Relevance of In Vitro Metabolism Models to PET Radiotracer Development: Prediction of In Vivo Clearance in Rats from Microsomal Stability Data

 , , , and
, , , and
Abstract
:1. Introduction
2. Results
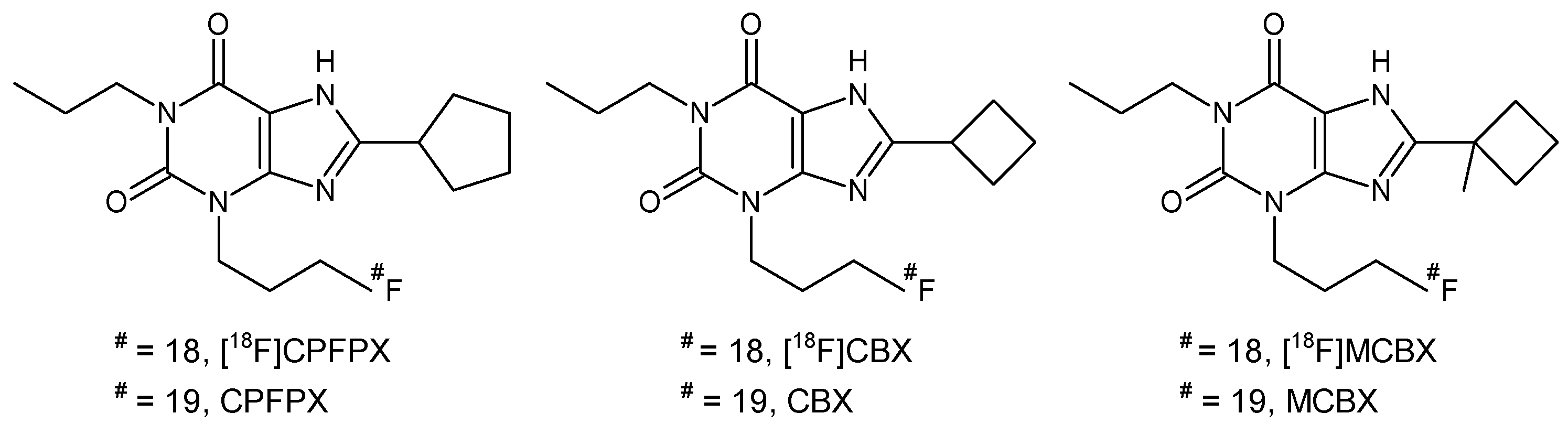
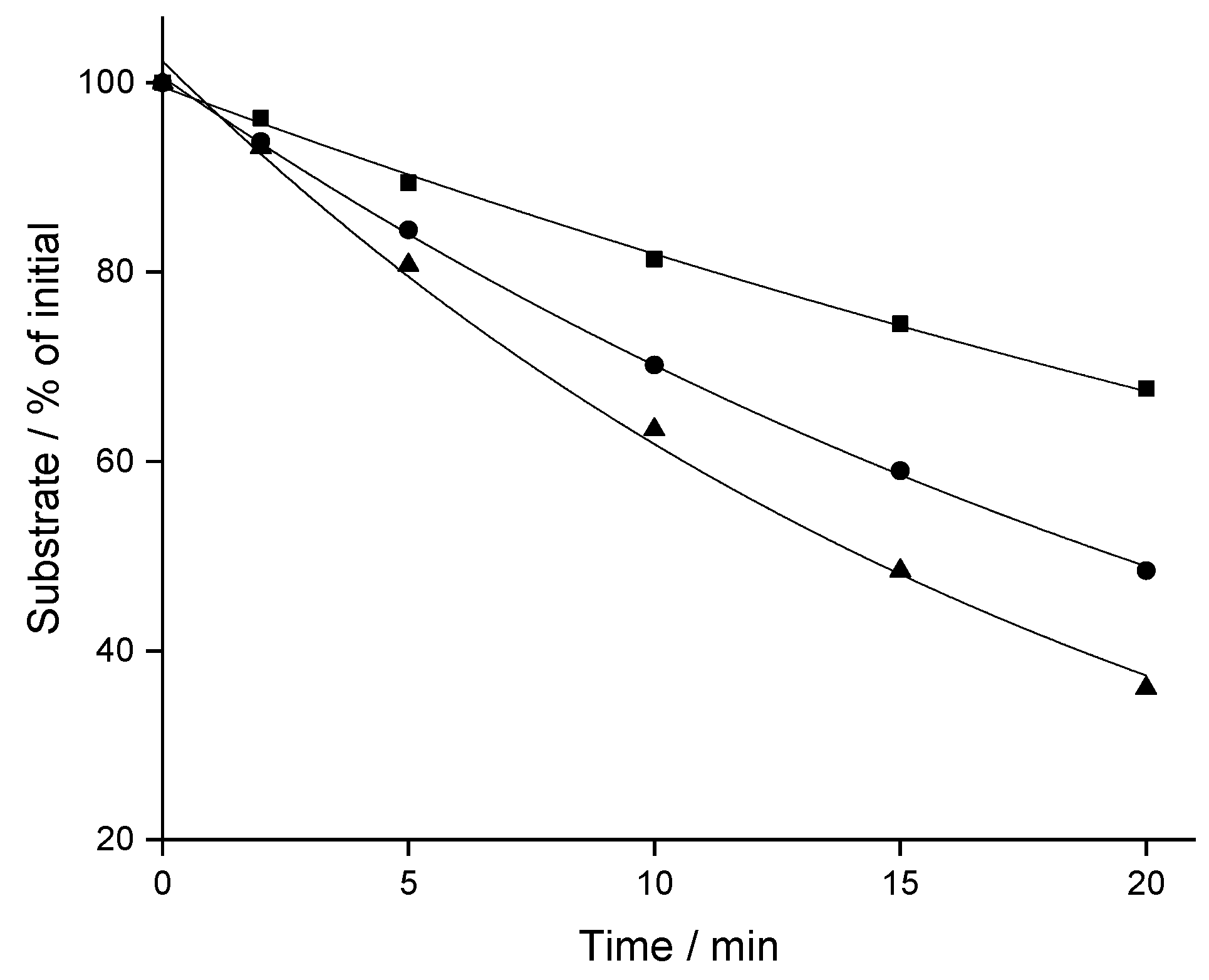
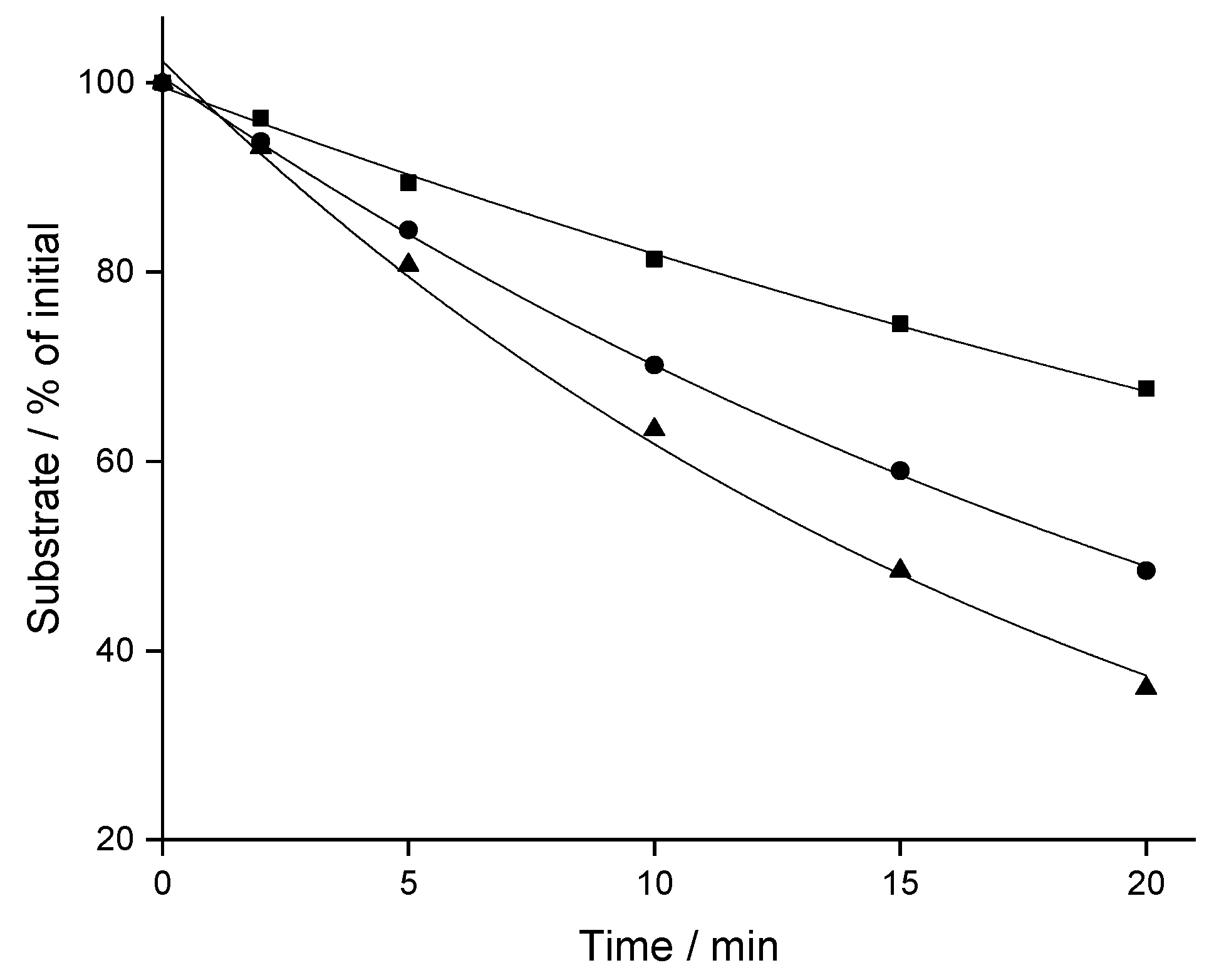
2.1. Stability in Liver Microsomes
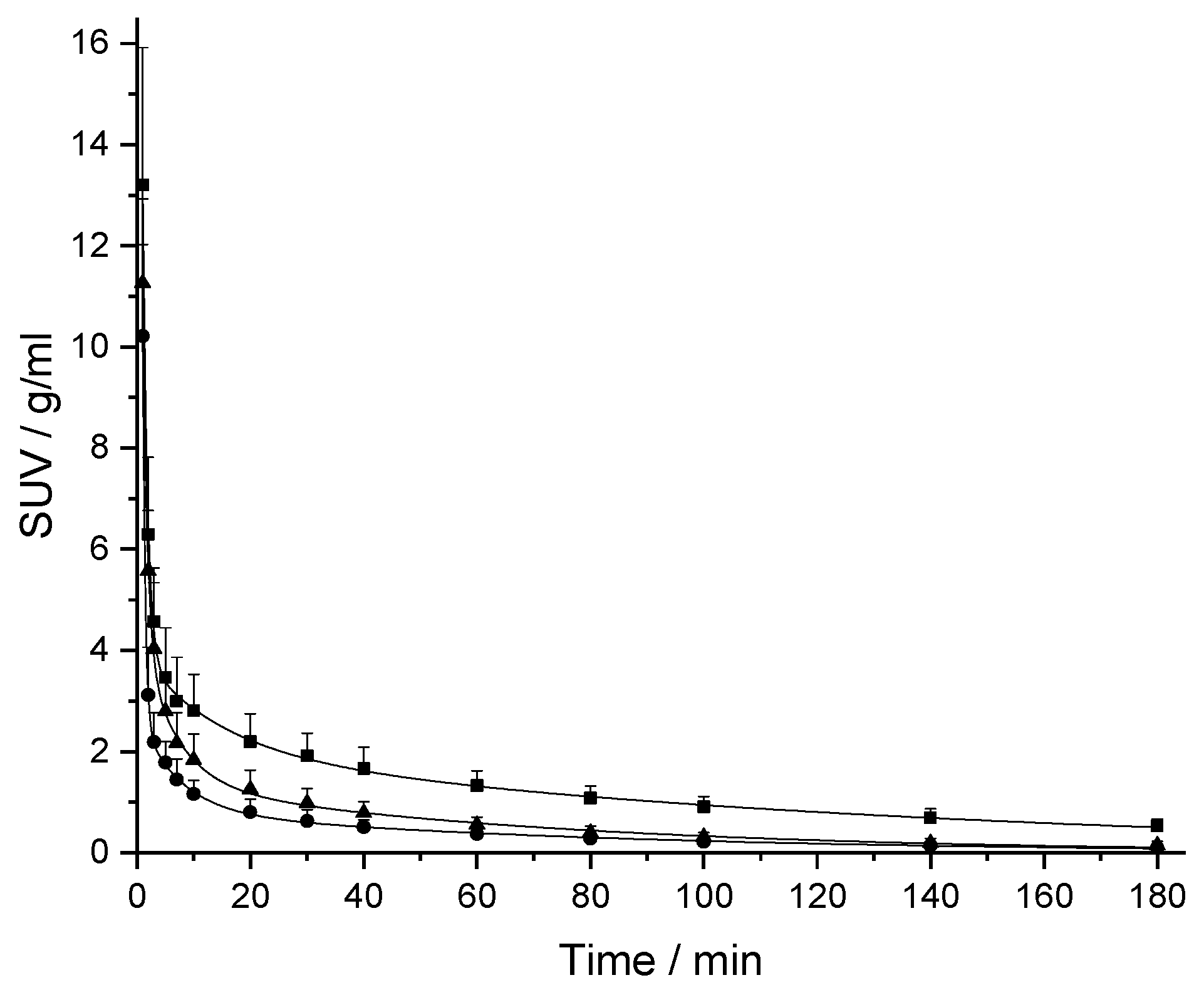
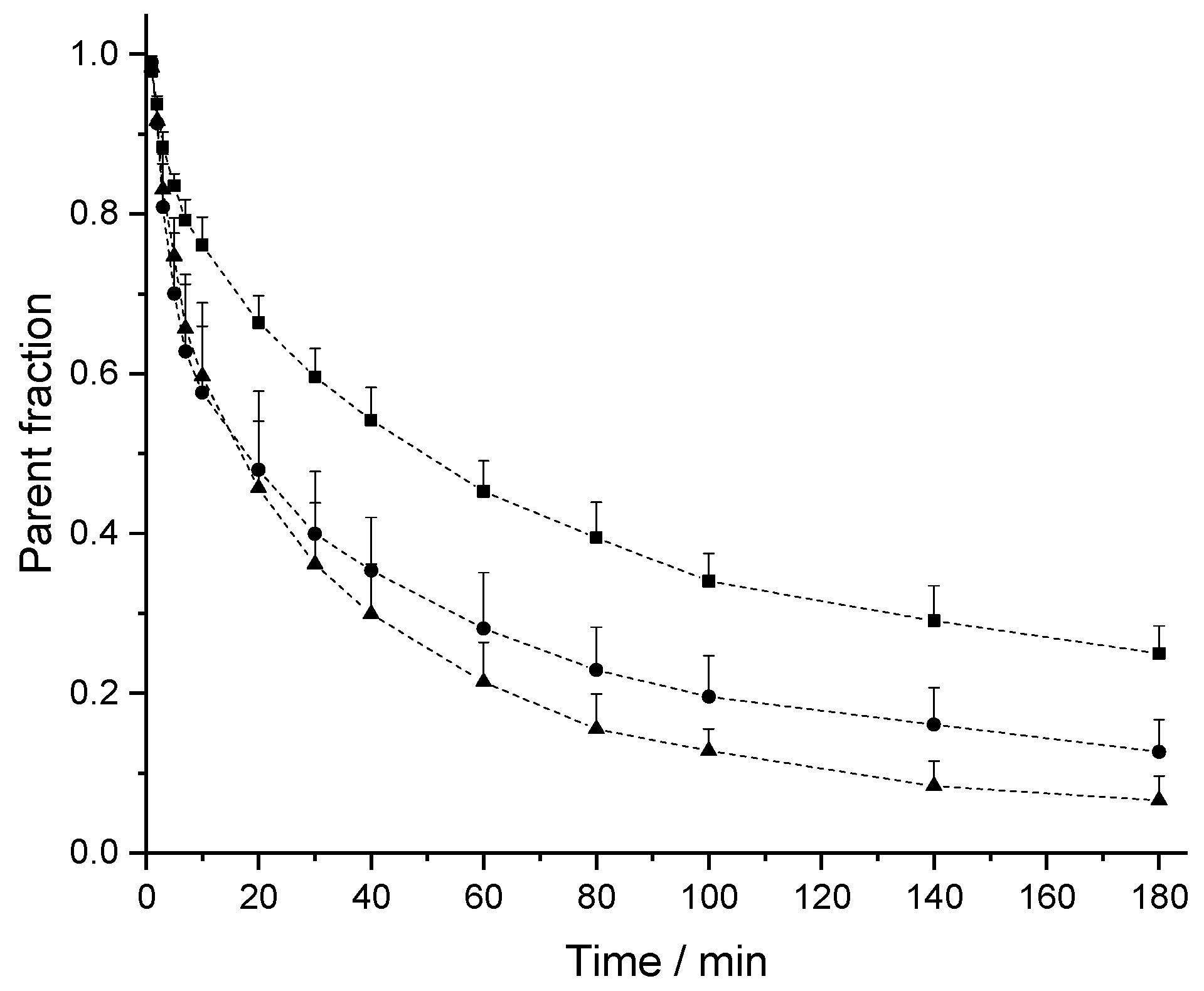
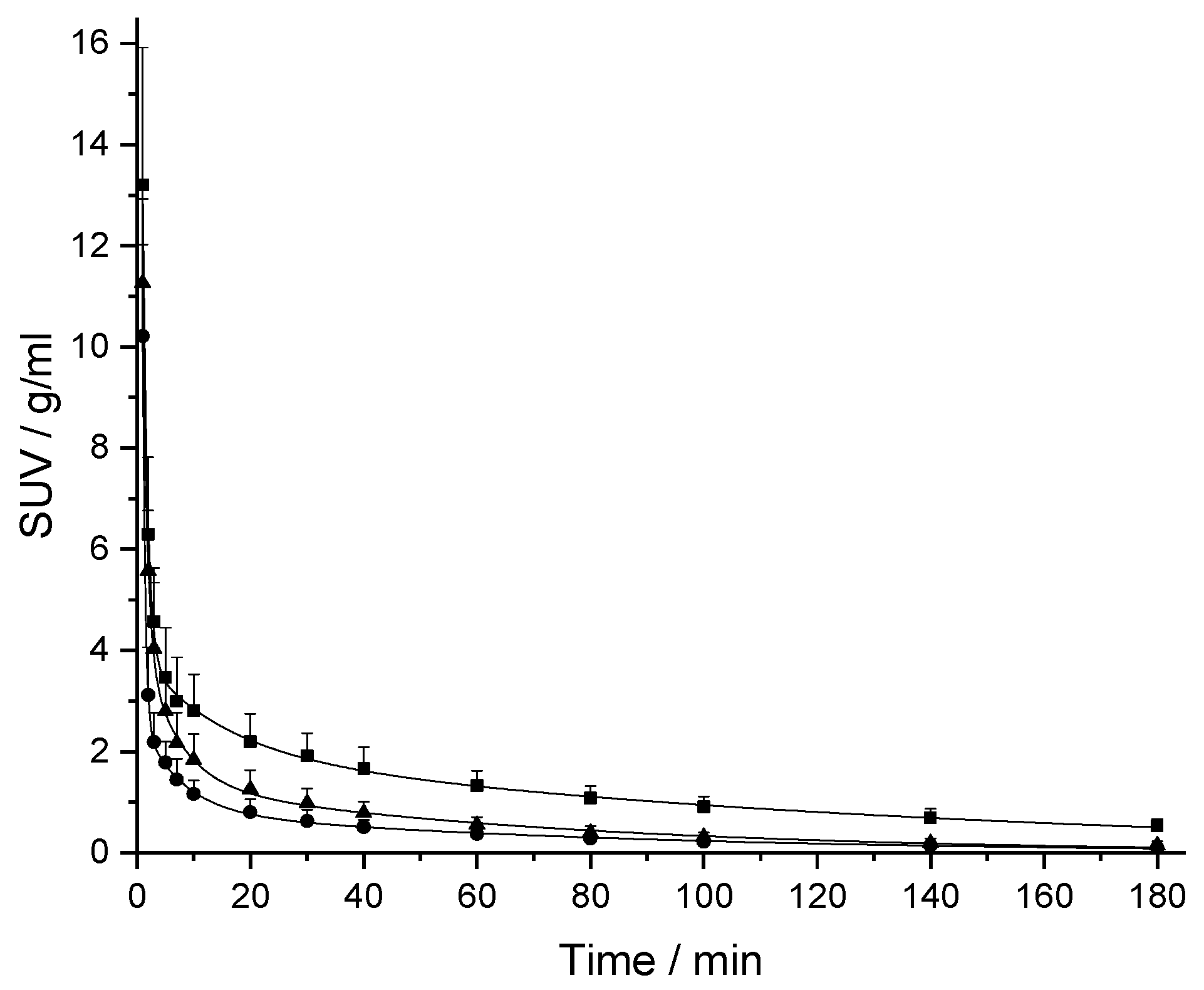
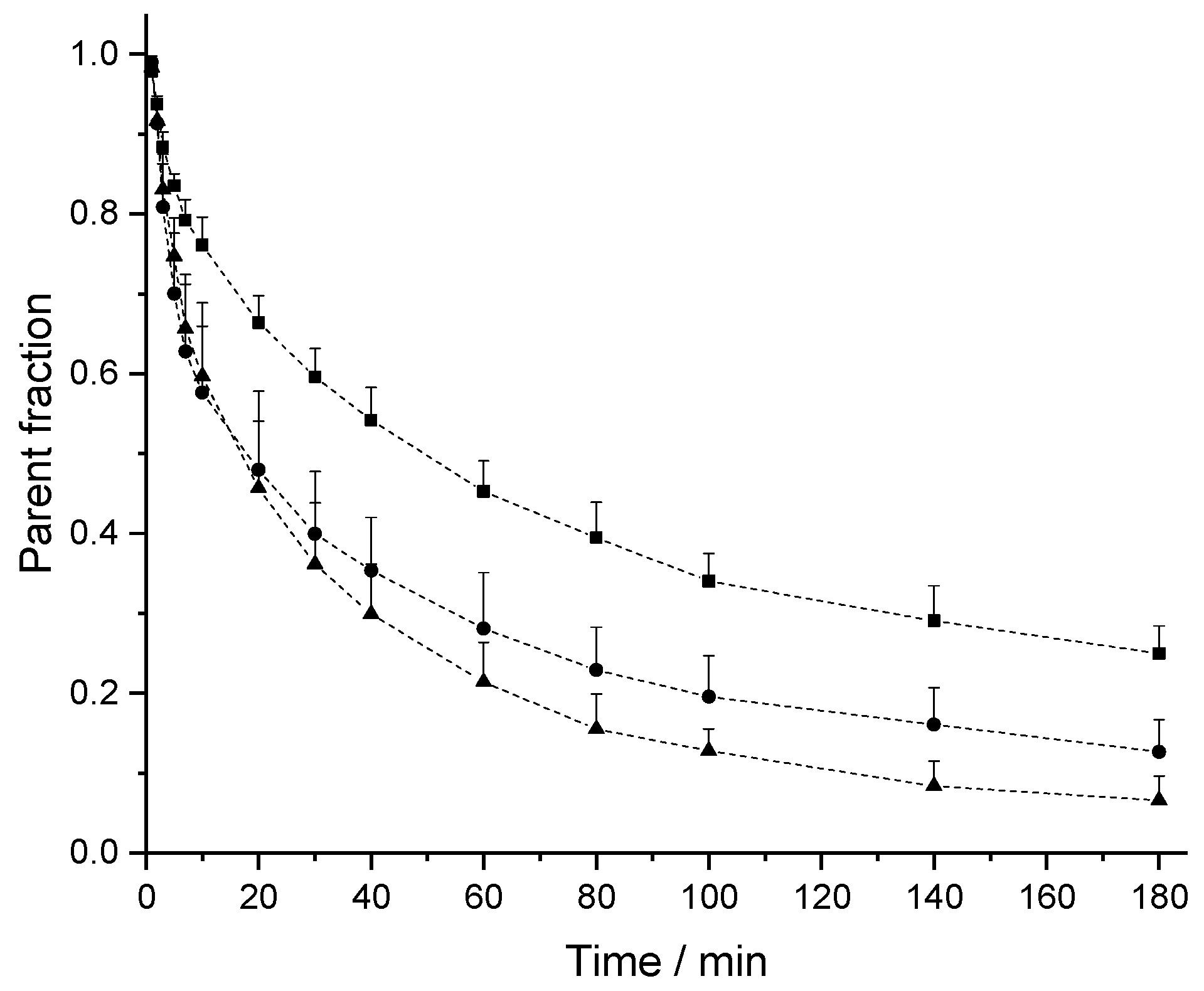
2.2. In Vivo Pharmacokinetic (PK) Studies
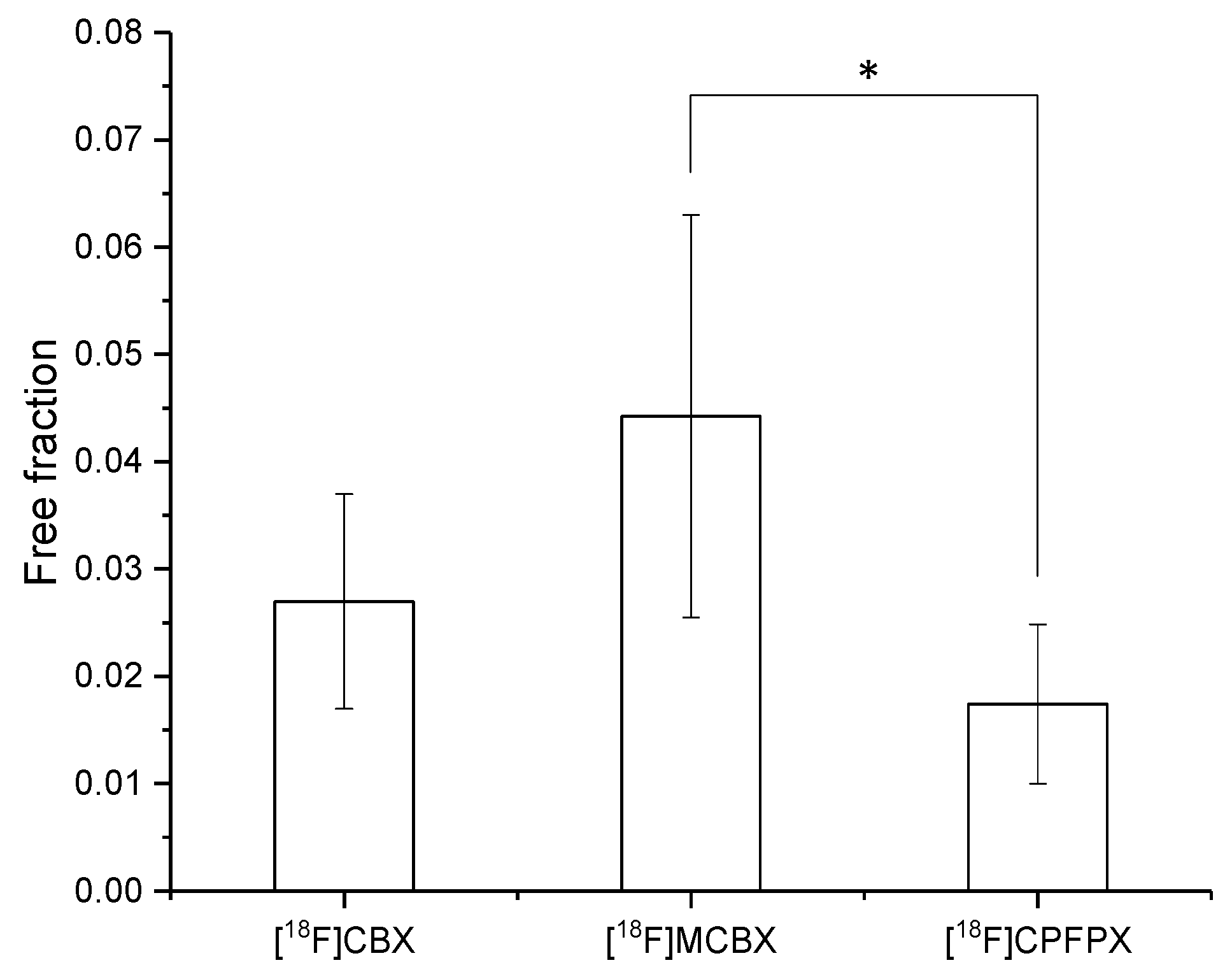
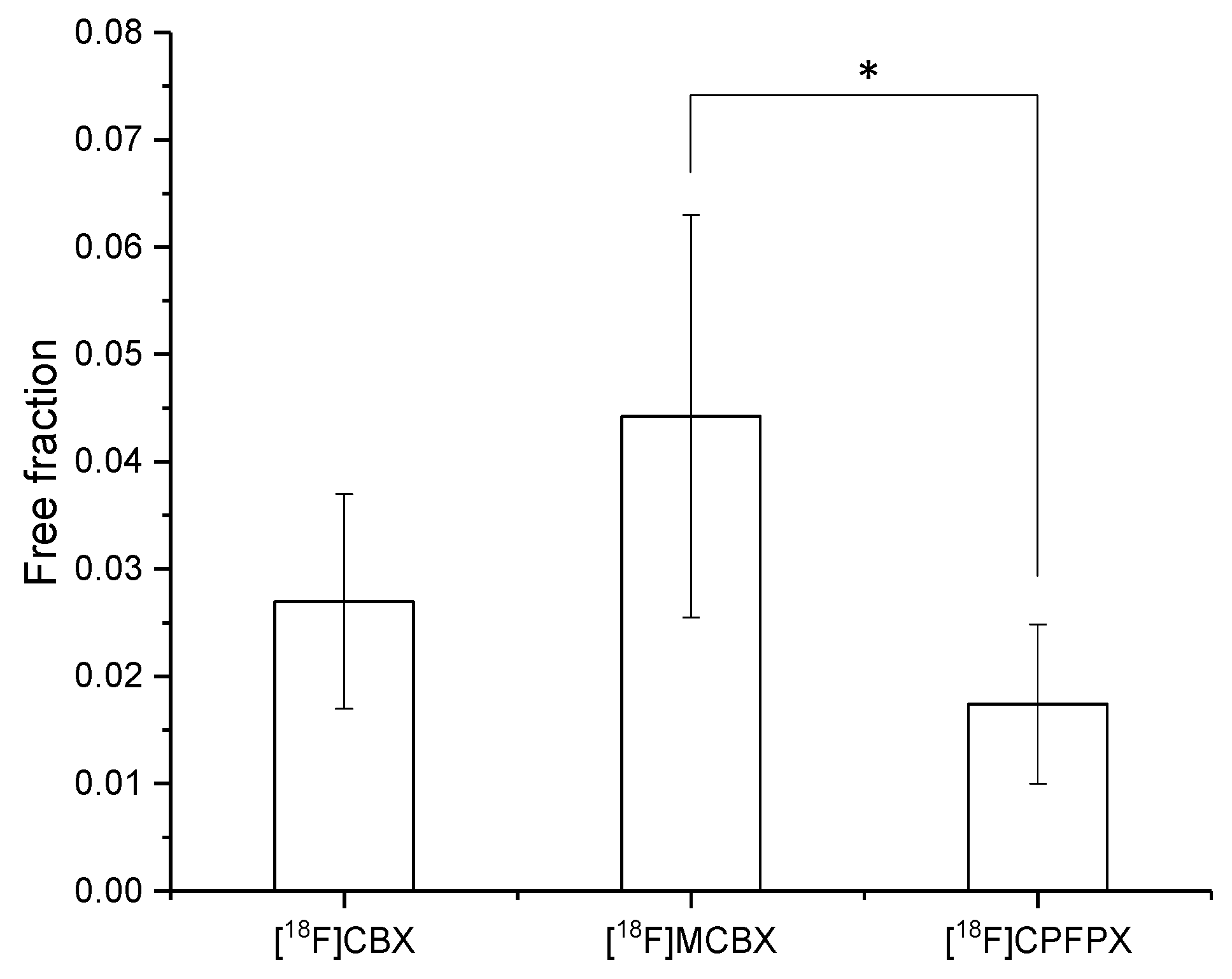
2.3. Plasma Protein Binding
2.4. Prediction of In Vivo Clearance from In Vitro Data
3. Discussion
4. Materials and Methods
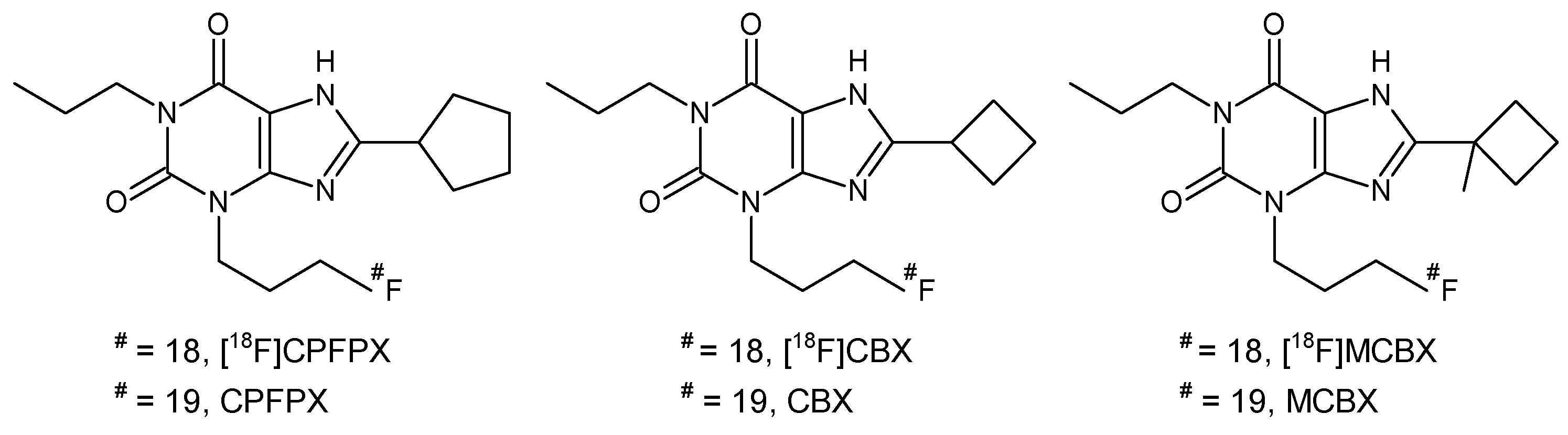
4.1. Compounds, Reagents and Solvents
4.2. Animals
4.3. Radiochemistry
4.4. Microsomal Stability Assays
4.5. Data Analysis
4.6. In Vivo Pharmacokinetics
4.7. Pharmacokinetic Analysis
4.8. Plasma Protein Binding
Author Contributions
Funding
Conflicts of Interest
References
- Pike, V.W. PET radiotracers: crossing the blood-brain barrier and surviving metabolism. Trends Pharmacol. Sci. 2009, 30, 431–440. [Google Scholar] [CrossRef]
- Huang, Y.; Hwang, D.R.; Narendran, R.; Sudo, Y.; Chatterjee, R.; Bae, S.A.; Mawlawi, O.; Kegeles, L.S.; Wilson, A.A.; Kung, H.F.; et al. Comparative evaluation in nonhuman primates of five PET radiotracers for imaging the serotonin transporters: [11C]McN 5652, [11C]ADAM, [11C]DASB, [11C]DAPA, and [11C]AFM. J. Cereb. Blood Flow Metab. 2002, 22, 1377–1398. [Google Scholar] [CrossRef] [PubMed]
- Laruelle, M.; Slifstein, M.; Huang, Y. Relationships between radiotracer properties and image quality in molecular imaging of the brain with positron emission tomography. Mol. Imaging Biol. 2003, 5, 363–375. [Google Scholar] [CrossRef] [PubMed]
- Brandon, E.F.; Raap, C.D.; Meijerman, I.; Beijnen, J.H.; Schellens, J.H. An update on in vitro test methods in human hepatic drug biotransformation research: Pros and cons. Toxicol. Appl. Pharmacol. 2003, 189, 233–246. [Google Scholar] [CrossRef]
- Jia, L.; Liu, X. The conduct of drug metabolism studies considered good practice (II): In vitro experiments. Curr. Drug Metab. 2007, 8, 822–829. [Google Scholar] [CrossRef]
- Lipscomb, J.C.; Poet, T.S. In vitro measurements of metabolism for application in pharmacokinetic modeling. Pharmacol. Ther. 2008, 118, 82–103. [Google Scholar] [CrossRef]
- Pelkonen, O.; Turpeinen, M. In vitro-in vivo extrapolation of hepatic clearance: biological tools, scaling factors, model assumptions and correct concentrations. Xenobiotica 2007, 37, 1066–1089. [Google Scholar] [CrossRef]
- Zhang, D.; Luo, G.; Ding, X.; Lu, C. Preclinical experimental models of drug metabolism and disposition in drug discovery and development. Acta Pharm. Sin. B 2012, 2, 549–561. [Google Scholar] [CrossRef]
- Müller, C.E.; Jacobson, K.A. Xanthines as adenosine receptor antagonists. Handb. Exp. Pharmacol. 2011, 200, 151–199. [Google Scholar] [CrossRef]
- Holschbach, M.H.; Olsson, R.A.; Bier, D.; Wutz, W.; Sihver, W.; Schüller, M.; Palm, B.; Coenen, H.H. Synthesis and Evaluation of No-Carrier-Added 8-Cyclopentyl-3-(3-[18F]fluoropropyl)-1-propylxanthine ([18F]CPFPX): A Potent and Selective A1-Adenosine Receptor Antagonist for in Vivo Imaging. J. Med. Chem. 2002, 45, 5150–5156. [Google Scholar] [CrossRef] [PubMed]
- Bauer, A.; Holschbach, M.H.; Cremer, M.; Weber, S.; Boy, C.; Shah, N.J.; Olsson, R.A.; Halling, H.; Coenen, H.H.; Zilles, K. Evaluation of 18F-CPFPX, a novel adenosine A1 receptor ligand: In vitro autoradiography and high-resolution small animal PET. J. Nucl. Med. 2003, 44, 1682–1689. [Google Scholar]
- Bauer, A.; Holschbach, M.H.; Meyer, P.T.; Boy, C.; Herzog, H.; Olsson, R.A.; Coenen, H.H.; Zilles, K. In vivo imaging of adenosine A1 receptors in the human brain with [18F]CPFPX and positron emission tomography. Neuroimage 2003, 19, 1760–1769. [Google Scholar] [CrossRef]
- Elmenhorst, D.; Elmenhorst, E.M.; Hennecke, E.; Kroll, T.; Matusch, A.; Aeschbach, D.; Bauer, A. Recovery sleep after extended wakefulness restores elevated A1 adenosine receptor availability in the human brain. Proc. Natl. Acad. Sci. USA 2017, 114, 4243–4248. [Google Scholar] [CrossRef] [PubMed]
- Elmenhorst, E.M.; Elmenhorst, D.; Benderoth, S.; Kroll, T.; Bauer, A.; Aeschbach, D. Cognitive impairments by alcohol and sleep deprivation indicate trait characteristics and a potential role for adenosine A1 receptors. Proc. Natl. Acad. Sci. USA 2018, 115, 8009–8014. [Google Scholar] [CrossRef] [PubMed]
- Kroll, T.; Elmenhorst, D.; Weisshaupt, A.; Beer, S.; Bauer, A. Reproducibility of non-invasive A1 adenosine receptor quantification in the rat brain using [18F]CPFPX and positron emission tomography. Mol. Imaging Biol. 2014, 16, 699–709. [Google Scholar] [CrossRef]
- Nabbi-Schroeter, D.; Elmenhorst, D.; Oskamp, A.; Laskowski, S.; Bauer, A.; Kroll, T. Effects of Long-Term Caffeine Consumption on the Adenosine A1 Receptor in the Rat Brain: an In Vivo PET Study with [18F]CPFPX. Mol. Imaging Biol. 2018, 20, 284–291. [Google Scholar] [CrossRef] [PubMed]
- Bier, D.; Holschbach, M.H.; Wutz, W.; Olsson, R.A.; Coenen, H.H. Metabolism of the A1 adenosine receptor positron emission tomography ligand [18F]8-cyclopentyl-3-(3-fluoropropyl)-1-propylxanthine ([18F]CPFPX) in rodents and humans. Drug Metab. Dispos. 2006, 34, 570–576. [Google Scholar] [CrossRef] [PubMed]
- Matusch, A.; Meyer, P.T.; Bier, D.; Holschbach, M.H.; Woitalla, D.; Elmenhorst, D.; Winz, O.H.; Zilles, K.; Bauer, A. Metabolism of the A1 adenosine receptor PET ligand [18F]CPFPX by CYP1A2: implications for bolus/infusion PET studies. Nucl. Med. Biol. 2006, 33, 891–898. [Google Scholar] [CrossRef]
- Kreft, S.; Bier, D.; Holschbach, M.H.; Schulze, A.; Coenen, H.H. New potent A1 adenosine receptor radioligands for positron emission tomography. Nucl. Med. Biol. 2017, 44, 69–77. [Google Scholar] [CrossRef]
- Klopf, W.; Worboys, P. Scaling in vivo pharmacokinetics from in vitro metabolic stability data in drug discovery. Comb. Chem. High Throughput Screen. 2010, 13, 159–169. [Google Scholar] [CrossRef]
- Bergström, M.; Grahnén, A.; Långström, B. Positron emission tomography microdosing: A new concept with application in tracer and early clinical drug development. Eur. J. Clin. Pharmacol. 2003, 59, 357–366. [Google Scholar] [CrossRef] [PubMed]
- Balani, S.K.; Nagaraja, N.V.; Qian, M.G.; Costa, A.O.; Daniels, J.S.; Yang, H.; Shimoga, P.R.; Wu, J.T.; Gan, L.S.; Lee, F.W.; et al. Evaluation of microdosing to assess pharmacokinetic linearity in rats using liquid chromatography-tandem mass spectrometry. Drug. Metab. Dispos. 2006, 34, 384–388. [Google Scholar] [CrossRef] [PubMed]
- Ni, J.; Ouyang, H.; Aiello, M.; Seto, C.; Borbridge, L.; Sakuma, T.; Ellis, R.; Welty, D.; Acheampong, A. Microdosing assessment to evaluate pharmacokinetics and drug metabolism in rats using liquid chromatography-tandem mass spectrometry. Pharm. Res. 2008, 25, 1572–1582. [Google Scholar] [CrossRef] [PubMed]
- Lappin, G.; Noveck, R.; Burt, T. Microdosing and drug development: past, present and future. Expert Opin. Drug Metab. Toxicol. 2013, 9, 817–834. [Google Scholar] [CrossRef]
- Varma, M.V.; Steyn, S.J.; Allerton, C.; El-Kattan, A.F. Predicting Clearance Mechanism in Drug Discovery: Extended Clearance Classification System (ECCS). Pharm. Res. 2015, 32, 3785–3802. [Google Scholar] [CrossRef]
- Ward, L.C.; Battersby, K.J. Assessment of body composition of rats by bioimpedance spectroscopy: Validation against dual-energy x-ray absorptiometry. Scand. J. Lab. Anim. Sci. 2009, 36, 253–261. [Google Scholar]
- Cornish, B.H.; Ward, L.C.; Thomas, B.J. Measurement of Extracellular and Total Body Water of Rats Using Multiple Frequency Bioelectrical Impedance Analysis. Nutr. Res. 1992, 12, 657–666. [Google Scholar] [CrossRef]
- Obach, R.S. Prediction of human clearance of twenty-nine drugs from hepatic microsomal intrinsic clearance data: An examination of in vitro half-life approach and nonspecific binding to microsomes. Drug. Metab. Dispos. 1999, 27, 1350–1359. [Google Scholar]
- Wood, F.L.; Houston, J.B.; Hallifax, D. Clearance Prediction Methodology Needs Fundamental Improvement: Trends Common to Rat and Human Hepatocytes/Microsomes and Implications for Experimental Methodology. Drug. Metab. Dispos. 2017, 45, 1178–1188. [Google Scholar] [CrossRef]
- Bowman, C.M.; Benet, L.Z. Hepatic Clearance Predictions from In Vitro-In Vivo Extrapolation and the Biopharmaceutics Drug Disposition Classification System. Drug. Metab. Dispos. 2016, 44, 1731–1735. [Google Scholar] [CrossRef]
- Ring, B.J.; Chien, J.Y.; Adkison, K.K.; Jones, H.M.; Rowland, M.; Jones, R.D.; Yates, J.W.; Ku, M.S.; Gibson, C.R.; He, H.; et al. PhRMA CPCDC initiative on predictive models of human pharmacokinetics, part 3: Comparative assessement of prediction methods of human clearance. J. Pharm. Sci. 2011, 100, 4090–4110. [Google Scholar] [CrossRef] [PubMed]
- Ye, M.; Nagar, S.; Korzekwa, K. A physiologically based pharmacokinetic model to predict the pharmacokinetics of highly protein-bound drugs and the impact of errors in plasma protein binding. Biopharm. Drug Dispos. 2016, 37, 123–141. [Google Scholar] [CrossRef] [PubMed]
- Hallifax, D.; Houston, J.B. Evaluation of hepatic clearance prediction using in vitro data: Emphasis on fraction unbound in plasma and drug ionisation using a database of 107 drugs. J. Pharm. Sci. 2012, 101, 2645–2652. [Google Scholar] [CrossRef]
- Baker, M.; Parton, T. Kinetic determinants of hepatic clearance: Plasma protein binding and hepatic uptake. Xenobiotica. 2007, 37, 1110–1134. [Google Scholar] [CrossRef]
- Bohnert, T.; Gan, L.S. Plasma protein binding: From discovery to development. J. Pharm. Sci. 2013, 102, 2953–2994. [Google Scholar] [CrossRef] [PubMed]
- Smith, D.A.; Di, L.; Kerns, E.H. The effect of plasma protein binding on in vivo efficacy: Misconceptions in drug discovery. Nat. Rev. Drug Discov. 2010, 9, 929–939. [Google Scholar] [CrossRef]
- Schneider, D.; Bier, D.; Bauer, A.; Neumaier, B.; Holschbach, M. Influence of incubation conditions on microsomal metabolism of xanthine-derived A1 adenosine receptor ligands. J. Pharmacol. Toxicol. Methods 2019, 95, 16–26. [Google Scholar] [CrossRef]
- Obach, R.S.; Baxter, J.G.; Liston, T.E.; Silber, B.M.; Jones, B.C.; MacIntyre, F.; Rance, D.J.; Wastall, P. The prediction of human pharmacokinetic parameters from preclinical and in vitro metabolism data. J. Pharmacol. Exp. Ther. 1997, 283, 46–58. [Google Scholar] [PubMed]
- Carlile, D.J.; Zomorodi, K.; Houston, J.B. Scaling factors to relate drug metabolic clearance in hepatic microsomes, isolated hepatocytes, and the intact liver: Studies with induced livers involving diazepam. Drug Metab. Dispos. 1997, 25, 903–911. [Google Scholar] [PubMed]
- Davies, B.; Morris, T. Physiological parameters in laboratory animals and humans. Pharm. Res. 1993, 10, 1093–1095. [Google Scholar] [CrossRef]
- Hallifax, D.; Houston, J.B. Binding of drugs to hepatic microsomes: comment and assessment of current prediction methodology with recommendation for improvement. Drug Metab. Dispos. 2006, 34, 724–726. [Google Scholar] [CrossRef] [PubMed]
- Rowland, M.; Benet, L.Z.; Graham, G.G. Clearance concepts in pharmacokinetics. J. Pharmacokinet. Biopharm. 1973, 1, 123–136. [Google Scholar] [CrossRef] [PubMed]
- Wilkinson, G.R.; Shand, D.G. A physiological approach to hepatic drug clearance. Clin. Pharmacol. Ther. 1975, 18, 377–390. [Google Scholar] [CrossRef] [PubMed]
- Yamaoka, K.; Nakagawa, T.; Uno, T. Statistical moments in pharmacokinetics. J. Pharmacokinet. Biopharm. 1978, 6, 547–558. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | Log P | fmic Calculated | In vitro t1/2 (SD) min | CLint (SD) mL/min/kg | CLp Predicted (SD) mL/min/kg |
|---|---|---|---|---|---|
| CBX | 2.19 | 0.90 | 35.1 (1.7) | 106.1 (5.4) | 2.72 (0.13) |
| MCBX | 2.82 | 0.82 | 19.8 (0.7) | 204.1 (6.8) | 7.76 (0.22) |
| CPFPX | 2.93 | 0.80 | 14.0 (0.2) | 295.0 (5.0) | 4.70 (0.07) |
| Compound | Vd ml/kg | t1/2,term min | CLp ml/min/kg | Fold Error CL | Fold Underprediction CL |
|---|---|---|---|---|---|
| [18F]CBX | 356 | 76.5 | 3.22 | 0.84 | 1.2 |
| [18F]MCBX | 715 | 54.5 | 9.10 | 0.85 | 1.2 |
| [18F]CPFPX | 523 | 51.0 | 7.10 | 0.66 | 1.5 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Schneider, D.; Oskamp, A.; Holschbach, M.; Neumaier, B.; Bauer, A.; Bier, D. Relevance of In Vitro Metabolism Models to PET Radiotracer Development: Prediction of In Vivo Clearance in Rats from Microsomal Stability Data. Pharmaceuticals 2019, 12, 57. https://doi.org/10.3390/ph12020057
Schneider D, Oskamp A, Holschbach M, Neumaier B, Bauer A, Bier D. Relevance of In Vitro Metabolism Models to PET Radiotracer Development: Prediction of In Vivo Clearance in Rats from Microsomal Stability Data. Pharmaceuticals. 2019; 12(2):57. https://doi.org/10.3390/ph12020057
Chicago/Turabian StyleSchneider, Daniela, Angela Oskamp, Marcus Holschbach, Bernd Neumaier, Andreas Bauer, and Dirk Bier. 2019. "Relevance of In Vitro Metabolism Models to PET Radiotracer Development: Prediction of In Vivo Clearance in Rats from Microsomal Stability Data" Pharmaceuticals 12, no. 2: 57. https://doi.org/10.3390/ph12020057
APA StyleSchneider, D., Oskamp, A., Holschbach, M., Neumaier, B., Bauer, A., & Bier, D. (2019). Relevance of In Vitro Metabolism Models to PET Radiotracer Development: Prediction of In Vivo Clearance in Rats from Microsomal Stability Data. Pharmaceuticals, 12(2), 57. https://doi.org/10.3390/ph12020057