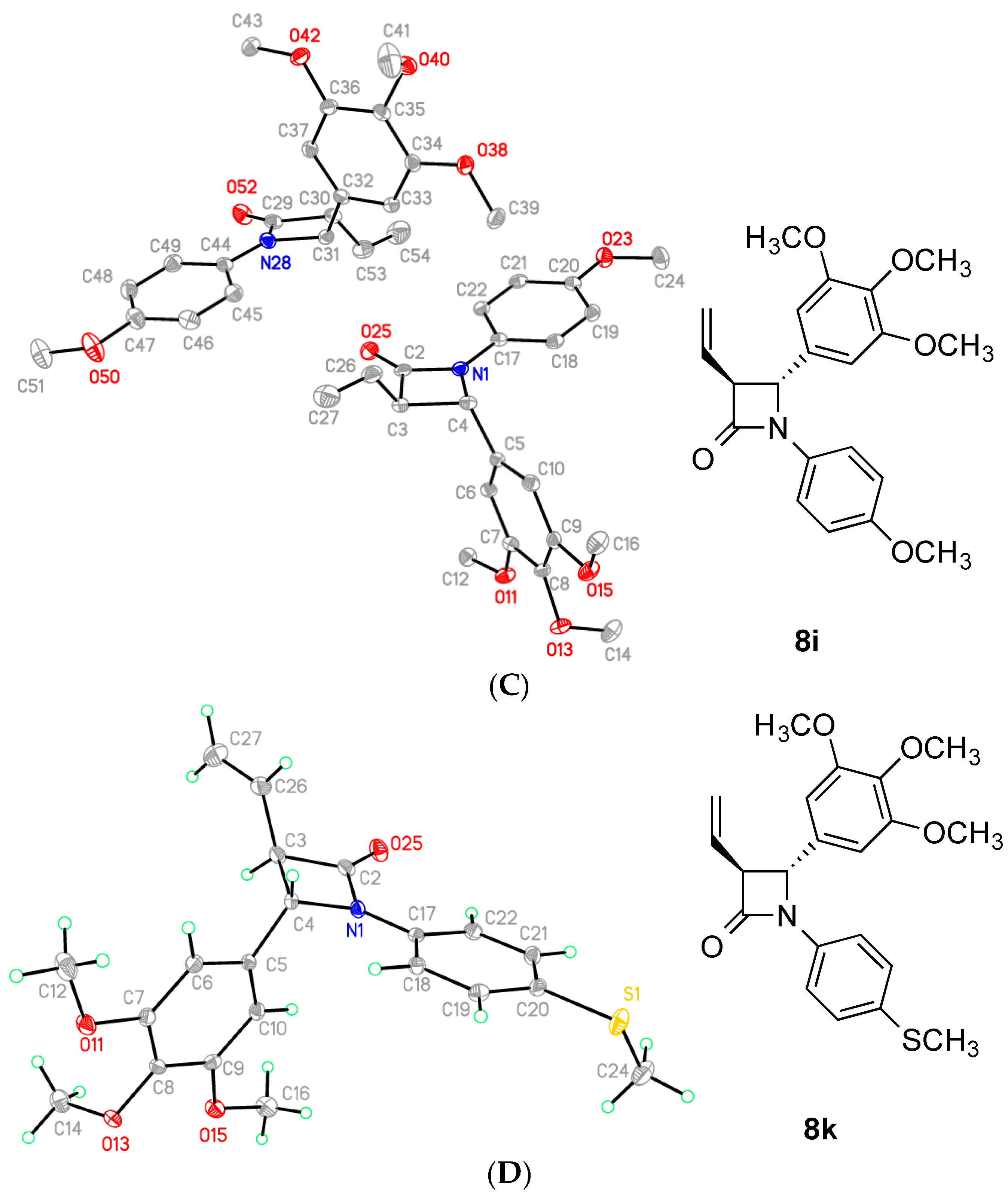
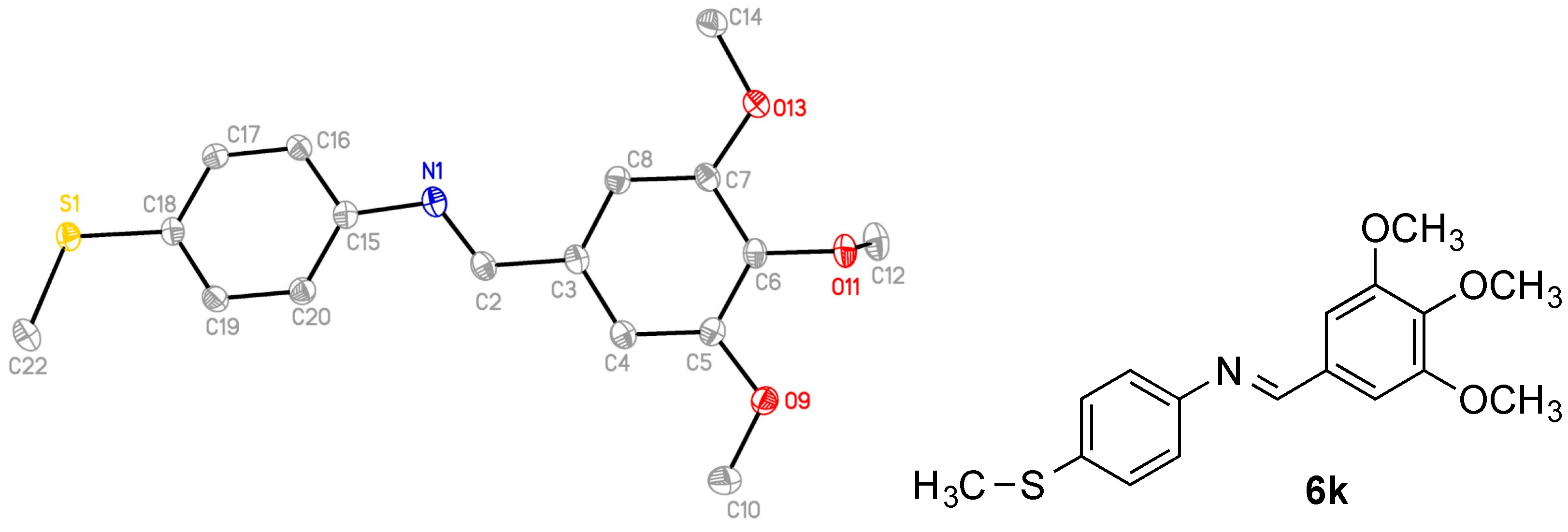
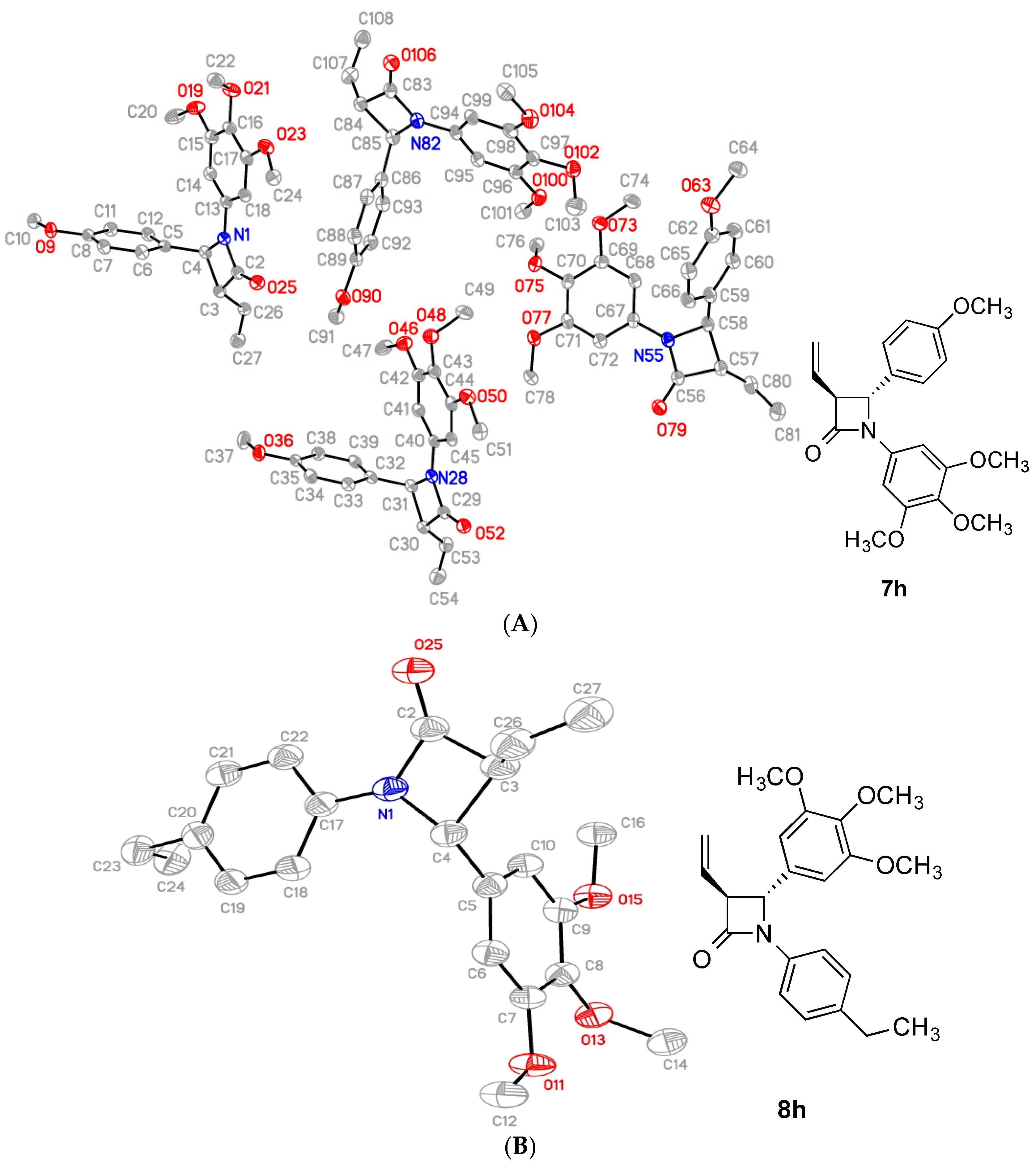
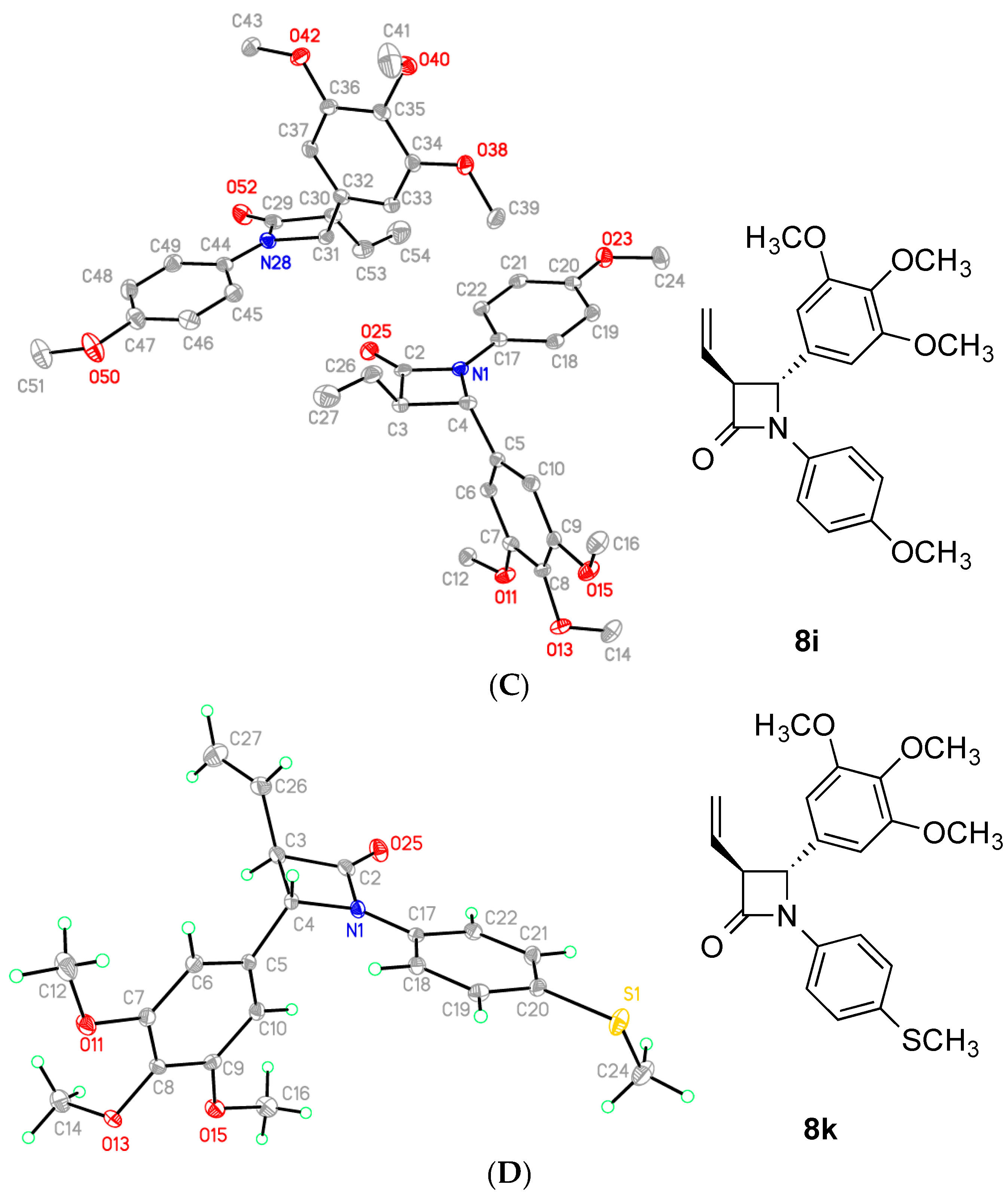
2.2. X-Ray Structural Study
2.3. Biological Results and Discussion
2.3.1. In vitro Antiproliferative Activities
2.3.2. Evaluation of β-Lactams in the NCI60 Cell Line Screen
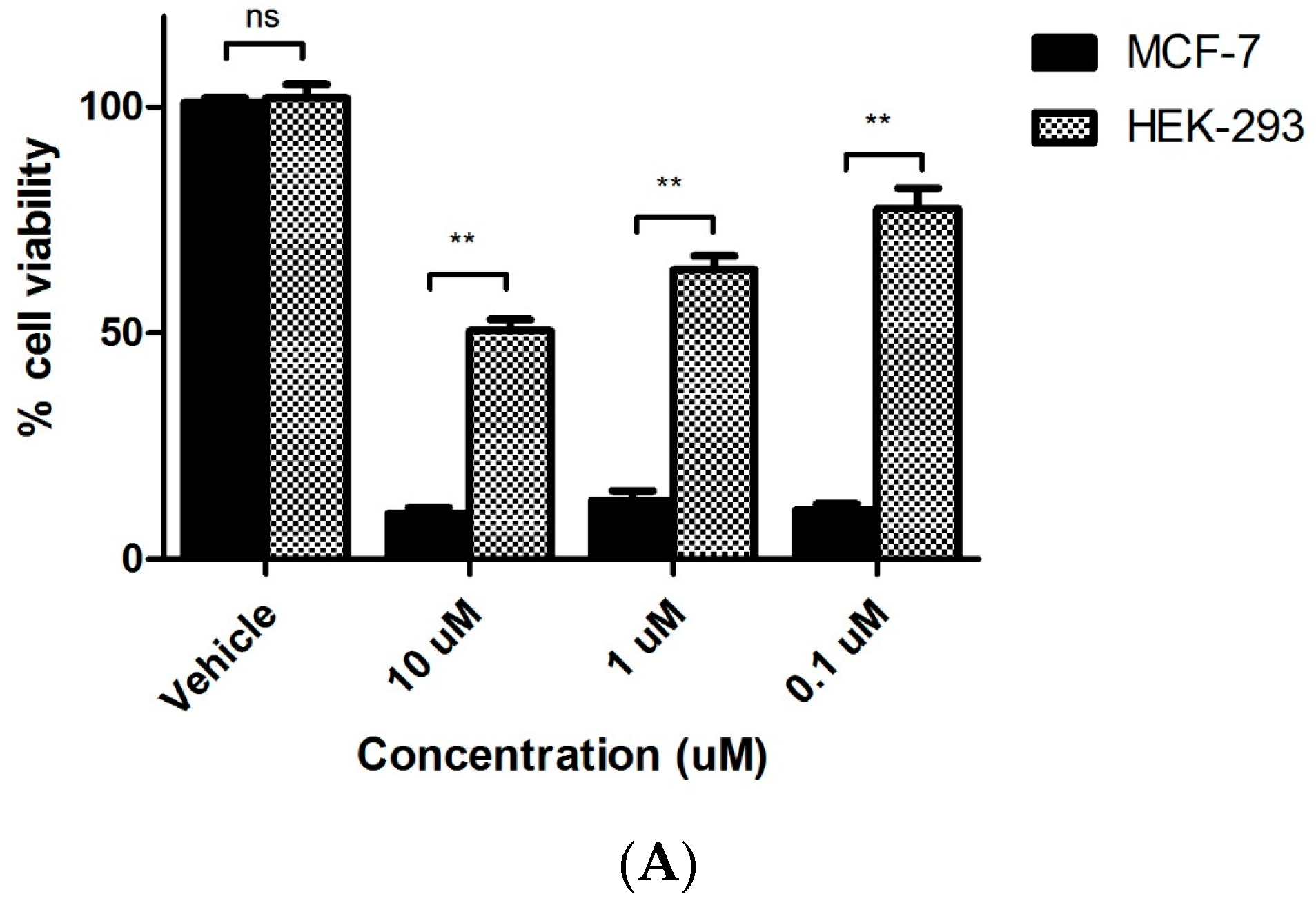
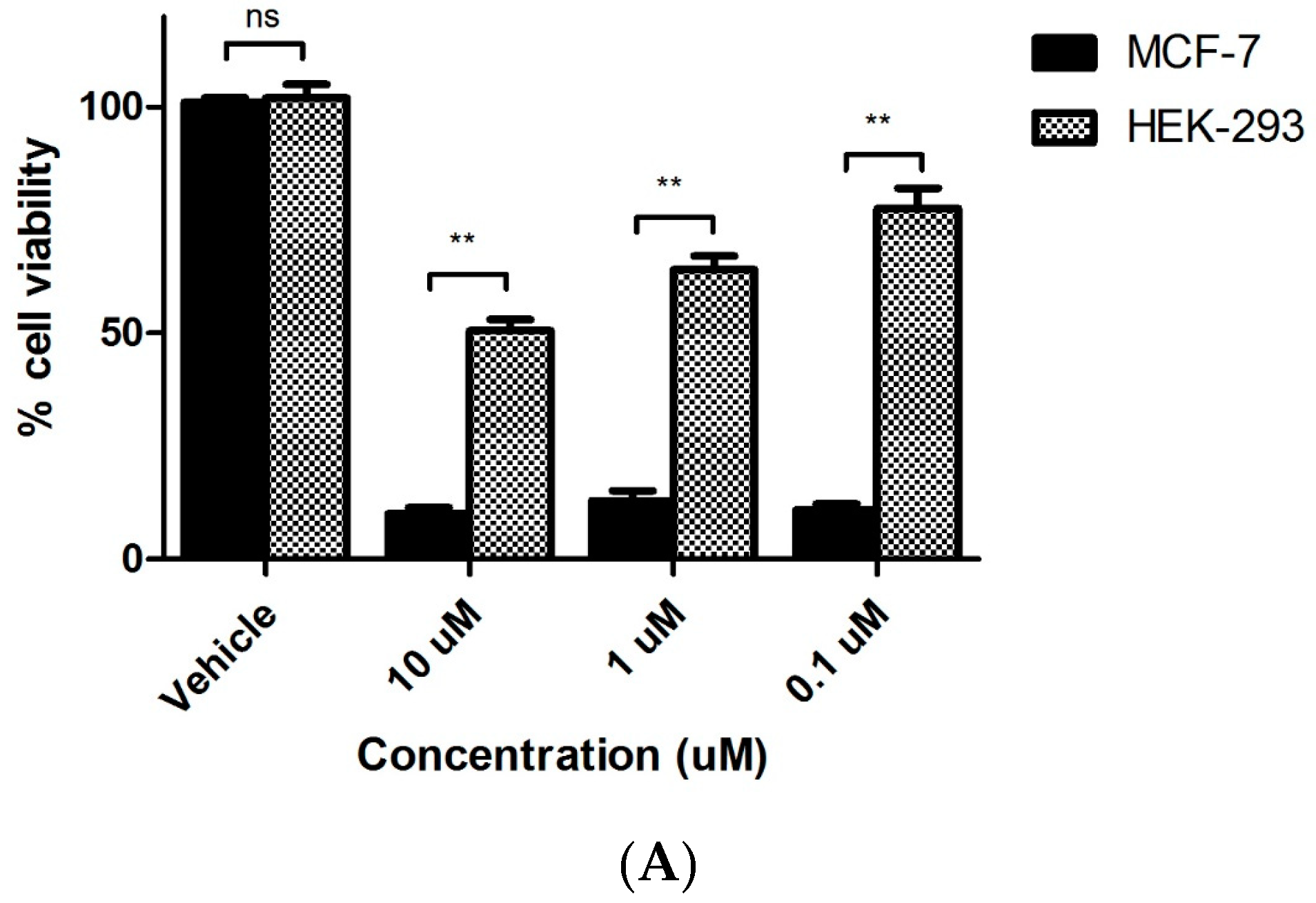
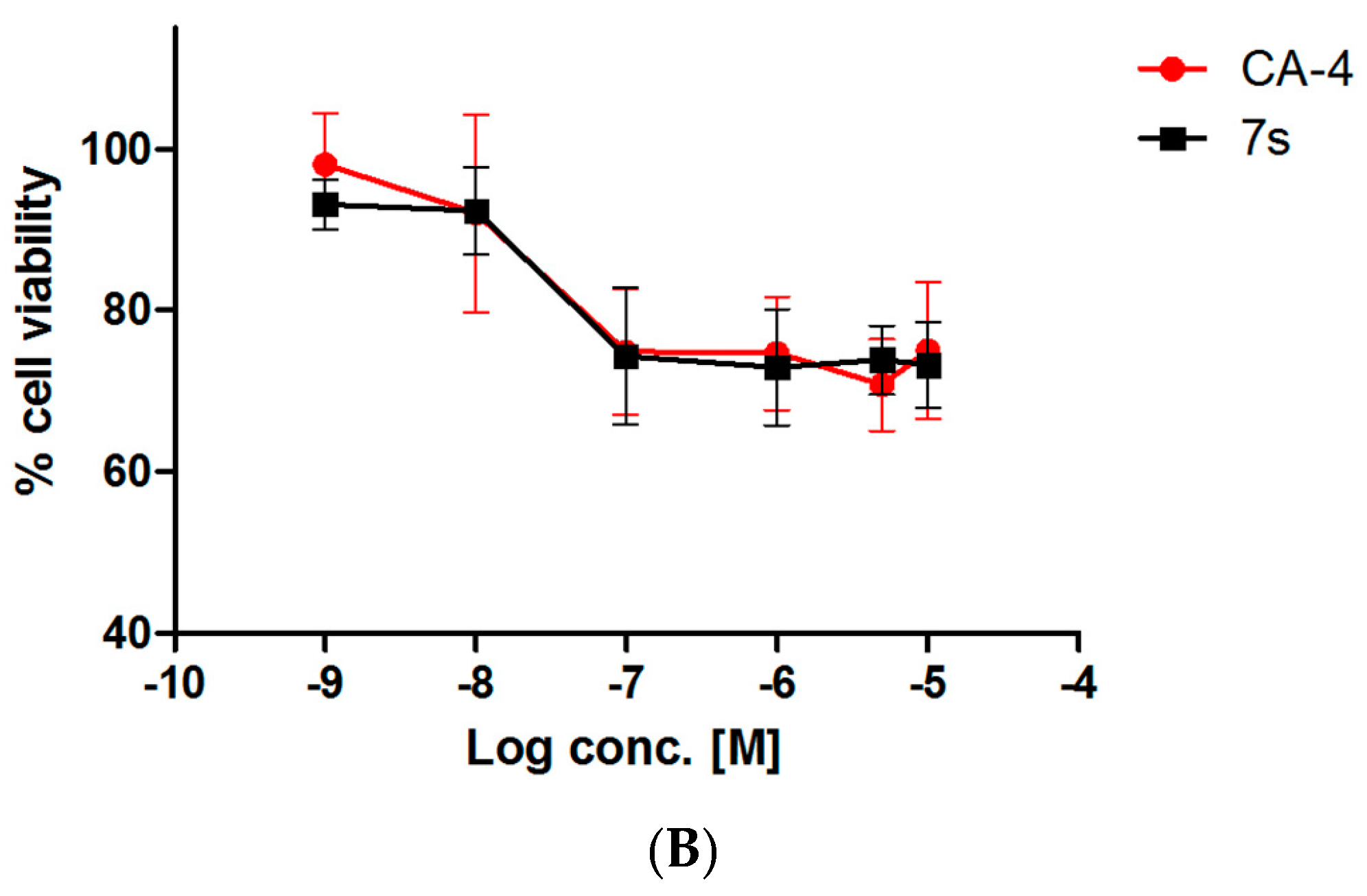
2.3.3. Evaluation of Toxicity of 7s in Normal Murine Mammary Epithelial Cells
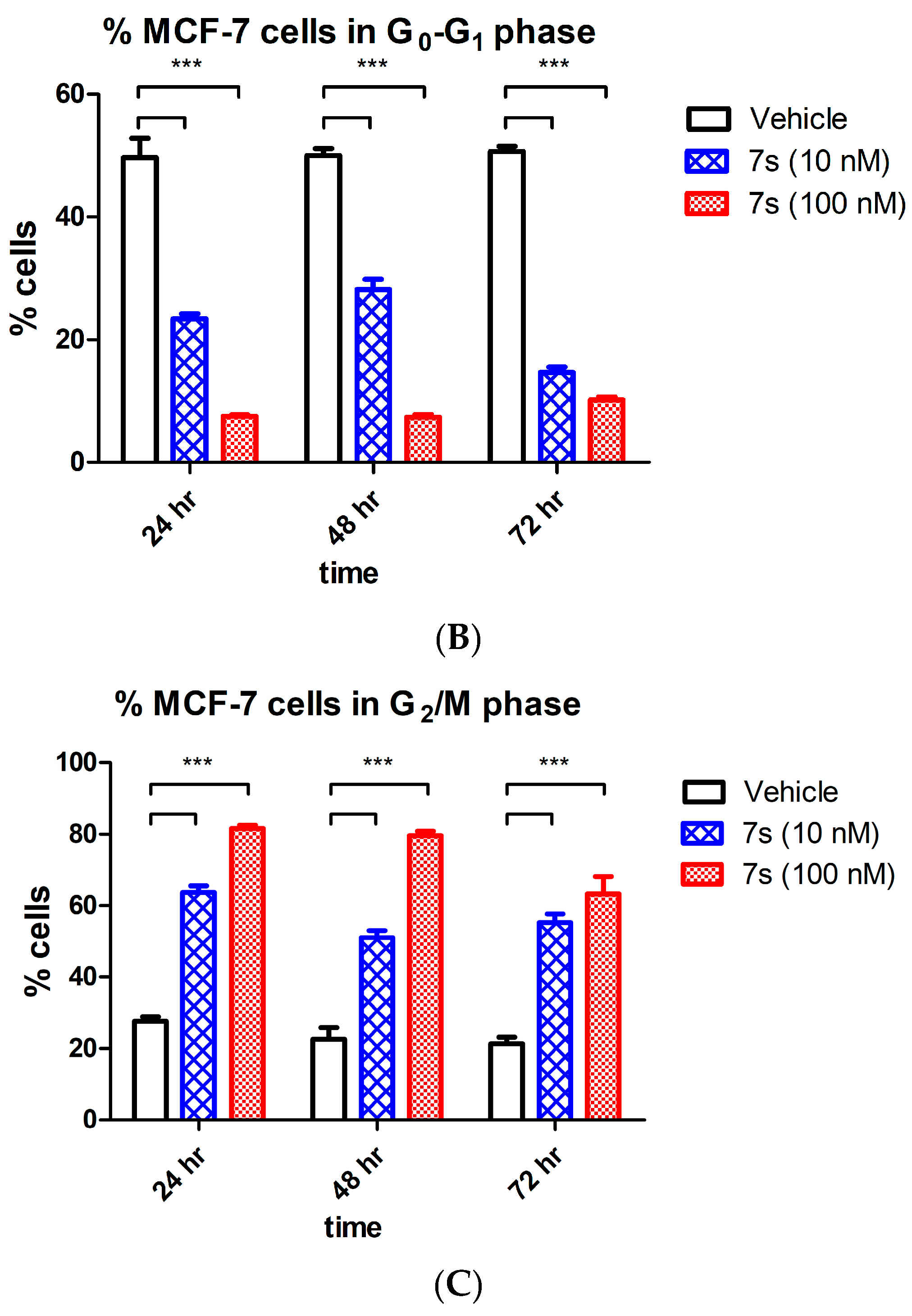
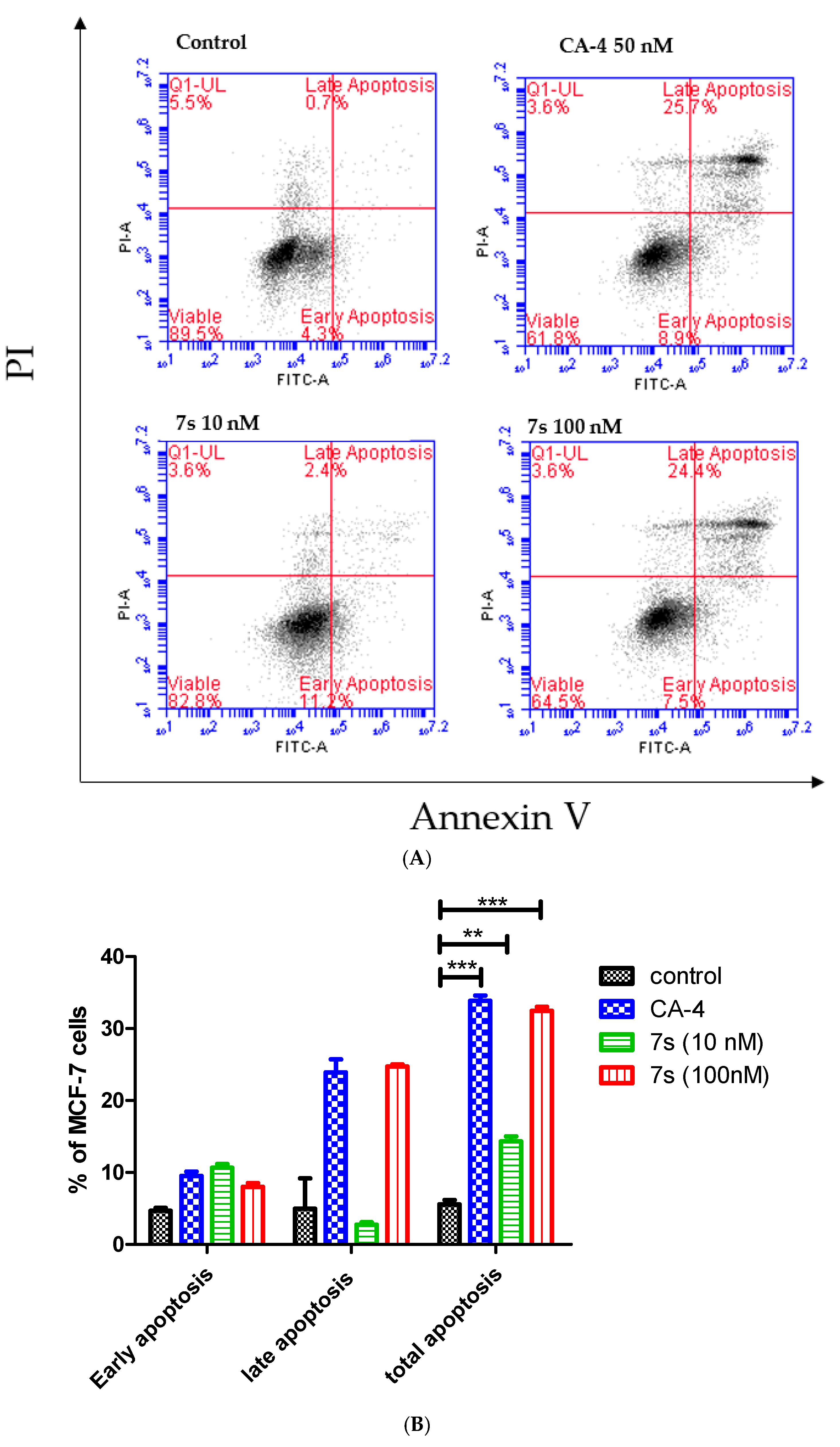
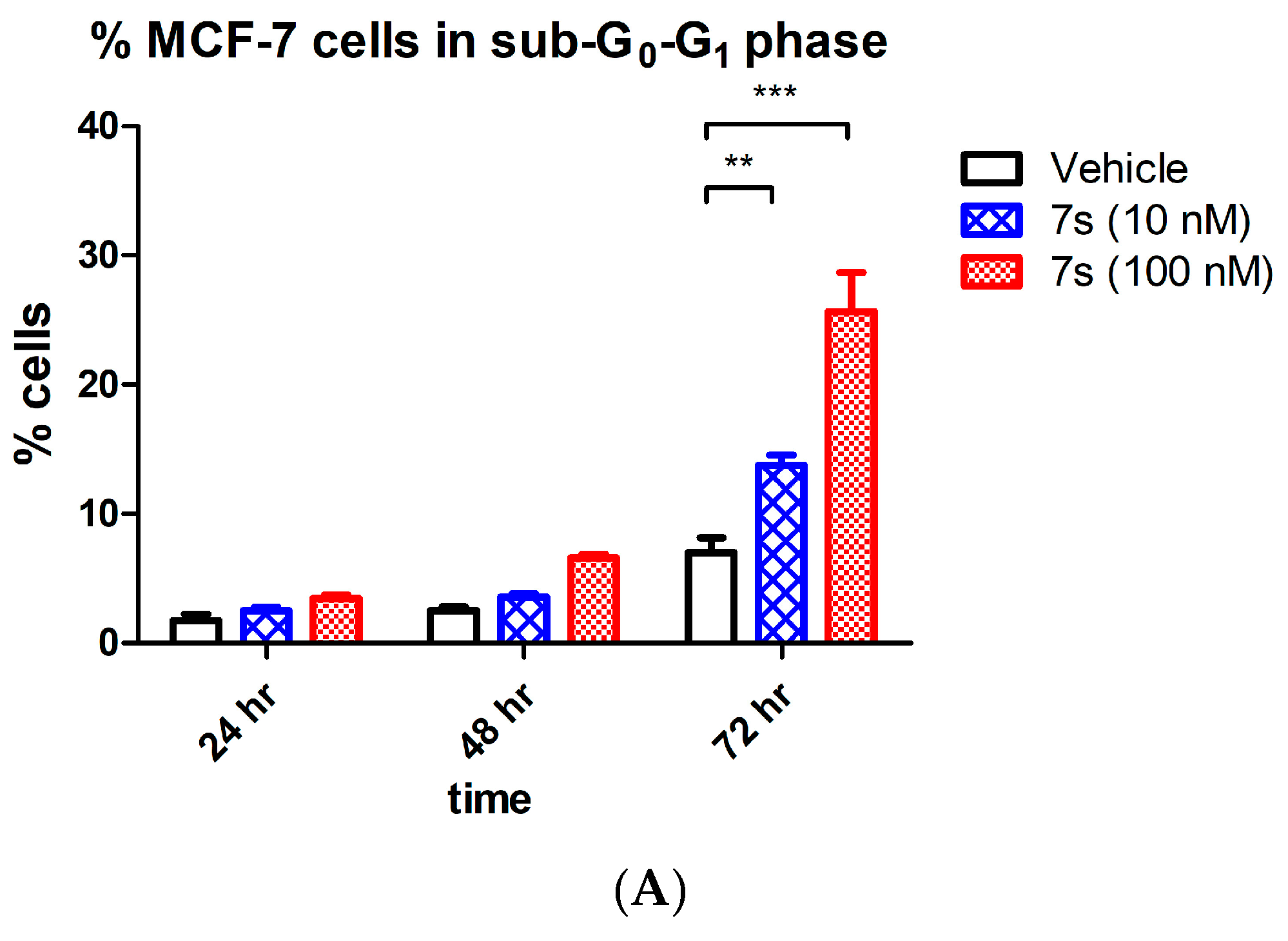
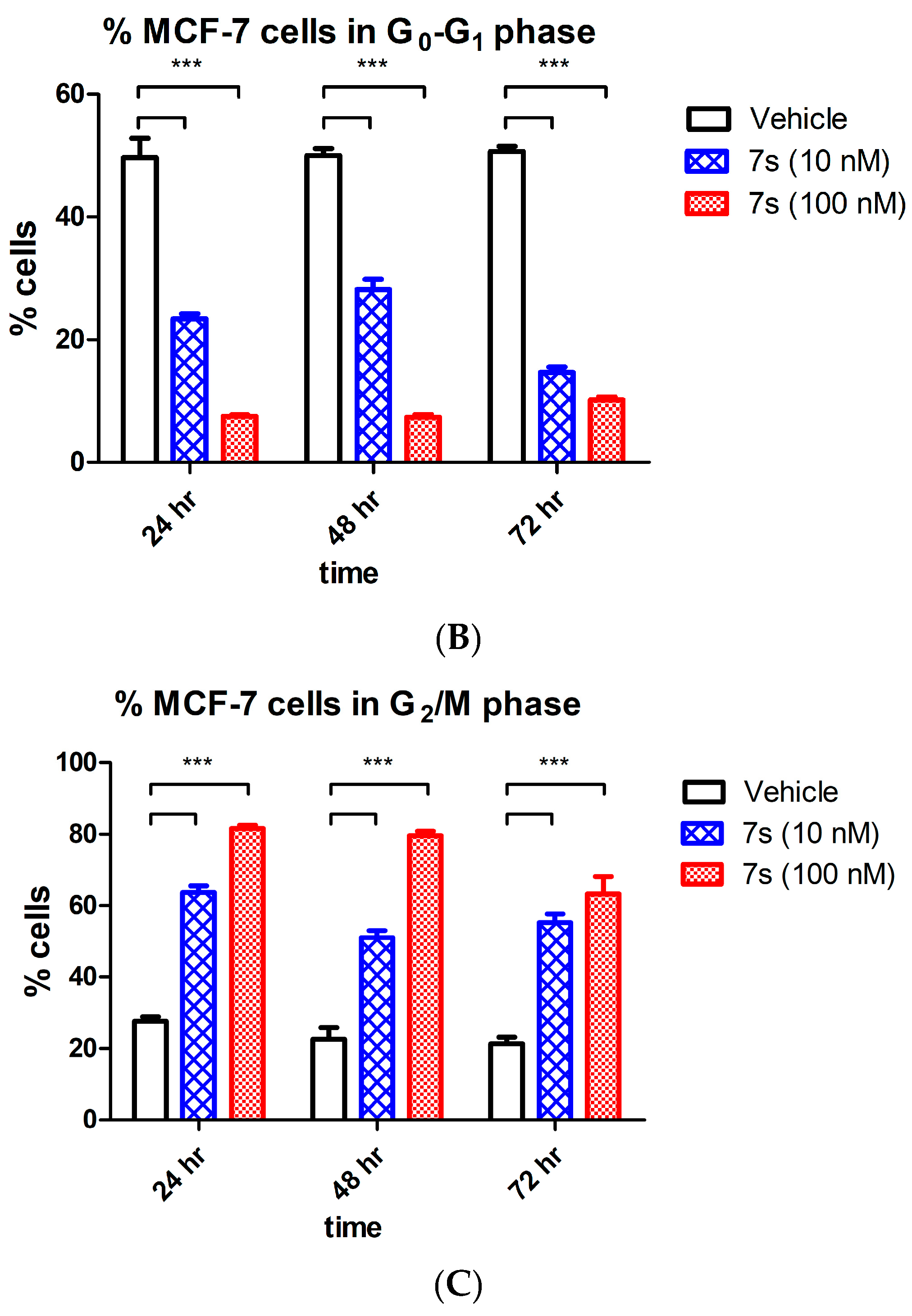
2.3.4. Effect of β-Lactam 7s on Cell Cycle and Apoptosis
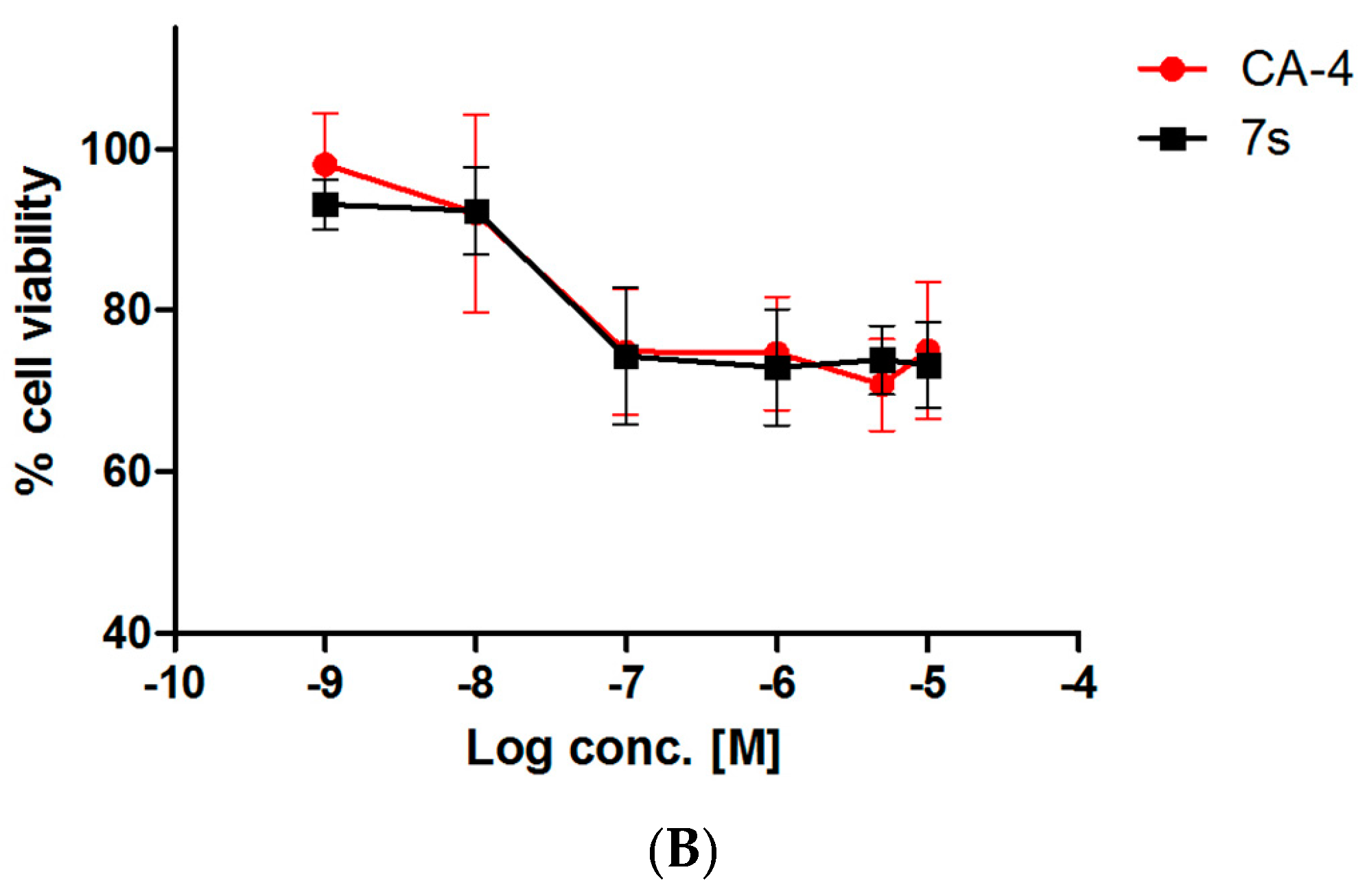
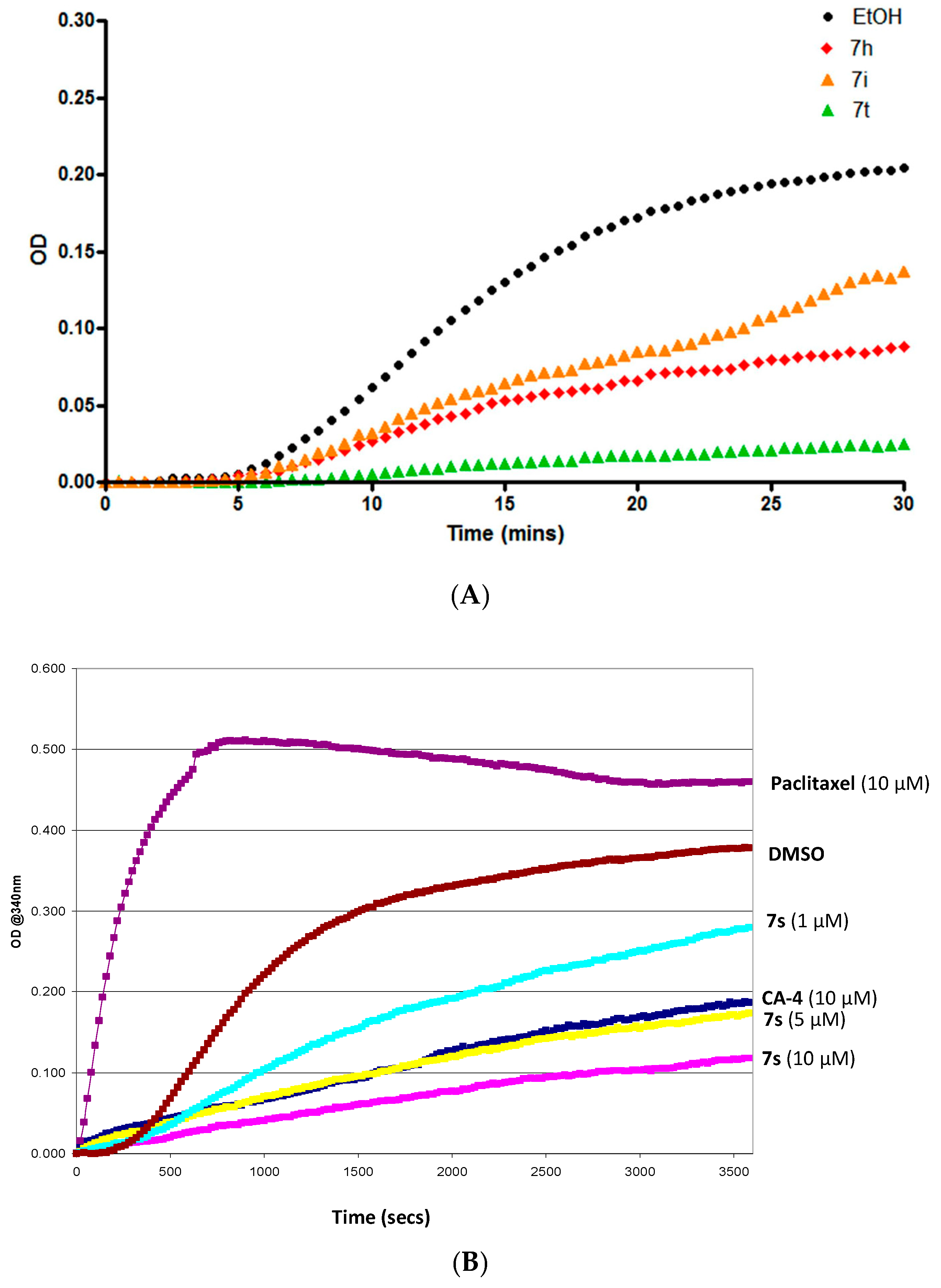
2.3.5. Tubulin Polymerization Studies
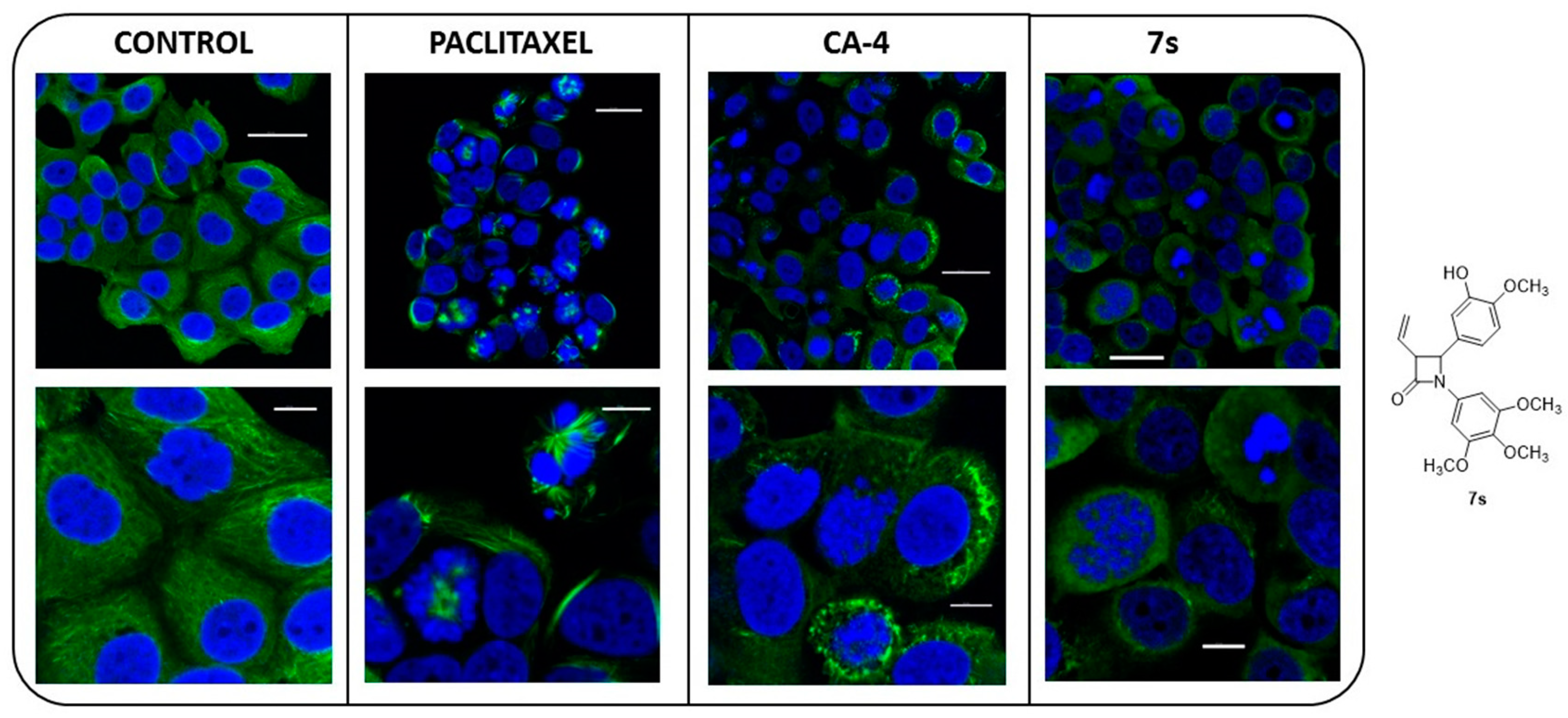
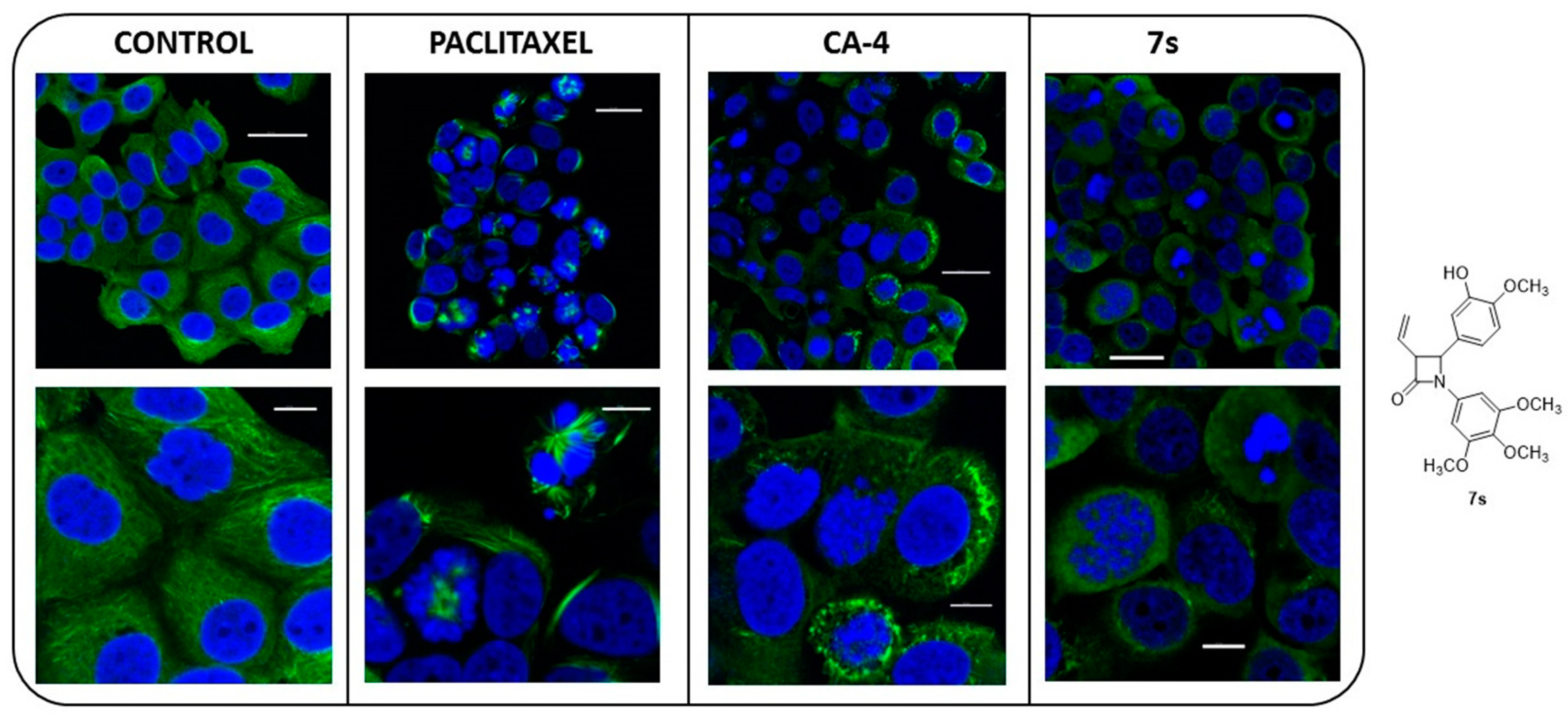
2.3.6. Immunofluorescence Microscopy
2.3.7. Interaction of β-Lactam 7s with Colchicine Binding Site of Tubulin
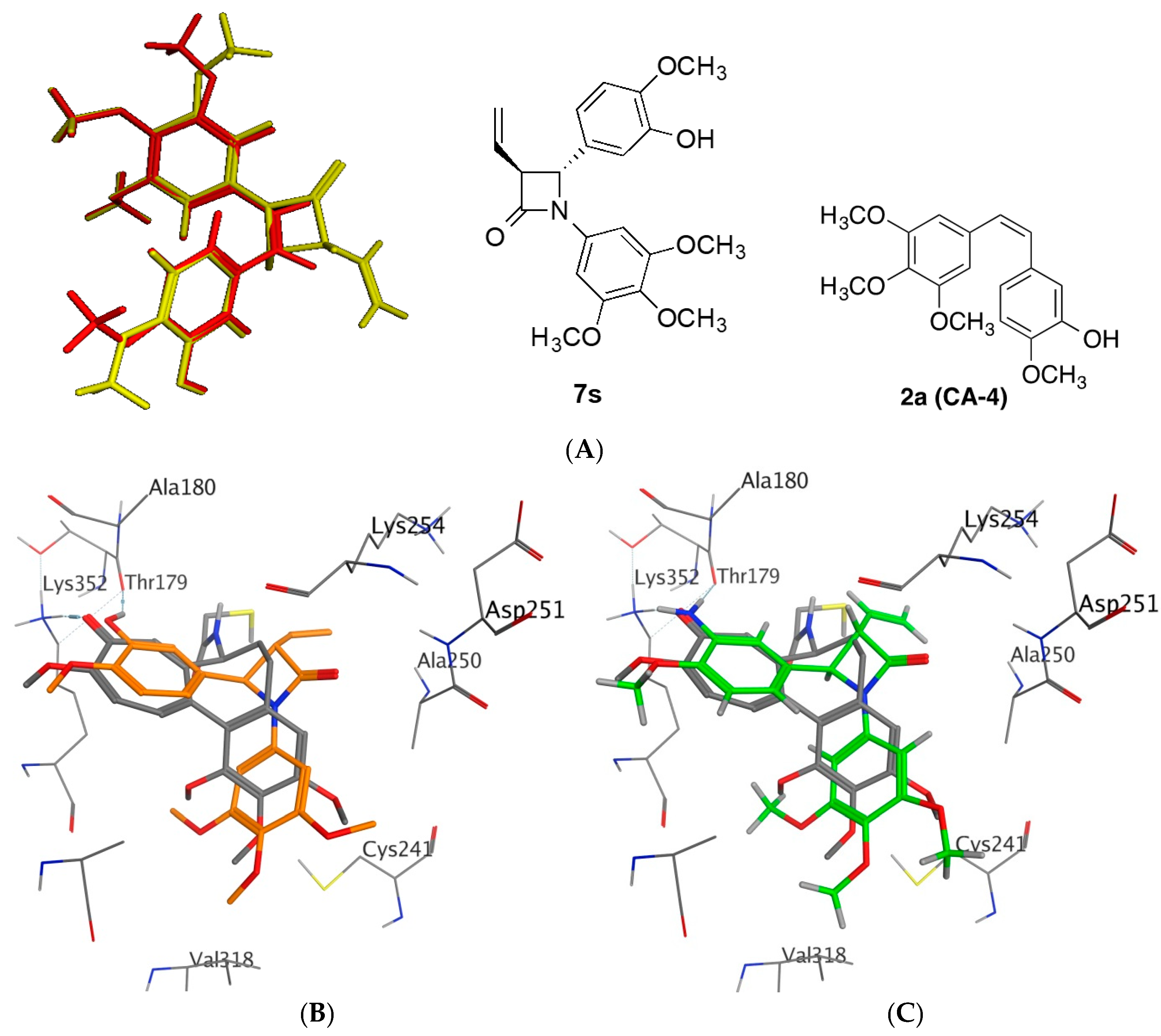
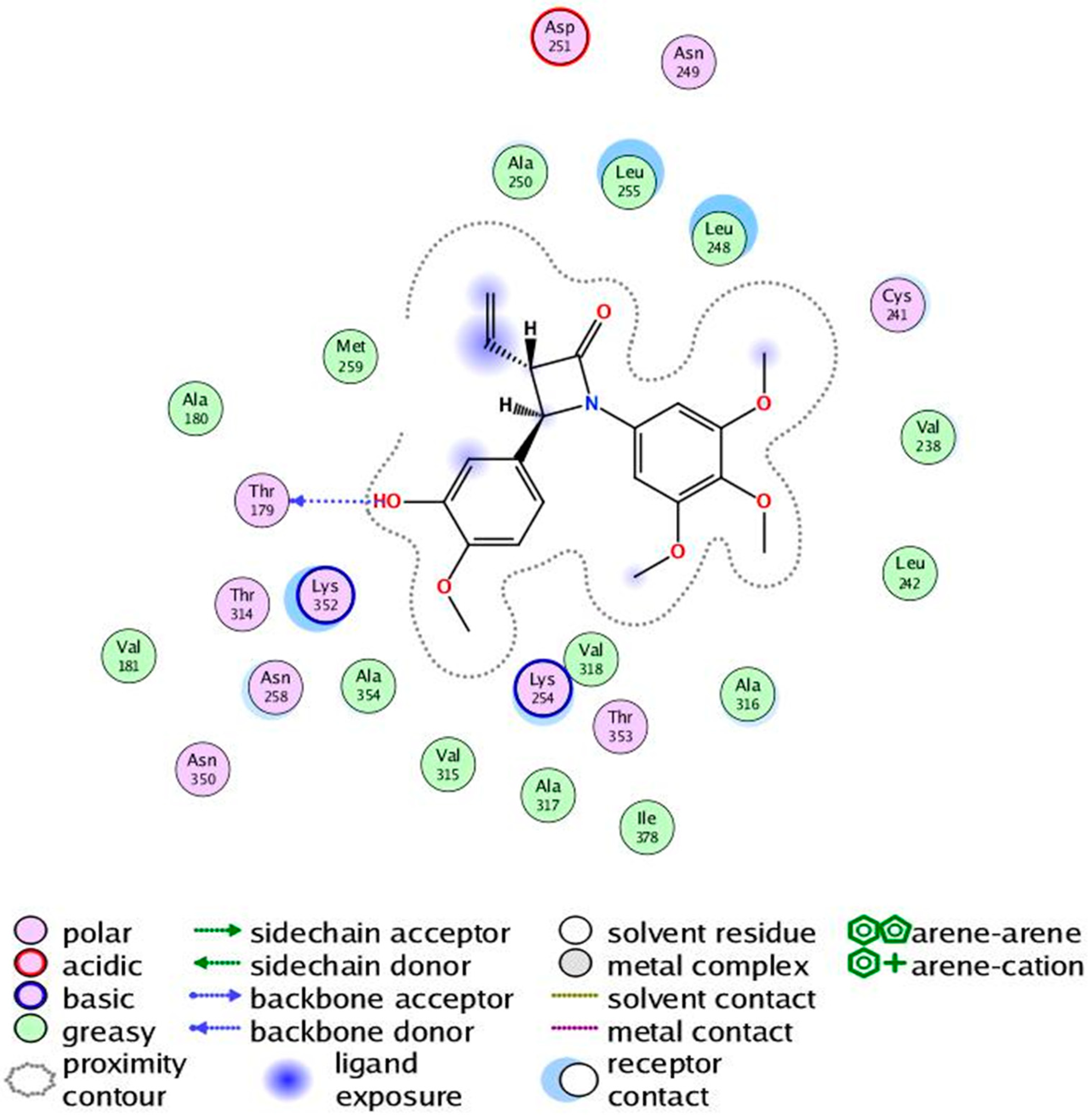
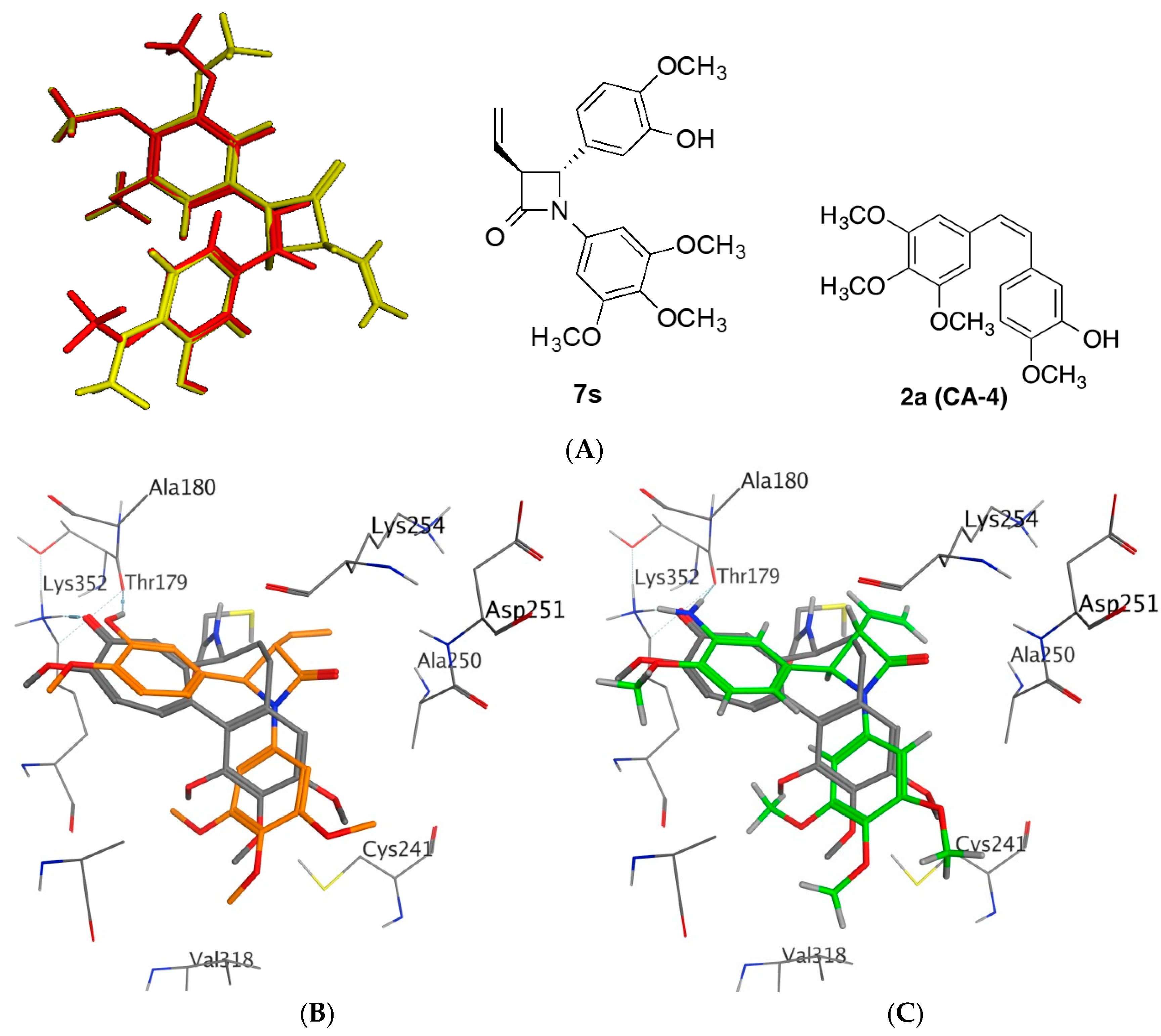
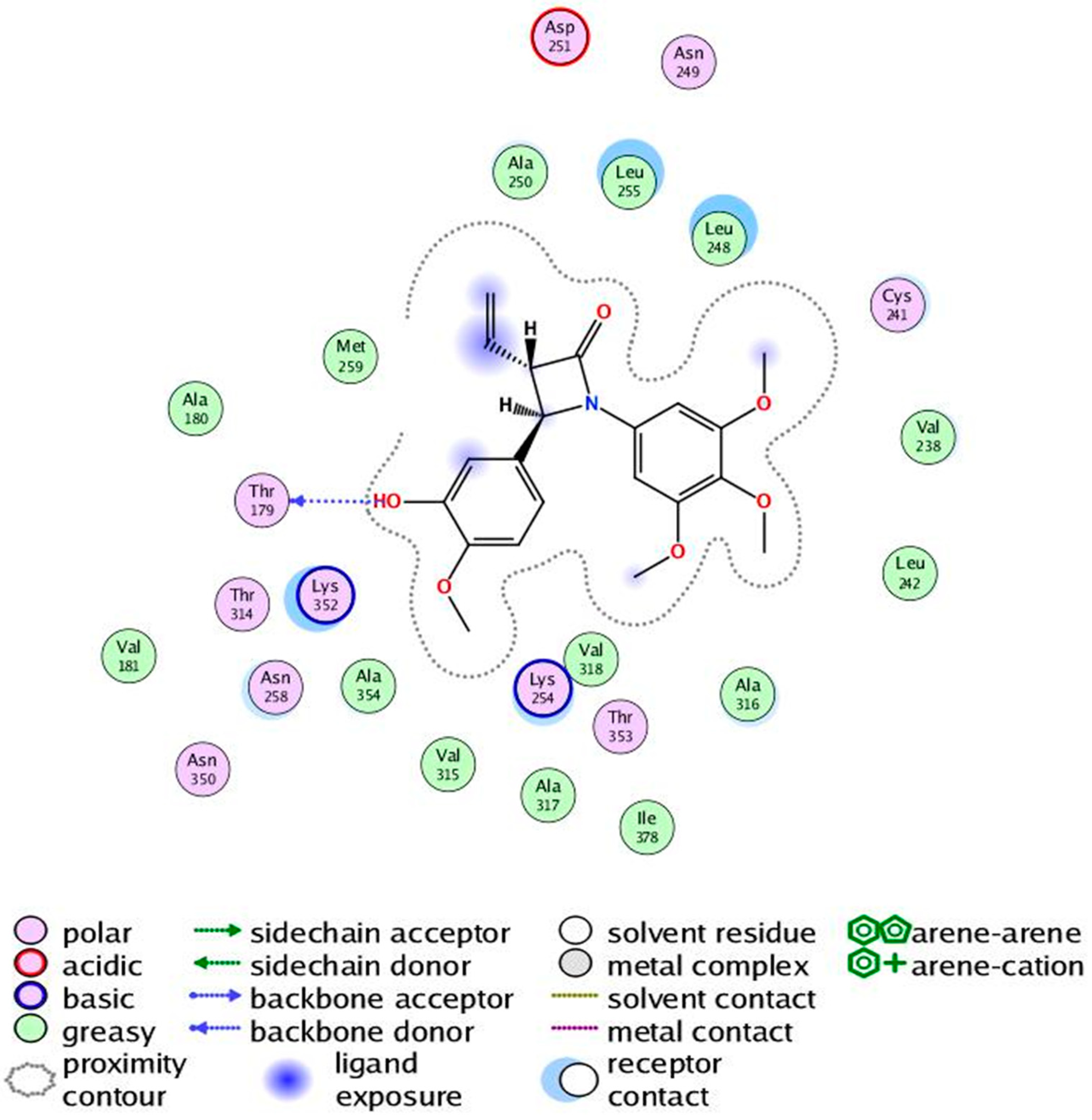
2.4. Molecular Modelling Studies
3. Conclusions
4. Experimental Section
4.1. Chemistry
4.1.1. 3-(tert-Butyldimethylsilyloxy)-4-methoxybenzaldehyde
4.1.2. General Method I: Preparation of Imines 5a–5s, 6a–6k
(E)-1-(4-Butoxyphenyl)-N-(3,4,5-trimethoxyphenyl)methanimine (5j)
(E)-1-(4-Phenoxyphenyl)-N-(3,4,5-trimethoxyphenyl)methanimine (5k)
(E)-1-(4-(Benzyloxy)phenyl)-N-(3,4,5-trimethoxyphenyl)methanimine (5l)
(E)-N-(4-Nitrophenyl)-1-(3,4,5-trimethoxyphenyl)methanimine (6d)
(E)-N-(4-(tert-Butyl)phenyl)-1-(3,4,5-trimethoxyphenyl)methanimine (6e)
4.1.3. General method II: Preparation of 2-azetidinones 7a–7u, 8a–8k
4-(4-Fluorophenyl)-1-(3,4,5-trimethoxyphenyl)-3-vinylazetidin-2-one (7a)
4-(4-Chlorophenyl)-1-(3,4,5-trimethoxyphenyl)-3-vinylazetidin-2-one (7b)
4-(4-Bromophenyl)-1-(3,4,5-trimethoxyphenyl)-3-vinylazetidin-2-one (7c)
4-(4-Nitrophenyl)-1-(3,4,5-trimethoxyphenyl)-3-vinylazetidin-2-one (7d)
4-(4-Dimethylaminophenyl)-1-(3,4,5-trimethoxyphenyl)-3-vinylazetidin-2-one (7e)
4-Phenyl-1-(3,4,5-trimethoxyphenyl)-3-vinylazetidin-2-one (7f)
4-p-Tolyl-1-(3,4,5-trimethoxyphenyl)-3-vinylazetidin-2-one (7g)
4-(4-Methoxyphenyl)-1-(3,4,5-trimethoxyphenyl)-3-vinylazetidin-2-one (7h)
4-(4-Ethoxyphenyl)-1-(3,4,5-trimethoxyphenyl)-3-vinylazetidin-2-one (7i)
4-(4-Butoxyphenyl)-1-(3,4,5-trimethoxyphenyl)-3-vinylazetidin-2-one (7j)
4-(4-Phenoxyphenyl)-1-(3,4,5-trimethoxyphenyl)-3-vinylazetidin-2-one (7k)
4-(4-Benzyloxyphenyl)-1-(3,4,5-trimethoxyphenyl)-3-vinylazetidin-2-one (7l)
4-Naphthalen-1-yl-1-(3,4,5-trimethoxyphenyl)-3-vinylazetidin-2-one (7m)
4-Naphthalen-2-yl-1-(3,4,5-trimethoxyphenyl)-3-vinylazetidin-2-one (7n)
4-(4-Methoxy-3-nitrophenyl)-1-(3,4,5-trimethoxyphenyl)-3-vinylazetidin-2-one (7p)
4-[4-Oxo-1-(3,4,5-trimethoxyphenyl)-3-vinyl-azetidin-2-yl]benzonitrile (7q)
4-(4-Methylsulfanylphenyl)-1-(3,4,5-trimethoxyphenyl)-3-vinylazetidin-2-one (7r)
1-(3,5-Dimethoxyphenyl)-4-(4-methoxyphenyl)-3-vinylazetidin-2-one (7u)
4-(3,4,5-Trimethoxyphenyl)-1-(4-fluorophenyl)-3-vinylazetidin-2-one (8a)
4-(3,4,5-Trimethoxyphenyl)-1-(4-chlorophenyl)-3-vinylazetidin-2-one (8b)
4-(3,4,5-Trimethoxyphenyl)-1-(4-bromophenyl)-3-vinylazetidin-2-one (8c)
4-(3,4,5-Trimethoxyphenyl)-1-(4-nitrophenyl)-3-vinylazetidin-2-one (8d)
4-(3,4,5-Trimethoxyphenyl)-1-(4-tert-butylphenyl)-3-vinylazetidin-2-one (8e)
4-(3,4,5-Trimethoxyphenyl)-1-phenyl-3-vinylazetidin-2-one (8f)
4-(3,4,5-Trimethoxyphenyl)-1-p-tolyl-3-vinylazetidin-2-one (8g)
4-(3,4,5-Trimethoxyphenyl)-1-(4-ethylphenyl)-3-vinylazetidin-2-one (8h)
4-(3,4,5-Trimethoxyphenyl)-1-(4-methoxyphenyl)-3-vinylazetidin-2-one (8i)
N-(4-(2-(3,4,5-Trimethoxyphenyl)-4-oxo-3-vinylazetidin-1-yl)phenyl)acetamide (8j)
1-(4-(Methylthio)phenyl)-4-(3,4,5-trimethoxyphenyl)-3-vinylazetidin-2-one (8k)
4.1.4. 4-[3-Hydroxy-4-methoxyphenyl]-1-(3,4,5-trimethoxyphenyl)-3-vinylazetidin-2-one (7s)
4.1.5. 4-(3-Amino-4-methoxyphenyl)-1-(3,4,5-trimethoxyphenyl)-3-vinylazetidin-2-one (7t)
4.1.6. 4-(4-Methoxyphenyl)-1-(3,4,5-trimethoxyphenyl)azetidin-2-one (9a)
4.1.7. 4-(3-Hydroxy-4-methoxyphenyl)-1-(3,4,5-trimethoxyphenyl)-azetidin-2-one (9c)
4.1.8. 3-(1-Hydroxyethyl)-4-(3-hydroxy-4-methoxyphenyl)-1-(3,4,5-trimethoxyphenyl) azetidin-2-one (10a)
4.1.9. 3-((E)-1-Hydroxybut-2-enyl)-1-(3,4,5-trimethoxyphenyl)-4-(4-methoxyphenyl) azetidin-2-one (10b)
4.1.10. 3-(1-Hydroxybut-2-enyl)-4-(3-hydroxy-4-methoxyphenyl)-1-(3,4,5-trimethoxyphenyl) azetidin-2-one (10c)
4.1.11. 4-(3-Hydroxy-4-methoxyphenyl)-3-(1-hydroxy-1-methylethyl)-1-(3,4,5-trimethoxy-phenyl)azetidin-2-one (10d)
4.1.12. 3-((E)-1-Hydroxy-3-phenylallyl)-1-(3,4,5-trimethoxyphenyl)-4-(4-methoxyphenyl) azetidin-2-one (10e)
4.1.13. 4-(3-Hydroxy-4-methoxy-phenyl)-3-(1-hydroxy-3-phenyl-allyl)-1-(3,4,5-trimethoxy-phenyl)-azetidin-2-one (10f)
4.1.14. 4-(3-Hydroxy-4-methoxyphenyl)-3-(1-hydroxyallyl)-1-(3,4,5-trimethoxy phenyl) azetidin-2-one (10g)
4.1.15. 4-(3-Hydroxy-4-methoxy-phenyl)-3-(1-hydroxy-1-methylallyl)-1-(3,4,5-trimethoxy-phenyl)-azetidin-2-one (10h)
4.1.16. 3-Acetyl-4-(3-hydroxy-4-methoxyphenyl)-1-(3,4,5-trimethoxyphenyl)azetidin-2-one (11)
4.1.17. 3-Ethylidene-4-(3-hydroxy-4-methoxy-phenyl)-1-(3,4,5-trimethoxy-phenyl)-azetidin-2-one (12)
4.1.18. 3-(1,2-Dihydroxyethyl)-4-(3-hydroxy-4-methoxyphenyl)-1-(3,4,5-trimethoxyphenyl) azetidin-2-one (13)
4.1.19. (1-((2-Methoxy-5-(4-oxo-1-(3,4,5-trimethoxyphenyl)-3-vinylazetidin-2-yl)phenyl)-amino)-1-oxopropan-2-yl) carbamaic acid 9H-fluoren-9-ylmethyl ester (14)
4.1.20. 2-Amino-N-(2-methoxy-5-(1-(3,4,5-trimethoxyphenyl)-4-oxo-3-vinylazetidin-2-yl)phenyl)propanamide (15)
4.1.21. General procedure III: Preparation of dibenzyl phosphates 16a, 16b
2-Methoxy-5-(1-(3,4,5-trimethoxyphenyl)-4-oxoazetidin-2-yl)phenyl dibenzyl phosphate (16a)
2-Methoxy-5-(1-(3,4,5-trimethoxyphenyl)-4-oxo-3-vinylazetidin-2-yl)phenyl dibenzyl phosphate (16b)
4.1.22. Phosphoric acid 2-methoxy-5-[4-oxo-1-(3,4,5-trimethoxyphenyl)azetidin-2-yl]phenyl ester dimethyl ester (16c)
4.1.23. Phosphoric acid diethyl ester 2-methoxy-5-[4-oxo-1-(3,4,5-trimethoxyphenyl)azetidin-2-yl]phenyl ester (16d)
4.1.24. 2-Methoxy-5-(1-(3,4,5-trimethoxyphenyl)-4-oxoazetidin-2-yl)phenyl dihydrogen phosphate (17a)
4.1.25. 2-Methoxy-5-(1-(3,4,5-trimethoxyphenyl)-4-oxo-3-vinylazetidin-2-yl)phenyl dihydrogen phosphate (17b)
4.1.26. 5-(3-Ethyl-1-(3,4,5-trimethoxyphenyl)-4-oxoazetidin-2-yl)-2-methoxyphenyl dihydrogen phosphate (17c)
4.2. Biochemical Evaluation
4.2.1. Cell Culture
4.2.2. Cell Viability Assay
4.2.3. Lactate Dehydrogenase Assay for Cytotoxicity
4.2.4. Cytotoxicity Assay
4.2.5. Cell Cycle Analysis
4.2.6. Annexin V/PI Apoptotic Assay
4.2.7. Tubulin Polymerization Assay
4.2.8. Colchicine-Binding Site Assay
4.2.9. Immunofluorescence Microscopy
4.3. Stability Study of Compounds 7s, 17b and 17c
4.4. X-Ray Crystallography
4.5. Computational Procedure: Molecular Docking Study
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| CA-4 | Combretastatin A-4 |
| DBU | 1,8-Diazabicyclo[5.4.0]undec-7-ene |
| DCC | N,N′-Dicyclohexyl carbodiimide |
| DCM | Dichloromethane |
| DCTD | Division of Cancer Treatment and Diagnosis |
| DEAD | Diethyl azodicarboxylate |
| DIPEA | N,N-diisopropylethylamine |
| DMAP | 4-Dimethylaminopyridine |
| DMF | N,N-Dimethylformamide |
| DTP | Development Therapeutics Program |
| Et3N | Triethylamine |
| EBI | N,N′-Ethylene-bis(iodoacetamide) |
| ESI | Electrospray ionisation |
| FMOC | Fluorenylmethyloxycarbonyl |
| HPLC | High-performance liquid chromatography |
| HRMS | High Resolution Mass Spectrometry |
| IC | Inhibitory concentration |
| IR | Infrared |
| MIC | Minimum inhibitory concentration |
| MTD | Maximum tolerated dose |
| MS | Mass spectrometry |
| NCI | National Cancer Institute |
| NIH | National Institute of Health |
| NMR | Nuclear Magnetic Resonance |
| PBS | Phosphate-buffered saline |
| SAR | Structure-activity relationship |
| SERM | Selective Estrogen Receptor Modulator |
| TBAF | Tetrabutylammonium fluoride |
| TBDMS | tert-Butyldimethylchlorosilane |
| TEA | Triethylamine |
| TLC | Thin layer chromatography |
| TMS | Tetramethylsilane |
| TMCS | Tetramethylchlorosilane |
| UV | Ultraviolet |
| VDA | Vascular disrupting agent |
References
- Bates, D.; Eastman, A. Microtubule destabilising agents: Far more than just antimitotic anticancer drugs. Br. J. Clin. Pharmcol. 2017, 83, 255–268. [Google Scholar] [CrossRef] [PubMed]
- Rohena, C.C.; Mooberry, S.L. Recent progress with microtubule stabilizers: New compounds, binding modes and cellular activities. Nat. Prod. Rep. 2014, 31, 335–355. [Google Scholar] [CrossRef]
- Van Vuuren, R.J.; Visagie, M.H.; Theron, A.E.; Joubert, A.M. Antimitotic drugs in the treatment of cancer. Cancer Chemother. Pharmcol. 2015, 76, 1101–1112. [Google Scholar] [CrossRef]
- Gigant, B.; Wang, C.; Ravelli, R.B.; Roussi, F.; Steinmetz, M.O.; Curmi, P.A.; Sobel, A.; Knossow, M. Structural basis for the regulation of tubulin by vinblastine. Nature 2005, 435, 519–522. [Google Scholar] [CrossRef] [PubMed]
- Ravelli, R.B.; Gigant, B.; Curmi, P.A.; Jourdain, I.; Lachkar, S.; Sobel, A.; Knossow, M. Insight into tubulin regulation from a complex with colchicine and a stathmin-like domain. Nature 2004, 428, 198–202. [Google Scholar] [CrossRef] [PubMed]
- De Filippis, B.; Ammazzalorso, A.; Fantacuzzi, M.; Giampietro, L.; Maccallini, C.; Amoroso, R. Anticancer activity of stilbene-based derivatives. ChemMedChem 2017, 12, 558–570. [Google Scholar] [CrossRef] [PubMed]
- Tron, G.C.; Pirali, T.; Sorba, G.; Pagliai, F.; Busacca, S.; Genazzani, A.A. Medicinal chemistry of combretastatin a4: Present and future directions. J. Med. Chem. 2006, 49, 3033–3044. [Google Scholar] [CrossRef] [PubMed]
- Hsieh, H.P.; Liou, J.P.; Mahindroo, N. Pharmaceutical design of antimitotic agents based on combretastatins. Curr. Pharm. Des. 2005, 11, 1655–1677. [Google Scholar] [CrossRef]
- Tozer, G.M.; Kanthou, C.; Baguley, B.C. Disrupting tumour blood vessels. Nat. Rev. Cancer 2005, 5, 423–435. [Google Scholar] [CrossRef]
- Kanthou, C.; Greco, O.; Stratford, A.; Cook, I.; Knight, R.; Benzakour, O.; Tozer, G. The tubulin-binding agent combretastatin a-4-phosphate arrests endothelial cells in mitosis and induces mitotic cell death. Am. J. Pathol. 2004, 165, 1401–1411. [Google Scholar] [CrossRef]
- Rustin, G.J.; Shreeves, G.; Nathan, P.D.; Gaya, A.; Ganesan, T.S.; Wang, D.; Boxall, J.; Poupard, L.; Chaplin, D.J.; Stratford, M.R.; et al. A phase ib trial of ca4p (combretastatin a-4 phosphate), carboplatin, and paclitaxel in patients with advanced cancer. Br. J. Cancer 2010, 102, 1355–1360. [Google Scholar] [CrossRef]
- Bilenker, J.H.; Flaherty, K.T.; Rosen, M.; Davis, L.; Gallagher, M.; Stevenson, J.P.; Sun, W.; Vaughn, D.; Giantonio, B.; Zimmer, R.; et al. Phase i trial of combretastatin a-4 phosphate with carboplatin. Clin. Cancer Res. 2005, 11, 1527–1533. [Google Scholar] [CrossRef] [PubMed]
- Liu, P.; Qin, Y.; Wu, L.; Yang, S.; Li, N.; Wang, H.; Xu, H.; Sun, K.; Zhang, S.; Han, X.; et al. A phase i clinical trial assessing the safety and tolerability of combretastatin a4 phosphate injections. Anticancer Drugs 2014, 25, 462–471. [Google Scholar] [CrossRef] [PubMed]
- Grisham, R.; Ky, B.; Tewari, K.S.; Chaplin, D.J.; Walker, J. Clinical trial experience with ca4p anticancer therapy: Focus on efficacy, cardiovascular adverse events, and hypertension management. Gynecol. Oncol. Res. Pract. 2018, 5, 1. [Google Scholar] [CrossRef]
- Combretastatin A4 Phosphate in Treating Patients with Advanced Anaplastic Thyroid Cancer. Available online: https://clinicaltrials.Gov/ct2/show/nct00060242 (accessed on 16 January 2019).
- Pazofos: Phase IB and Phase II Trial of Pazopanib +/− Fosbretabulin in Advanced Recurrent Ovarian Cancer (pazofos)clinicaltrials.Gov; a Service of the U.S. National Institutes of Health. Available online: https://www.Clinicaltrials.Gov/ct2/show/nct02055690 (accessed on 16 January 2019).
- Nathan, P.; Zweifel, M.; Padhani, A.R.; Koh, D.M.; Ng, M.; Collins, D.J.; Harris, A.; Carden, C.; Smythe, J.; Fisher, N.; et al. Phase i trial of combretastatin a4 phosphate (ca4p) in combination with bevacizumab in patients with advanced cancer. Clin. Cancer Res. 2012, 18, 3428–3439. [Google Scholar] [CrossRef] [PubMed]
- Aboubakr, E.M.; Taye, A.; Aly, O.M.; Gamal-Eldeen, A.M.; El-Moselhy, M.A. Enhanced anticancer effect of combretastatin a-4 phosphate when combined with vincristine in the treatment of hepatocellular carcinoma. Biomed. Pharmcol. 2017, 89, 36–46. [Google Scholar] [CrossRef]
- Ng, Q.S.; Mandeville, H.; Goh, V.; Alonzi, R.; Milner, J.; Carnell, D.; Meer, K.; Padhani, A.R.; Saunders, M.I.; Hoskin, P.J. Phase ib trial of radiotherapy in combination with combretastatin-a4-phosphate in patients with non-small-cell lung cancer, prostate adenocarcinoma, and squamous cell carcinoma of the head and neck. Ann. Oncol. 2012, 23, 231–237. [Google Scholar] [CrossRef] [PubMed]
- Siemann, D.W.; Chaplin, D.J.; Walicke, P.A. A review and update of the current status of the vasculature-disabling agent combretastatin-a4 phosphate (ca4p). Expert Opin. Investig. Drugs 2009, 18, 189–197. [Google Scholar] [CrossRef] [PubMed]
- Greene, L.M.; Meegan, M.J.; Zisterer, D.M. Combretastatins: More than just vascular targeting agents? J. Pharmacol. Exp. Ther. 2015, 355, 212–227. [Google Scholar] [CrossRef]
- Pettit, G.R.; Toki, B.; Herald, D.L.; Verdier-Pinard, P.; Boyd, M.R.; Hamel, E.; Pettit, R.K. Antineoplastic agents. 379. Synthesis of phenstatin phosphate. J. Med. Chem. 1998, 41, 1688–1695. [Google Scholar] [CrossRef] [PubMed]
- Lu, Y.; Chen, J.; Xiao, M.; Li, W.; Miller, D.D. An overview of tubulin inhibitors that interact with the colchicine binding site. Pharm. Res. 2012, 29, 2943–2971. [Google Scholar] [CrossRef]
- Pettit, G.R.; Rhodes, M.R.; Herald, D.L.; Hamel, E.; Schmidt, J.M.; Pettit, R.K. Antineoplastic agents. 445. Synthesis and evaluation of structural modifications of (z)- and (e)-combretastatin a-41. J. Med. Chem. 2005, 48, 4087–4099. [Google Scholar] [CrossRef]
- Theeramunkong, S.; Caldarelli, A.; Massarotti, A.; Aprile, S.; Caprioglio, D.; Zaninetti, R.; Teruggi, A.; Pirali, T.; Grosa, G.; Tron, G.C.; et al. Regioselective suzuki coupling of dihaloheteroaromatic compounds as a rapid strategy to synthesize potent rigid combretastatin analogues. J. Med. Chem. 2011, 54, 4977–4986. [Google Scholar] [CrossRef]
- Hadimani, M.B.; Macdonough, M.T.; Ghatak, A.; Strecker, T.E.; Lopez, R.; Sriram, M.; Nguyen, B.L.; Hall, J.J.; Kessler, R.J.; Shirali, A.R.; et al. Synthesis of a 2-aryl-3-aroyl indole salt (oxi8007) resembling combretastatin a-4 with application as a vascular disrupting agent. J. Nat. Prod. 2013, 76, 1668–1678. [Google Scholar] [CrossRef]
- Macdonough, M.T.; Strecker, T.E.; Hamel, E.; Hall, J.J.; Chaplin, D.J.; Trawick, M.L.; Pinney, K.G. Synthesis and biological evaluation of indole-based, anti-cancer agents inspired by the vascular disrupting agent 2-(3′-hydroxy-4′-methoxyphenyl)-3-(3″,4″,5″-trimethoxybenzoyl)-6-methoxyindole (oxi8006). Bioorg. Med. Chem. 2013, 21, 6831–6843. [Google Scholar] [CrossRef]
- Romagnoli, R.; Baraldi, P.G.; Prencipe, F.; Oliva, P.; Baraldi, S.; Tabrizi, M.A.; Lopez-Cara, L.C.; Ferla, S.; Brancale, A.; Hamel, E.; et al. Design and synthesis of potent in vitro and in vivo anticancer agents based on 1-(3′,4′,5′-trimethoxyphenyl)-2-aryl-1h-imidazole. Sci. Rep. 2016, 6, 26602. [Google Scholar] [CrossRef]
- Lee, S.; Kim, J.N.; Lee, H.K.; Yoon, K.S.; Shin, K.D.; Kwon, B.M.; Han, D.C. Biological evaluation of kribb3 analogs as a microtubule polymerization inhibitor. Bioorg. Med. Chem. Lett. 2011, 21, 977–979. [Google Scholar] [CrossRef]
- Odlo, K.; Fournier-Dit-Chabert, J.; Ducki, S.; Gani, O.A.; Sylte, I.; Hansen, T.V. 1,2,3-triazole analogs of combretastatin a-4 as potential microtubule-binding agents. Bioorg. Med. Chem. 2010, 18, 6874–6885. [Google Scholar] [CrossRef]
- Romagnoli, R.; Baraldi, P.G.; Salvador, M.K.; Preti, D.; Aghazadeh Tabrizi, M.; Brancale, A.; Fu, X.H.; Li, J.; Zhang, S.Z.; Hamel, E.; et al. Synthesis and evaluation of 1,5-disubstituted tetrazoles as rigid analogues of combretastatin a-4 with potent antiproliferative and antitumor activity. J. Med. Chem. 2012, 55, 475–488. [Google Scholar] [CrossRef]
- Rasolofonjatovo, E.; Provot, O.; Hamze, A.; Rodrigo, J.; Bignon, J.; Wdzieczak-Bakala, J.; Lenoir, C.; Desravines, D.; Dubois, J.; Brion, J.D.; et al. Design, synthesis and anticancer properties of 5-arylbenzoxepins as conformationally restricted isocombretastatin a-4 analogs. Eur. J. Med. Chem. 2013, 62, 28–39. [Google Scholar] [CrossRef]
- Xu, Q.L.; Qi, H.; Sun, M.L.; Zuo, D.Y.; Jiang, X.W.; Wen, Z.Y.; Wang, Z.W.; Wu, Y.L.; Zhang, W.G. Synthesis and biological evaluation of 3-alkyl-1,5-diaryl-1h-pyrazoles as rigid analogues of combretastatin a-4 with potent antiproliferative activity. PLoS ONE 2015, 10, e0128710. [Google Scholar] [CrossRef]
- Zheng, S.; Zhong, Q.; Mottamal, M.; Zhang, Q.; Zhang, C.; Lemelle, E.; McFerrin, H.; Wang, G. Design, synthesis, and biological evaluation of novel pyridine-bridged analogues of combretastatin-a4 as anticancer agents. J. Med. Chem. 2014, 57, 3369–3381. [Google Scholar] [CrossRef]
- Prota, A.E.; Danel, F.; Bachmann, F.; Bargsten, K.; Buey, R.M.; Pohlmann, J.; Reinelt, S.; Lane, H.; Steinmetz, M.O. The novel microtubule-destabilizing drug bal27862 binds to the colchicine site of tubulin with distinct effects on microtubule organization. J. Mol. Biol. 2014, 426, 1848–1860. [Google Scholar] [CrossRef]
- Rajak, H.; Dewangan, P.K.; Patel, V.; Jain, D.K.; Singh, A.; Veerasamy, R.; Sharma, P.C.; Dixit, A. Design of combretastatin a-4 analogs as tubulin targeted vascular disrupting agent with special emphasis on their cis-restricted isomers. Curr. Pharm. Des. 2013, 19, 1923–1955. [Google Scholar] [CrossRef]
- Zhou, P.; Liu, Y.; Zhou, L.; Zhu, K.; Feng, K.; Zhang, H.; Liang, Y.; Jiang, H.; Luo, C.; Liu, M.; et al. Potent antitumor activities and structure basis of the chiral beta-lactam bridged analogue of combretastatin a-4 binding to tubulin. J. Med. Chem. 2016, 59, 10329–10334. [Google Scholar] [CrossRef]
- Zhou, P.L.; Liang, Y.; Zhang, H.; Jiang, H.; Feng, K.; Xu, P.; Wang, J.; Wang, X.; Ding, K.; Luo, C.; et al. Design, synthesis, biological evaluation and cocrystal structures with tubulin of chiral b-lactam bridged combretastatin a-4 analogues as potent antitumor agents. Eur. J. Med. Chem. 2018, 144, 817–842. [Google Scholar] [CrossRef]
- Galletti, P.; Soldati, R.; Pori, M.; Durso, M.; Tolomelli, A.; Gentilucci, L.; Dattoli, S.D.; Baiula, M.; Spampinato, S.; Giacomini, D. Targeting integrins alphavbeta3 and alpha5beta1 with new beta-lactam derivatives. Eur. J. Med. Chem. 2014, 83, 284–293. [Google Scholar] [CrossRef]
- Geesala, R.; Gangasani, J.K.; Budde, M.; Balasubramanian, S.; Vaidya, J.R.; Das, A. 2-azetidinones: Synthesis and biological evaluation as potential anti-breast cancer agents. Eur. J. Med. Chem. 2016, 124, 544–558. [Google Scholar] [CrossRef]
- Arya, N.; Jagdale, A.Y.; Patil, T.A.; Yeramwar, S.S.; Holikatti, S.S.; Dwivedi, J.; Shishoo, C.J.; Jain, K.S. The chemistry and biological potential of azetidin-2-ones. Eur. J. Med. Chem. 2014, 74, 619–656. [Google Scholar] [CrossRef]
- Fu, D.J.; Fu, L.; Liu, Y.C.; Wang, J.W.; Wang, Y.Q.; Han, B.K.; Li, X.R.; Zhang, C.; Li, F.; Song, J.; et al. Structure-activity relationship studies of beta-lactam-azide analogues as orally active antitumor agents targeting the tubulin colchicine site. Sci. Rep. 2017, 7, 12788. [Google Scholar] [CrossRef]
- Kamath, A.; Ojima, I. Advances in the chemistry of beta-lactam and its medicinal applications. Tetrahedron 2012, 68, 10640–10664. [Google Scholar] [CrossRef]
- O’Boyle, N.M.; Pollock, J.K.; Carr, M.; Knox, A.J.; Nathwani, S.M.; Wang, S.; Caboni, L.; Zisterer, D.M.; Meegan, M.J. Beta-lactam estrogen receptor antagonists and a dual-targeting estrogen receptor/tubulin ligand. J. Med. Chem. 2014, 57, 9370–9382. [Google Scholar] [CrossRef]
- O’Boyle, N.M.; Carr, M.; Greene, L.M.; Bergin, O.; Nathwani, S.M.; McCabe, T.; Lloyd, D.G.; Zisterer, D.M.; Meegan, M.J. Synthesis and evaluation of azetidinone analogues of combretastatin a-4 as tubulin targeting agents. J. Med. Chem. 2010, 53, 8569–8584. [Google Scholar] [CrossRef] [PubMed]
- Greene, T.F.; Wang, S.; Greene, L.M.; Nathwani, S.M.; Pollock, J.K.; Malebari, A.M.; McCabe, T.; Twamley, B.; O’Boyle, N.M.; Zisterer, D.M.; et al. Synthesis and biochemical evaluation of 3-phenoxy-1,4-diarylazetidin-2-ones as tubulin-targeting antitumor agents. J. Med. Chem. 2016, 59, 90–113. [Google Scholar] [CrossRef] [PubMed]
- Nathwani, S.M.; Hughes, L.; Greene, L.M.; Carr, M.; O’Boyle, N.M.; McDonnell, S.; Meegan, M.J.; Zisterer, D.M. Novel cis-restricted beta-lactam combretastatin a-4 analogues display anti-vascular and anti-metastatic properties in vitro. Oncol. Rep. 2013, 29, 585–594. [Google Scholar] [CrossRef] [PubMed]
- Singh, G.S.; Sudheesh, S. Advances in synthesis of monocyclic beta-lactams. Arkivoc 2014, 337–385. [Google Scholar] [CrossRef]
- Gaspari, R.; Prota, A.E.; Bargsten, K.; Cavalli, A.; Steinmetz, M.O. Structural basis of cis- and trans-combretastatin binding to tubulin. Chem 2017, 2, 102–113. [Google Scholar] [CrossRef]
- Zamboni, R.; Just, G. Beta-lactams. 7. Synthesis of 3-vinyl and 3-isopropenyl 4-substituted azetidinones. Can. J. Chem. 1979, 57, 1945–1948. [Google Scholar] [CrossRef]
- Neary, A.D.; Burke, C.M.; O’Leary, A.C.; Meegan, M.J. Transformation of 4-acetoxy-3-vinylazetidin-2-ones to 3-(1-hydroxyethyl)azetidin-2-ones and 3-ethylideneazetidin-2-ones: Intermediates for carbapenem antibiotics. J. Chem. Res. 2001, 2001, 166–169. [Google Scholar] [CrossRef]
- Chang, J.Y.; Yang, M.F.; Chang, C.Y.; Chen, C.M.; Kuo, C.C.; Liou, J.P. 2-amino and 2′-aminocombretastatin derivatives as potent antimitotic agents. J. Med. Chem. 2006, 49, 6412–6415. [Google Scholar] [CrossRef]
- Tripodi, F.; Pagliarin, R.; Fumagalli, G.; Bigi, A.; Fusi, P.; Orsini, F.; Frattini, M.; Coccetti, P. Synthesis and biological evaluation of 1,4-diaryl-2-azetidinones as specific anticancer agents: Activation of adenosine monophosphate activated protein kinase and induction of apoptosis. J. Med. Chem. 2012, 55, 2112–2124. [Google Scholar] [CrossRef]
- O’Boyle, N.M.; Greene, L.M.; Bergin, O.; Fichet, J.B.; McCabe, T.; Lloyd, D.G.; Zisterer, D.M.; Meegan, M.J. Synthesis, evaluation and structural studies of antiproliferative tubulin-targeting azetidin-2-ones. Bioorg. Med. Chem. 2011, 19, 2306–2325. [Google Scholar] [CrossRef]
- Mayrhofer, R.; Otto, H.H. Simple preparation of 3-benzylidene-2-azetidinones. Synthesis 1980, 1980, 247–248. [Google Scholar] [CrossRef]
- Kano, S.; Ebata, T.; Funaki, K.; Shibuya, S. New and facile synthesis of 3-alkylideneazetidin-2-ones by reactions of 3-trimethylsilylazetidin-2-one with carbonyl-compounds. Synthesis 1978, 1978, 746–747. [Google Scholar] [CrossRef]
- Plantan, I.; Selic, L.; Mesar, T.; Anderluh, P.S.; Oblak, M.; Prezelj, A.; Hesse, L.; Andrejasic, M.; Vilar, M.; Turk, D.; et al. 4-substituted trinems as broad spectrum beta-lactamase inhibitors: Structure-based design, synthesis, and biological activity. J. Med. Chem. 2007, 50, 4113–4121. [Google Scholar] [CrossRef]
- O’Boyle, N.M.; Greene, L.M.; Keely, N.O.; Wang, S.; Cotter, T.S.; Zisterer, D.M.; Meegan, M.J. Synthesis and biochemical activities of antiproliferative amino acid and phosphate derivatives of microtubule-disrupting beta-lactam combretastatins. Eur. J. Med. Chem. 2013, 62, 705–721. [Google Scholar] [CrossRef]
- Spek, A.L.; Vandersteen, F.H.; Jastrzebski, J.T.B.H.; Vankoten, G. Trans-3-amino-1-methyl-4-phenyl-2-azetidinone, C10H12N2O. Acta Crystallogr. C 1994, 50, 1933–1935. [Google Scholar] [CrossRef]
- Kabak, M.; Senoz, H.; Elmali, A.; Adar, V.; Svoboda, I.; Dusek, M.; Fejfarova, K. Synthesis and x-ray crystal structure determination of n-p-methylphenyl-4-benzoyl-3,4-diphenyl-2-azetidinone. Crystallogr. Rep. 2010, 55, 1220–1222. [Google Scholar] [CrossRef]
- Lara-Ochoa, F.; Espinosa-Perez, G. A new synthesis of combretastatins a-4 and ave-8062a. Tetrahedron Lett. 2007, 48, 7007–7010. [Google Scholar] [CrossRef]
- Combes, S.; Barbier, P.; Douillard, S.; McLeer-Florin, A.; Bourgarel-Rey, V.; Pierson, J.T.; Fedorov, A.Y.; Finet, J.P.; Boutonnat, J.; Peyrot, V. Synthesis and biological evaluation of 4-arylcoumarin analogues of combretastatins. Part 2. J. Med. Chem. 2011, 54, 3153–3162. [Google Scholar] [CrossRef]
- Messaoudi, S.; Treguier, B.; Hamze, A.; Provot, O.; Peyrat, J.F.; De Losada, J.R.; Liu, J.M.; Bignon, J.; Wdzieczak-Bakala, J.; Thoret, S.; et al. Isocombretastatins a versus combretastatins a: The forgotten isoca-4 isomer as a highly promising cytotoxic and antitubulin agent. J. Med. Chem. 2009, 52, 4538–4542. [Google Scholar] [CrossRef]
- Cushman, M.; Nagarathnam, D.; Gopal, D.; He, H.M.; Lin, C.M.; Hamel, E. Synthesis and evaluation of analogues of (z)-1-(4-methoxyphenyl)-2-(3,4,5-trimethoxyphenyl)ethene as potential cytotoxic and antimitotic agents. J. Med. Chem. 1992, 35, 2293–2306. [Google Scholar] [CrossRef]
- Flynn, B.L.; Flynn, G.P.; Hamel, E.; Jung, M.K. The synthesis and tubulin binding activity of thiophene-based analogues of combretastatin a-4. Bioorg. Med. Chem. Lett. 2001, 11, 2341–2343. [Google Scholar] [CrossRef]
- Chaudhary, A.; Pandeya, S.N.; Kumar, P.; Sharma, P.P.; Gupta, S.; Soni, N.; Verma, K.K.; Bhardwaj, G. Combretastatin a-4 analogs as anticancer agents. Mini Rev. Med. Chem. 2007, 7, 1186–1205. [Google Scholar] [CrossRef]
- Devkota, L.; Lin, C.M.; Strecker, T.E.; Wang, Y.; Tidmore, J.K.; Chen, Z.; Guddneppanavar, R.; Jelinek, C.J.; Lopez, R.; Liu, L.; et al. Design, synthesis, and biological evaluation of water-soluble amino acid prodrug conjugates derived from combretastatin, dihydronaphthalene, and benzosuberene-based parent vascular disrupting agents. Bioorg. Med. Chem. 2016, 24, 938–956. [Google Scholar] [CrossRef]
- Mousset, C.; Giraud, A.; Provot, O.; Hamze, A.; Bignon, J.; Liu, J.M.; Thoret, S.; Dubois, J.; Brion, J.D.; Alami, M. Synthesis and antitumor activity of benzils related to combretastatin a-4. Bioorg. Med. Chem. Lett. 2008, 18, 3266–3271. [Google Scholar] [CrossRef]
- Hughes, L.; Malone, C.; Chumsri, S.; Burger, A.M.; McDonnell, S. Characterisation of breast cancer cell lines and establishment of a novel isogenic subclone to study migration, invasion and tumourigenicity. Clin. Exp. Metastasis 2008, 25, 549–557. [Google Scholar] [CrossRef]
- National Cancer Institute Division of Cancer Treatment and Diagnosis. Available online: https://dtp.Cancer.Gov (accessed on 25 February 2019).
- National Cancer Institute Biological Testing Branch; National Cancer Institute: Bethesda, MD, USA. Available online: https://dtp.Nci.Nih.Gov/branches/btb/hfa.Html (accessed on 16 January 2019).
- Vichai, V.; Kirtikara, K. Sulforhodamine b colorimetric assay for cytotoxicity screening. Nat. Protoc. 2006, 1, 1112–1116. [Google Scholar] [CrossRef]
- Smith, S.M.; Wunder, M.B.; Norris, D.A.; Shellman, Y.G. A simple protocol for using a ldh-based cytotoxicity assay to assess the effects of death and growth inhibition at the same time. PLoS ONE 2011, 6, e26908. [Google Scholar] [CrossRef]
- Furlong, E.E.; Keon, N.K.; Thornton, F.D.; Rein, T.; Martin, F. Expression of a 74-kda nuclear factor 1 (nf1) protein is induced in mouse mammary gland involution. Involution-enhanced occupation of a twin nf1 binding element in the testosterone-repressed prostate message-2/clusterin promoter. J. Biol. Chem. 1996, 271, 29688–29697. [Google Scholar] [CrossRef]
- Murtagh, J.; McArdle, E.; Gilligan, E.; Thornton, L.; Furlong, F.; Martin, F. Organization of mammary epithelial cells into 3d acinar structures requires glucocorticoid and jnk signaling. J. Cell Biol. 2004, 166, 133–143. [Google Scholar] [CrossRef]
- Shen, C.H.; Shee, J.J.; Wu, J.Y.; Lin, Y.W.; Wu, J.D.; Liu, Y.W. Combretastatin a-4 inhibits cell growth and metastasis in bladder cancer cells and retards tumour growth in a murine orthotopic bladder tumour model. Br. J. Pharmcol. 2010, 160, 2008–2027. [Google Scholar] [CrossRef]
- Greene, L.M.; O’Boyle, N.M.; Nolan, D.P.; Meegan, M.J.; Zisterer, D.M. The vascular targeting agent combretastatin-a4 directly induces autophagy in adenocarcinoma-derived colon cancer cells. Biochem. Pharmcol. 2012, 84, 612–624. [Google Scholar] [CrossRef]
- Tubulin Polymerization Assay Kit Manual (CDS03 and BK006); Cytoskeleton: Denver, CO, USA, 2009; pp. 1–18.
- Barbier, P.; Tsvetkov, P.O.; Breuzard, G.; Devred, F. Deciphering the molecular mechanisms of anti-tubulin plant derived drugs. Phytochem. Rev. 2014, 13, 157–169. [Google Scholar] [CrossRef]
- Castedo, M.; Perfettini, J.-L.; Roumier, T.; Andreau, K.; Medema, R.; Kroemer, G. Cell death by mitotic catastrophe: A molecular definition. Oncogene 2004, 23, 2825–2837. [Google Scholar] [CrossRef]
- Vitale, I.; Antoccia, A.; Cenciarelli, C.; Crateri, P.; Meschini, S.; Arancia, G.; Pisano, C.; Tanzarella, C. Combretastatin ca-4 and combretastatin derivative induce mitotic catastrophe dependent on spindle checkpoint and caspase-3 activation in non-small cell lung cancer cells. Apoptosis 2007, 12, 155–166. [Google Scholar] [CrossRef]
- Cenciarelli, C.; Tanzarella, C.; Vitale, I.; Pisano, C.; Crateri, P.; Meschini, S.; Arancia, G.; Antoccia, A. The tubulin-depolymerising agent combretastatin-4 induces ectopic aster assembly and mitotic catastrophe in lung cancer cells h460. Apoptosis 2008, 13, 659–669. [Google Scholar] [CrossRef]
- Simoni, D.; Romagnoli, R.; Baruchello, R.; Rondanin, R.; Rizzi, M.; Pavani, M.G.; Alloatti, D.; Giannini, G.; Marcellini, M.; Riccioni, T.; et al. Novel combretastatin analogues endowed with antitumor activity. J. Med. Chem. 2006, 49, 3143–3152. [Google Scholar] [CrossRef]
- O’Boyle, N.M.; Carr, M.; Greene, L.M.; Knox, A.J.S.; Lloyd, D.G.; Zisterer, D.M.; Meegan, M.J. Synthesis, biochemical and molecular modelling studies of antiproliferative azetidinones causing microtubule disruption and mitotic catastrophe. Eur. J. Med. Chem. 2011, 46, 4595–4607. [Google Scholar] [CrossRef]
- Fortin, S.; Lacroix, J.; Cote, M.F.; Moreau, E.; Petitclerc, E.; Gaudreault, R.C. Quick and simple detection technique to assess the binding of antimicrotubule agents to the colchicine-binding site. Biol. Proced. Online 2010, 12, 113–117. [Google Scholar] [CrossRef]
- Canela, M.D.; Perez-Perez, M.J.; Noppen, S.; Saez-Calvo, G.; Diaz, J.F.; Camarasa, M.J.; Liekens, S.; Priego, E.M. Novel colchicine-site binders with a cyclohexanedione scaffold identified through a ligand-based virtual screening approach. J. Med. Chem. 2014, 57, 3924–3938. [Google Scholar] [CrossRef]
- Carr, M.; Greene, L.M.; Knox, A.J.; Lloyd, D.G.; Zisterer, D.M.; Meegan, M.J. Lead identification of conformationally restricted beta-lactam type combretastatin analogues: Synthesis, antiproliferative activity and tubulin targeting effects. Eur. J. Med. Chem. 2010, 45, 5752–5766. [Google Scholar] [CrossRef]
- Molecular Operating Environment (MOE); C.C.G.I.; (1010 Sherbooke St. West, Suite #910, Montreal, QC, Canada). Personal communications, 2016.
- Elmeligie, S.; Taher, A.T.; Khalil, N.A.; El-Said, A.H. Synthesis and cytotoxic activity of certain trisubstituted azetidin-2-one derivatives as a cis-restricted combretastatin a-4 analogues. Arch. Pharm. Res. 2017, 40, 13–24. [Google Scholar] [CrossRef]
- Georg, G.I.; He, P.; Kant, J.; Mudd, J. N-vinyl and n-unsubstituted beta-lactams from 1-substituted 2-aza-1,3-butadienes. Tetrahedron Lett. 1990, 31, 451–454. [Google Scholar] [CrossRef]
- Arroyo, Y.; Sanz-Tejedor, M.A.; Alonso, I.; Garcia-Ruano, J.L. Synthesis of optically pure vic-sulfanyl amines mediated by a remote sulfinyl group. Org. Lett. 2011, 13, 4534–4537. [Google Scholar] [CrossRef]
- Sandhar, R.K.; Sharma, J.R.; Manrao, M.R. Reaction of acetylacetone with benzal-4-fluoroanilines and antifungal potential of the products. J. Indian Counc. Chem. 2005, 22, 32–34. [Google Scholar]
- Dehno Khalaji, A.; Fejfarova, K.; Dusek, M. N,N′-bis(3,4-dimethoxy-benzyl-idene)butane-1,4-diamine. Acta Crystallogr. Sect. E Struct. Rep. Online 2009, 65, o1773. [Google Scholar] [CrossRef]
- Khalaji, A.D.; Weil, M.; Gotoh, K.; Ishida, H. 4-bromo-N-(3,4,5-trimethoxy-benzyl-idene)aniline. Acta Crystallogr. Sect. E Struct. Rep. Online 2009, 65, o436. [Google Scholar] [CrossRef]
- Yang, Z. Synthesis and in vitro biological activity evaluation of the derivatives of combretastatin a-4. Lett. Drug Des. Discov. 2006 3, 544–546. [CrossRef]
- Cushman, M.; He, H.M.; Lin, C.M.; Hamel, E. Synthesis and evaluation of a series of benzylaniline hydrochlorides as potential cytotoxic and antimitotic agents acting by inhibition of tubulin polymerization. J. Med. Chem. 1993, 36, 2817–2821. [Google Scholar] [CrossRef]
- Gaidhane, M.K.; Ghatole, A.M.; Lanjewar, K.R. Novel synthesis and antimicrobial activity of novel schiff base derived quinoline and their beta-lactam derivatives. Int. J. Pharm. Pharm. Sci. 2013, 5, 421–426. [Google Scholar]
- Malebari, A.M.; Greene, L.M.; Nathwani, S.M.; Fayne, D.; O’Boyle, N.M.; Wang, S.; Twamley, B.; Zisterer, D.M.; Meegan, M.J. Beta-lactam analogues of combretastatin a-4 prevent metabolic inactivation by glucuronidation in chemoresistant ht-29 colon cancer cells. Eur. J. Med. Chem. 2017, 130, 261–285. [Google Scholar] [CrossRef]
- Meegan, M.J.; Zisterer, D.M.; Carr, M.; Greene, T.; O’Boyle, N.; Greene, L. Combretastatin Derivatives and Uses Therefor. European Patent WO 2011073211, 23 June 2011. [Google Scholar]
- Wang, Y.L.; Liu, M.; Zhou, P.; Feng, K.; Ding, K.; Wang, X. Diaryl-Β-Lactam Compound and Preparation Method and Pharmaceutical Use Thereof. Chinese Patent WO 2017167183, 5 October 2017. [Google Scholar]
- Promega Corporation. Cytotox 96® Non-Radioactive Cytotoxicity Assay; Promega Cytotox 96 Nonradioactive Cytotoxicity Assay Protocol; Promega Corporation: Fitchburg, WI, USA, 2016. [Google Scholar]
- Bruker Apex2 v2012.12-0; Bruker Axs Inc.: Madison, Wi, USA, 2012.
- Sheldrick, G.M. A short history of shelx. Acta Crystallogr. A 2008, 64, 112–122. [Google Scholar] [CrossRef]
- Sheldrick, G.M. Shelxt—Integrated space-group and crystal-structure determination. Acta Crystallogr. A Found. Adv. 2015, 71, 3–8. [Google Scholar] [CrossRef]
- Sheldrick, G.M. Crystal structure refinement with shelxl. Acta Crystallogr. C Struct. Chem. 2015, 71, 3–8. [Google Scholar] [CrossRef]
- Dolomanov, O.V.; Bourhis, L.J.; Gildea, R.J.; Howard, J.A.K.; Puschmann, H. Olex2: A complete structure solution, refinement and analysis program. J. Appl. Crystallogr. 2009, 42, 339–341. [Google Scholar] [CrossRef]
- Sadabs; (Bruker Axs Inc., Madison, WI, USA); Sheldrick, G.M.; (University of Göttingen, Göttingen, Germany). Personal communications, 2014.
- Crystalclear Rigaku Molecular Structure Corporation Inc.; (The Woodlands, TX, USA). Personal communications, 2000.
- Pflugrath, J.W. The finer things in x-ray diffraction data collection. Acta Crystallogr. D Biol. Crystallogr. 1999, 55, 1718–1725. [Google Scholar] [CrossRef]
- Molecular Operating Environment (MOE) Version 2015.10; Chemical Computing Group Inc.: Montreal, QC, Canada, 2016.

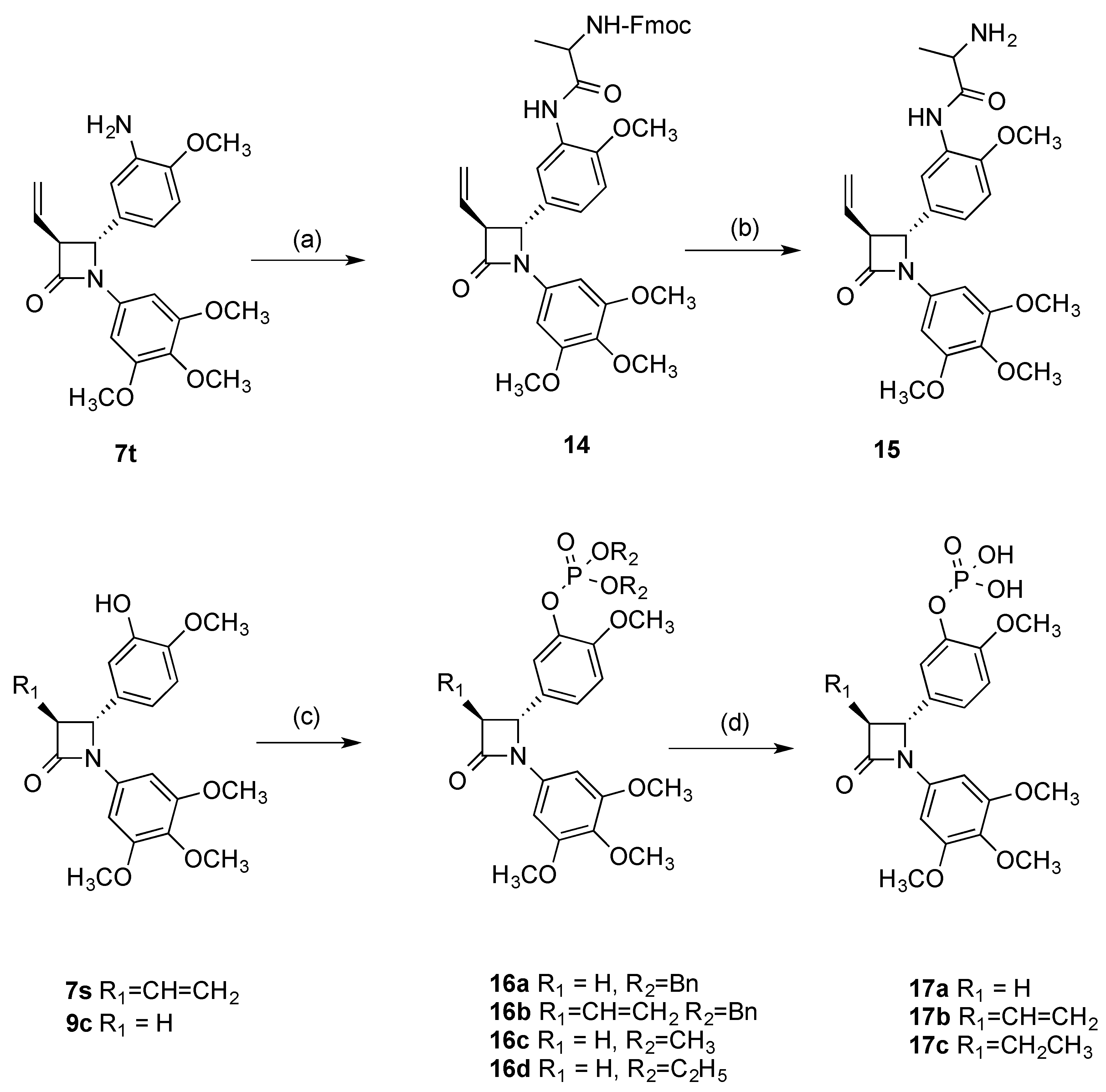




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | 6k | 7h | 8h | 8i | 8k |
|---|---|---|---|---|---|
| Empirical formula | C17H19NO3S | C21H23NO5 | C22H25NO4 | C21H23NO5 | C21H23NO4S |
| M (g/mol) | 317.39 | 369.40 | 367.43 | 369.40 | 385.46 |
| Crystal System | monoclinic | monoclinic | monoclinic | triclinic | triclinic |
| SG | P21 (No. 4) | P21 (No. 4) | P21/n (No. 14) | P (No. 2) | P (No. 2) |
| a (Å) | 7.8282(3) | 20.1106(7) | 7.2135(14) | 10.9022(5) | 8.2131(4) |
| b (Å) | 7.7880(3) | 9.1481(3) | 26.440(5) | 13.0315(6) | 10.5047(5) |
| c (Å) | 13.2937(6) | 22.6378(8) | 10.389(2) | 14.9787(8) | 12.5704(6) |
| α (°) | - | - | - | 94.994(2) | 107.9218(16)° |
| β (°) | 106.4320(10) | 110.6238(14) | 101.41(3) | 105.024(2)° | 96.7759(18)° |
| γ (°) | - | - | - | 108.284(2)° | 103.3909(18)° |
| V (Å3) | 777.36(6) | 3897.9(2) | 1942.3(7) | 1918.37(16) | 982.64(8) |
| T (K) | 100(2) | 100(2) | 150(2) | 100(2) | 100(2) |
| Z | 2 | 8 | 4 | 4 | 2 |
| Dcalc (g/cm3) | 1.356 | 1.259 | 1.256 | 1.279 | 1.303 |
| μ (mm−1) | 0.220 (Mo Kα) | 0.738 (Cu Kα) | 0.086 (Mo Kα) | 0.091 (Mo Kα) | 0.191 (Mo Kα) |
| Total reflns | 27644 | 59468 | 14461 | 29738 | 14916 |
| Indep. reflns | 4394 | 14180 | 3492 | 9673 | 4903 |
| R(int) | 0.0258 | 0.0602 | 0.0565 | 0.0532 | 0.0304 |
| S | 1.058 | 1.047 | 1.188 | 1.006 | 1.038 |
| R1 * [I > 2σ(I)] | 0.0251 | 0.0364 | 0.0666 | 0.0509 | 0.0388 |
| wR2 * [all data] | 0.0676 | 0.0945 | 0.1509 | 0.1191 | 0.0918 |
| Flack | 0.027(12) | ||||
| CCDC number | 1820354 | 1820355 | 1820358 | 1820356 | 1820357 |
| Compound | Ring Plane Normal AB Angle(°) | Ring A to Central Torsion (°) a | Ring B to Central Torsion (°) b | RingAB Torsion (°) c | Ring B Vinyl Torsion (°) d |
|---|---|---|---|---|---|
| 7h * | 82.79(8) | 177.0(2) | 137.7(2) | −59.5(3) | 124.8(2) |
| 75.19(8) | 179.4(2) | 137.1(2) | −53.8(5) | 123.8(2) | |
| 100.51(8) | 177.6(2) | −136.1(2) | 58.1(3) | −126.1(2) | |
| 85.35(8) | −167.5(2) | −138.9(2) | 59.7(3) | −120.9(2) | |
| 8h | 93.14(7) | −171.6(3) | −130.1(2) | 59.7(3) | −124.4(2) |
| 8i§ | 86.28(5) | 170.67(17) | 150.21(14) | −73.5(2) | 127.75(18) |
| 85.68(6) | 165.62(18) | 155.26(16) | −73.2(2) | 127.49(16) | |
| 8k | 85.60(5) | 162.50(17) | 156.94(13) | −77.0(2) | 122.95(15) |
| a | b | c | d |
| C14-C13-N1-C2 | C12-C5-C4-N1 | C13-N1-C4-C5 | C5-C4-C3-C26 |
| C18-C17-N1-C2 | C10-C5-C4-N1 | C17-N1-C4-C5 | C5-C4-C3-C26 |
| C18-C17-N1-C2 | C6-C5-C4-N1 | C17-N1-C4-C5 | C5-C4-C3-C26 |
| C18-C17-N1-C2 | C6-C5-C4-N1 | C17-N1-C4-C5 | C5-C4-C3-C26 |
| C14-C13-N1-C2 | C10-C5-C4-N1 | C13-N1-C4-C5 | C5-C4-C3-C26 |
| Compound Number | Antiproliferative Activity IC50 (µM) a |
|---|---|
| 7a | 8.314 ± 1.40 |
| 7b | 0.690 ± 0.11 |
| 7c | 0.445 ± 0.07 |
| 7d | 3.827 ± 0.53 |
| 7e | 4.047 ± 0.45 |
| 7f | 4.034 ± 0.42 |
| 7g | 0.355 ± 0.03 |
| 7h | 0.020 ± 0.0025 |
| 7i | 0.037 ± 0.0033 |
| 7j | 13.990 ± 1.81 |
| 7k | 57.041 ± 3.72 |
| 7l | 8.015 ± 0.63 |
| 7m | 1.738 ± 0.17 |
| 7n | 0.068 ± 0.01 |
| 7p | 0.618 ± 0.10 |
| 7q | 6.251 ± 5.05 |
| 7r | 0.051 ± 0.001 |
| 7s | 0.008 ± 0.00071 |
| 7t | 0.017 ± 0.0018 |
| 7u | 0.170 ± 0.07. |
| 8a | 1.066 ± 0.14 |
| 8b | 29.150 ± 1.14 |
| 8c | 10.400 ± 0.87 |
| 8d | 59.150 ± 4.16 |
| 8e | 68.840 ± 3.63 |
| 8f | 50.460 ± 4.25 |
| 8g | 43.130 ± 2.16 |
| 8h | 36.400 ± 2.13 |
| 8i | 65.120 ± 5.55 |
| 8j | 4.024 ± 0.64 |
| 8k | >50 |
| CA-4 b | 0.0039 ± 0.00032 |
| Compound Number | Antiproliferative Activity IC50 (nM) a |
|---|---|
| 10a | 65 ± 15 |
| 10b | 292 ± 50 |
| 10c | 5701 ± 246 |
| 10d | 544 ± 310 |
| 10e | 537 ± 80 |
| 10f | 46 ± 41 |
| 10g | 288 ± 76 |
| 10h | 467 ± 0.253 |
| 11 | 414 ± 132 |
| 12 | 502 ± 212 |
| 13 | 69 ± 29 |
| 15 | 3251 ± 270 |
| 17a | 22 ± 1.5 |
| 17b | 27 ± 2 |
| 17c | 21 ± 1.5 |
| CA-4 b | 39 ± 3.2 |
| Compound Number | Antiproliferative Activity IC50 (nM) a |
|---|---|
| 7b | 191 ± 16 |
| 7h b | 31.7 |
| 7i | 61 ± 7 |
| 7n | 77 ± 9 |
| 7s b | 10 |
| 7t | 30 ± 2 |
| 17a | 30 ± 4 |
| 17b b | 48.6 |
| 17c | 44 ± 7 |
| CA-4 c | 43 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wang, S.; Malebari, A.M.; Greene, T.F.; O’Boyle, N.M.; Fayne, D.; Nathwani, S.M.; Twamley, B.; McCabe, T.; Keely, N.O.; Zisterer, D.M.; et al. 3-Vinylazetidin-2-Ones: Synthesis, Antiproliferative and Tubulin Destabilizing Activity in MCF-7 and MDA-MB-231 Breast Cancer Cells. Pharmaceuticals 2019, 12, 56. https://doi.org/10.3390/ph12020056
Wang S, Malebari AM, Greene TF, O’Boyle NM, Fayne D, Nathwani SM, Twamley B, McCabe T, Keely NO, Zisterer DM, et al. 3-Vinylazetidin-2-Ones: Synthesis, Antiproliferative and Tubulin Destabilizing Activity in MCF-7 and MDA-MB-231 Breast Cancer Cells. Pharmaceuticals. 2019; 12(2):56. https://doi.org/10.3390/ph12020056
Chicago/Turabian StyleWang, Shu, Azizah M. Malebari, Thomas F. Greene, Niamh M. O’Boyle, Darren Fayne, Seema M. Nathwani, Brendan Twamley, Thomas McCabe, Niall O. Keely, Daniela M. Zisterer, and et al. 2019. "3-Vinylazetidin-2-Ones: Synthesis, Antiproliferative and Tubulin Destabilizing Activity in MCF-7 and MDA-MB-231 Breast Cancer Cells" Pharmaceuticals 12, no. 2: 56. https://doi.org/10.3390/ph12020056
APA StyleWang, S., Malebari, A. M., Greene, T. F., O’Boyle, N. M., Fayne, D., Nathwani, S. M., Twamley, B., McCabe, T., Keely, N. O., Zisterer, D. M., & Meegan, M. J. (2019). 3-Vinylazetidin-2-Ones: Synthesis, Antiproliferative and Tubulin Destabilizing Activity in MCF-7 and MDA-MB-231 Breast Cancer Cells. Pharmaceuticals, 12(2), 56. https://doi.org/10.3390/ph12020056