Proximal Pathway Enrichment Analysis for Targeting Comorbid Diseases via Network Endopharmacology

 , , ,
, , ,
Abstract
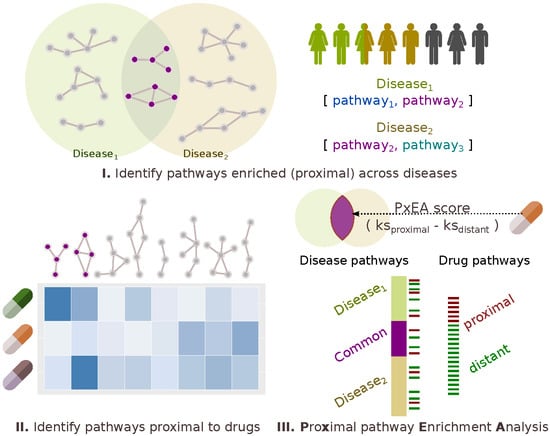
1. Introduction
2. Results
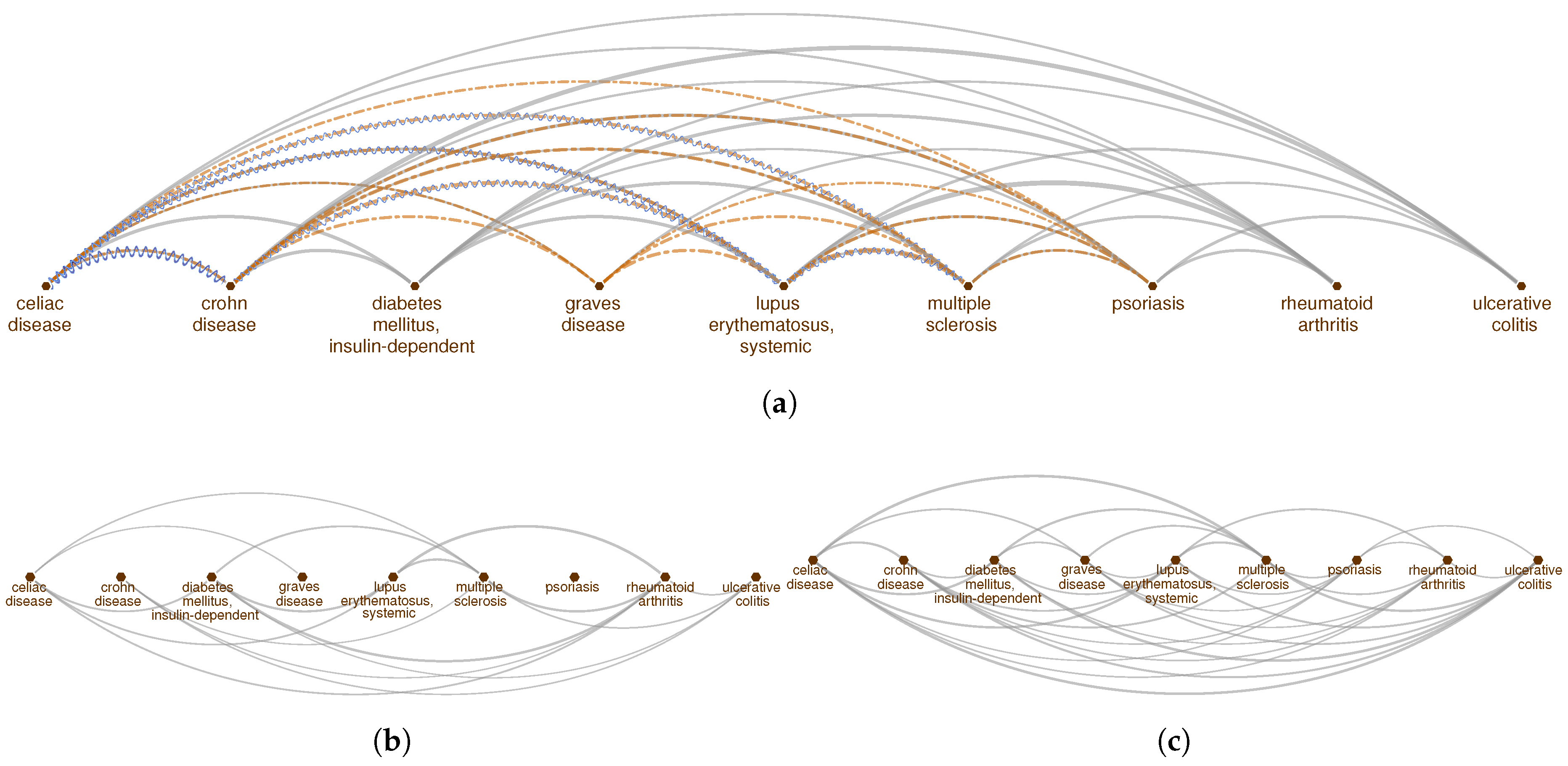
2.1. Pathway Proximity Captures the Similarities between Autoimmune Disorders
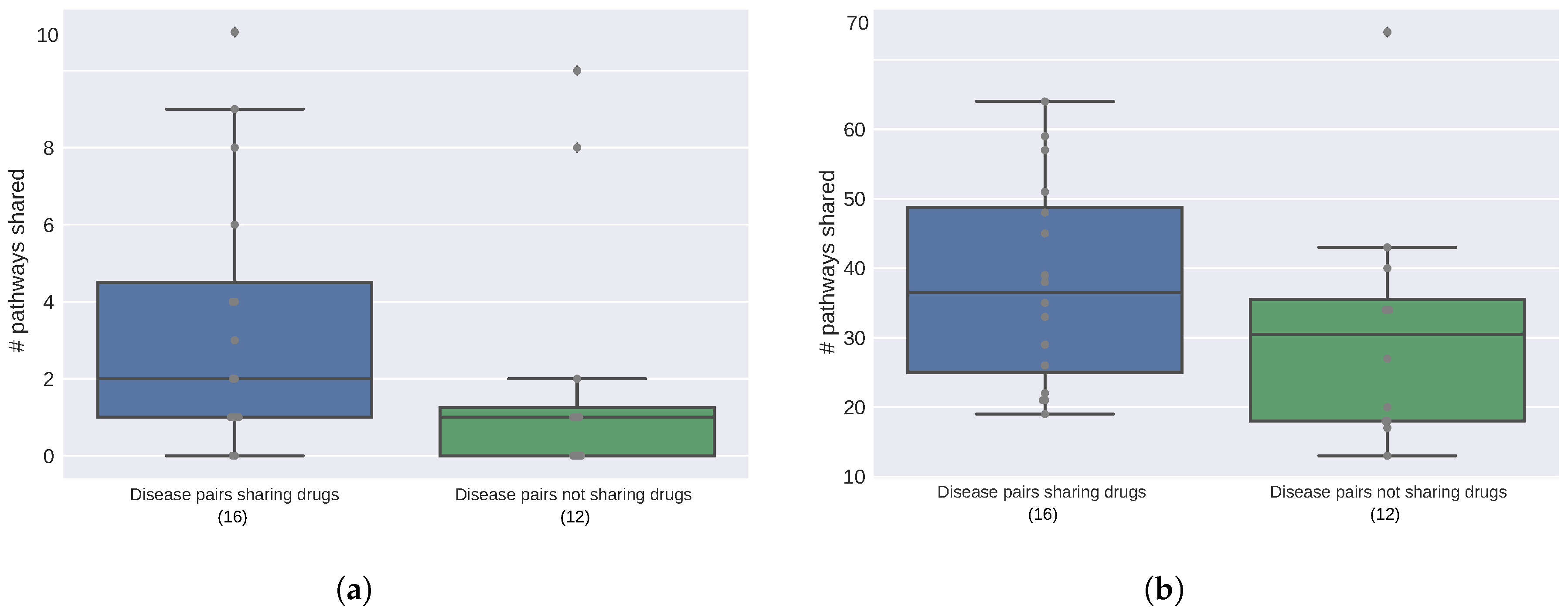
2.2. Diseases Targeted by the Same Drugs Exhibit Functional Similarities
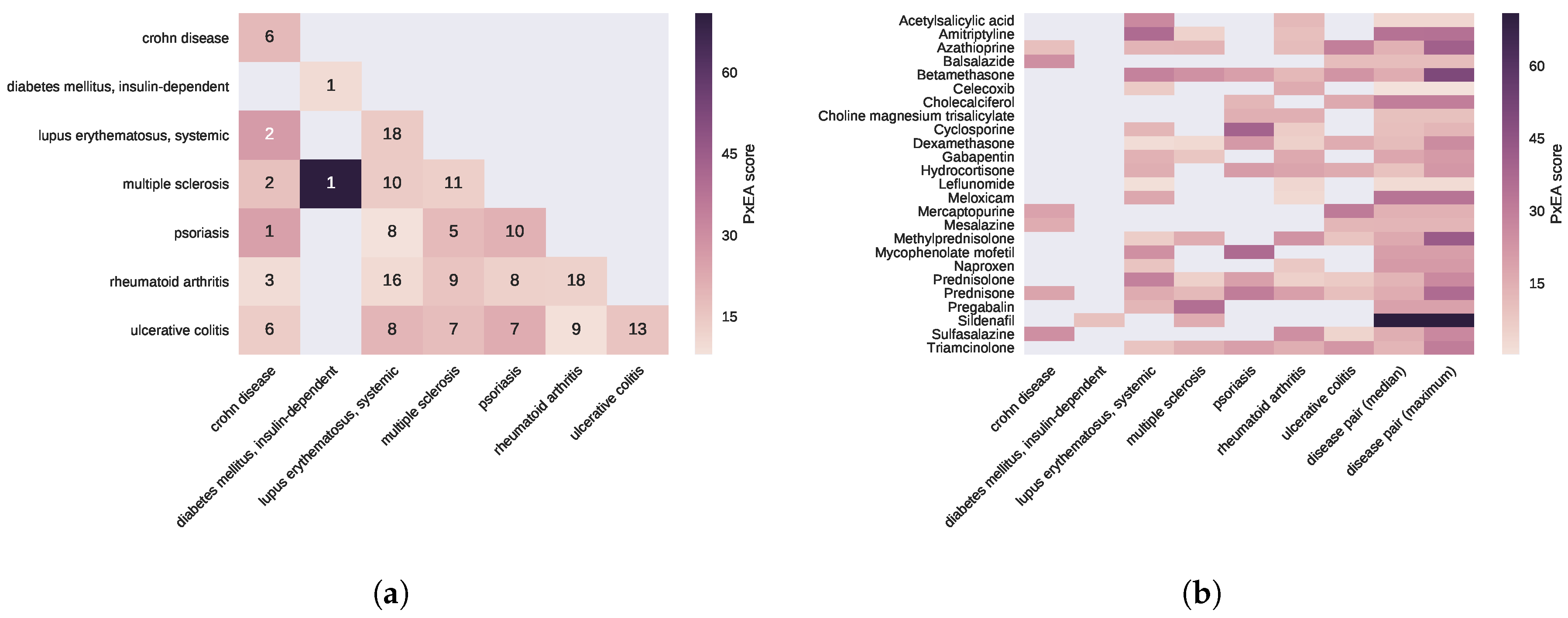
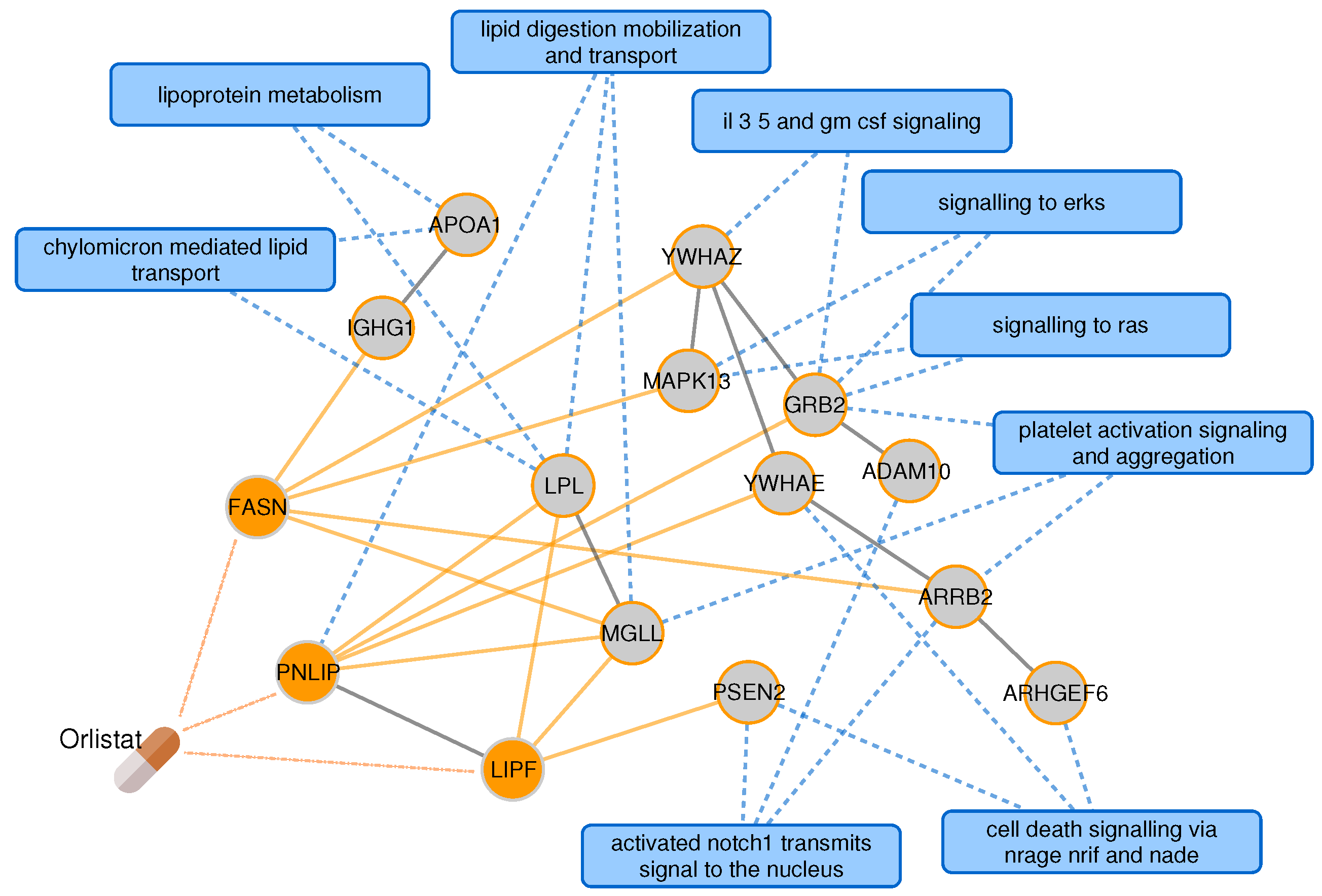
2.3. Proximal Pathway Enrichment Analysis Reveals Drugs Targeting the Autoimmune Endophenotypes
2.4. Targeting the Common Pathology of Type 2 Diabetes and Alzheimer’s Disease
3. Discussion
4. Materials and Methods
4.1. Protein Interaction Data and Interactome-Based Proximity
4.2. Disease-Gene, Drug and Pathway Information
4.3. Genetic, Phenotypic and Functional Relationships across Diseases
4.4. PxEA: Proximal Pathway Enrichment Analysis
4.5. Implementation Details and Code Availability
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| AD | Alzheimer’s disease |
| ATC | Anatomical Therapeutic Chemical |
| GSEA | Gene set enrichment analysis |
| PxEA | Proximal pathway enrichment analysis |
| T2D | Type 2 diabetes |
| TF-IDF | Time frequency-inverse document frequency approach |
References
- Strebhardt, K.; Ullrich, A. Paul Ehrlich’s magic bullet concept: 100 years of progress. Nat. Rev. Cancer 2008, 8, 473–480. [Google Scholar] [CrossRef] [PubMed]
- Csermely, P.; Korcsmáros, T.; Kiss, H.J.; London, G.; Nussinov, R. Structure and dynamics of molecular networks: A novel paradigm of drug discovery: A comprehensive review. Pharmacol. Ther. 2013, 138, 333–408. [Google Scholar] [CrossRef] [PubMed]
- Allison, M. Reinventing clinical trials. Nat. Biotechnol. 2012, 30, 41–49. [Google Scholar] [CrossRef] [PubMed]
- Hay, M.; Thomas, D.W.; Craighead, J.L.; Economides, C.; Rosenthal, J. Clinical development success rates for investigational drugs. Nat. Biotechnol. 2014, 32, 40–51. [Google Scholar] [CrossRef] [PubMed]
- Hopkins, A.L. Network pharmacology: The next paradigm in drug discovery. Nat. Chem. Biol. 2008, 4, 682–690. [Google Scholar] [CrossRef] [PubMed]
- Pujol, A.; Mosca, R.; Farres, J.; Aloy, P. Unveiling the role of network and systems biology in drug discovery. Trends Pharmacol. Sci. 2010, 31, 115–123. [Google Scholar] [CrossRef] [PubMed]
- Guney, E.; Menche, J.; Vidal, M.; Barábasi, A.L. Network-based in silico drug efficacy screening. Nat. Commun. 2016, 7, 10331. [Google Scholar] [CrossRef] [PubMed]
- Jaeger, S.; Igea, A.; Arroyo, R.; Alcalde, V.; Canovas, B.; Orozco, M.; Nebreda, A.R.; Aloy, P. Quantification of Pathway Cross-talk Reveals Novel Synergistic Drug Combinations for Breast Cancer. Cancer Res. 2017, 77, 459–469. [Google Scholar] [CrossRef] [PubMed]
- Jin, G.; Wong, S.T.C. Toward better drug repositioning: Prioritizing and integrating existing methods into efficient pipelines. Drug Discov. Today 2014, 19, 637–644. [Google Scholar] [CrossRef] [PubMed]
- Hodos, R.A.; Kidd, B.A.; Shameer, K.; Readhead, B.P.; Dudley, J.T. In silico methods for drug repurposing and pharmacology. Wiley Interdiscip. Rev. Syst. Biol. Med. 2016, 8, 186–210. [Google Scholar] [CrossRef] [PubMed]
- Vilar, S.; Hripcsak, G. The role of drug profiles as similarity metrics: Applications to repurposing, adverse effects detection and drug–drug interactions. Brief. Bioinform. 2016, 18, 670–681. [Google Scholar] [CrossRef] [PubMed]
- Loscalzo, J.; Kohane, I.; Barabasi, A.L. Human disease classification in the postgenomic era: A complex systems approach to human pathobiology. Mol. Syst. Biol. 2007, 3, 124. [Google Scholar] [CrossRef] [PubMed]
- Duran-Frigola, M.; Mateo, L.; Aloy, P. Drug repositioning beyond the low-hanging fruits. Curr. Opin. Syst. Biol. 2017, 3, 95–102. [Google Scholar] [CrossRef]
- Li, Y.; Agarwal, P.; Rajagopalan, D. A global pathway crosstalk network. Bioinformatics 2008, 24, 1442–1447. [Google Scholar] [CrossRef] [PubMed]
- Garcia-Garcia, J.; Guney, E.; Aragues, R.; Planas-Iglesias, J.; Oliva, B. Biana: A software framework for compiling biological interactions and analyzing networks. BMC Bioinform. 2010, 11, 56. [Google Scholar] [CrossRef] [PubMed]
- Menche, J.; Sharma, A.; Kitsak, M.; Ghiassian, S.D.; Vidal, M.; Loscalzo, J.; Barabási, A.L. Disease networks. Uncovering disease-disease relationships through the incomplete interactome. Science 2015, 347, 1257601. [Google Scholar] [CrossRef] [PubMed]
- Ko, Y.; Cho, M.; Lee, J.S.; Kim, J. Identification of disease comorbidity through hidden molecular mechanisms. Sci. Rep. 2016, 6, 39433. [Google Scholar] [CrossRef] [PubMed]
- Rubio-Perez, C.; Guney, E.; Aguilar, D.; Piñero, J.; Garcia-Garcia, J.; Iadarola, B.; Sanz, F.; Fernandez-Fuentes, N.; Furlong, L.I.; Oliva, B. Genetic and functional characterization of disease associations explains comorbidity. Sci. Rep. 2017, 7, 6207. [Google Scholar] [CrossRef] [PubMed]
- Cuadrado, A.; Manda, G.; Hassan, A.; Alcaraz, M.J.; Barbas, C.; Daiber, A.; Ghezzi, P.; León, R.; López, M.G.; Oliva, B.; et al. Transcription Factor NRF2 as a Therapeutic Target for Chronic Diseases: A Systems Medicine Approach. Pharmacol. Rev. 2018, 70, 348–383. [Google Scholar] [CrossRef] [PubMed]
- Toro-Domínguez, D.; Carmona-Sáez, P.; Alarcón-Riquelme, M.E. Shared signatures between rheumatoid arthritis, systemic lupus erythematosus and Sjögren’s syndrome uncovered through gene expression meta-analysis. Arthritis Res. Ther. 2014, 16, 489. [Google Scholar] [CrossRef] [PubMed]
- Luan, M.; Shang, Z.; Teng, Y.; Chen, X.; Zhang, M.; Lv, H.; Zhang, R. The shared and specific mechanism of four autoimmune diseases. Oncotarget 2017, 8, 108355–108374. [Google Scholar] [CrossRef] [PubMed]
- American Autoimmune Related Diseases Association. Autoimmune Disease Statistics. Available online: www.aarda.org/news-information/statistics (accessed on 13 June 2018).
- Baranzini, S.E. Chapter 70—Autoimmune Disorders. In Genomic and Personalized Medicine, 2nd ed.; Ginsburg, G.S., Willard, H.F., Eds.; Academic Press: Cambridge, MA, USA, 2013; pp. 822–838. [Google Scholar] [CrossRef]
- Gottesman, I.I.; Gould, T.D. The Endophenotype Concept in Psychiatry: Etymology and Strategic Intentions. Am. J. Psychiatry 2003, 160, 636–645. [Google Scholar] [CrossRef] [PubMed]
- Ghiassian, S.D.; Menche, J.; Chasman, D.I.; Giulianini, F.; Wang, R.; Ricchiuto, P.; Aikawa, M.; Iwata, H.; Müller, C.; Zeller, T.; et al. Endophenotype Network Models: Common Core of Complex Diseases. Sci. Rep. 2016, 6, 27414. [Google Scholar] [CrossRef] [PubMed]
- Glaab, E.; Baudot, A.; Krasnogor, N.; Schneider, R.; Valencia, A. EnrichNet: Network-based gene set enrichment analysis. Bioinformatics 2012, 28, i451–i457. [Google Scholar] [CrossRef] [PubMed]
- Himmelstein, D.S.; Lizee, A.; Hessler, C.; Brueggeman, L.; Chen, S.L.; Hadley, D.; Green, A.; Khankhanian, P.; Baranzini, S.E. Systematic integration of biomedical knowledge prioritizes drugs for repurposing. eLife 2018, 6. [Google Scholar] [CrossRef]
- Lamb, J.; Crawford, E.D.; Peck, D.; Modell, J.W.; Blat, I.C.; Wrobel, M.J.; Lerner, J.; Brunet, J.P.; Subramanian, A.; Ross, K.N.; et al. The Connectivity Map: Using Gene-Expression Signatures to Connect Small Molecules, Genes, and Disease. Science 2006, 313, 1929–1935. [Google Scholar] [CrossRef] [PubMed]
- Sims-Robinson, C.; Kim, B.; Rosko, A.; Feldman, E.L. How does diabetes accelerate Alzheimer disease pathology? Nat. Rev. Neurol. 2010, 6, 551–559. [Google Scholar] [CrossRef] [PubMed]
- Hiltunen, M.; Khandelwal, V.K.M.; Yaluri, N.; Tiilikainen, T.; Tusa, M.; Koivisto, H.; Krzisch, M.; Vepsäläinen, S.; Mäkinen, P.; Kemppainen, S.; et al. Increased risk of type 2 diabetes in Alzheimer disease. J. Cell. Mol. Med. 2012, 16, 1206–1222. [Google Scholar] [CrossRef] [PubMed]
- Yarchoan, M.; Arnold, S.E. Repurposing Diabetes Drugs for Brain Insulin Resistance in Alzheimer Disease. Diabetes 2014, 63, 2253–2261. [Google Scholar] [CrossRef] [PubMed]
- Du, J.; Wang, Z. Therapeutic potential of lipase inhibitor orlistat in Alzheimer’s disease. Med. Hypotheses 2009, 73, 662–663. [Google Scholar] [CrossRef] [PubMed]
- Mahmoudiandehkordi, S.; Arnold, M.; Nho, K.; Ahmad, S.; Jia, W.; Xia, G.; Louie, G.; Kueider, A.; Moseley, M.A.; Thompson, J.W.; et al. Altered Bile Acid Profile Associates with Cognitive Impairment in Alzheimer’s Disease: An Emerging Role for Gut Microbiome. bioRxiv 2018, 281956. [Google Scholar] [CrossRef]
- Guerciolini, R. Mode of action of orlistat. Int. J. Obes. Relat. Metab. Disord. 1997, 21, S12–S23. [Google Scholar] [PubMed]
- Snowden, S.G.; Ebshiana, A.A.; Hye, A.; An, Y.; Pletnikova, O.; O’Brien, R.; Troncoso, J.; Legido-Quigley, C.; Thambisetty, M. Association between fatty acid metabolism in the brain and Alzheimer disease neuropathology and cognitive performance: A nontargeted metabolomic study. PLoS Med. 2017, 14, e1002266. [Google Scholar] [CrossRef] [PubMed]
- Daugherty, D.; Goldberg, J.; Fischer, W.; Dargusch, R.; Maher, P.; Schubert, D. A novel Alzheimer’s disease drug candidate targeting inflammation and fatty acid metabolism. Alzheimers Res. Ther. 2017, 9, 50. [Google Scholar] [CrossRef] [PubMed]
- Ramos, B.P.; Colgan, L.A.; Nou, E.; Arnsten, A.F. β2 adrenergic agonist, clenbuterol, enhances working memory performance in aging animals. Neurobiol. Aging 2008, 29, 1060–1069. [Google Scholar] [CrossRef] [PubMed]
- Chai, G.S.; Wang, Y.Y.; Yasheng, A.; Zhao, P. Beta 2-adrenergic receptor activation enhances neurogenesis in Alzheimer’s disease mice. Neural Regener. Res. 2016, 11, 1617–1624. [Google Scholar] [CrossRef]
- Pan, S.J.; Hancock, J.; Ding, Z.; Fogt, D.; Lee, M.; Ivy, J.L. Effects of clenbuterol on insulin resistance in conscious obese Zucker rats. Am. J. Physiol. Endocrinol. Metab. 2001, 280, E554–E561. [Google Scholar] [CrossRef] [PubMed]
- Ceol, A.; Aryamontri, A.C.; Licata, L.; Peluso, D.; Briganti, L.; Perfetto, L.; Castagnoli, L.; Cesareni, G. MINT, the molecular interaction database: 2009 update. Nucleic Acids Res. 2010, 38, D532–D539. [Google Scholar] [CrossRef] [PubMed]
- Stark, C.; Breitkreutz, B.J.; Chatr-aryamontri, A.; Boucher, L.; Oughtred, R.; Livstone, M.S.; Nixon, J.; Van Auken, K.; Wang, X.; Shi, X.; et al. The BioGRID Interaction Database: 2011 update. Nucleic Acids Res. 2010, 39, D698–D704. [Google Scholar] [CrossRef] [PubMed]
- Prasad, T.S.K.; Goel, R.; Kandasamy, K.; Keerthikumar, S.; Kumar, S.; Mathivanan, S.; Telikicherla, D.; Raju, R.; Shafreen, B.; Venugopal, A.; et al. Human Protein Reference Database—2009 update. Nucleic Acids Res. 2009, 37, D767–D772. [Google Scholar] [CrossRef] [PubMed]
- Kanehisa, M.; Goto, S.; Hattori, M.; Aoki-Kinoshita, K.F.; Itoh, M.; Kawashima, S.; Katayama, T.; Araki, M.; Hirakawa, M. From genomics to chemical genomics: New developments in KEGG. Nucleic Acids Res. 2006, 34, D354–D357. [Google Scholar] [CrossRef] [PubMed]
- Duarte, N.C.; Becker, S.A.; Jamshidi, N.; Thiele, I.; Mo, M.L.; Vo, T.D.; Srivas, R.; Palsson, B.O. Global reconstruction of the human metabolic network based on genomic and bibliomic data. Proc. Natl. Acad. Sci. USA 2007, 104, 1777–1782. [Google Scholar] [CrossRef] [PubMed]
- Ruepp, A.; Brauner, B.; Dunger-Kaltenbach, I.; Frishman, G.; Montrone, C.; Stransky, M.; Waegele, B.; Schmidt, T.; Doudieu, O.N.; Stümpflen, V.; et al. CORUM: The comprehensive resource of mammalian protein complexes. Nucleic Acids Res. 2008, 36, D646–D650. [Google Scholar] [CrossRef] [PubMed]
- Hornbeck, P.V.; Kornhauser, J.M.; Tkachev, S.; Zhang, B.; Skrzypek, E.; Murray, B.; Latham, V.; Sullivan, M. PhosphoSitePlus: A comprehensive resource for investigating the structure and function of experimentally determined post-translational modifications in man and mouse. Nucleic Acids Res. 2012, 40, D261–D270. [Google Scholar] [CrossRef] [PubMed]
- Leskovec, J.; Faloutsos, C. Sampling from Large Graphs. In Proceedings of the 12th ACM SIGKDD International Conference on Knowledge Discovery and Data Mining, Philadelphia, PA, USA, 20–23 August 2006; ACM: New York, NY, USA, 2006; pp. 631–636. [Google Scholar] [CrossRef]
- Erten, S.; Bebek, G.; Ewing, R.; Koyuturk, M. DADA: Degree-Aware Algorithms for Network-Based Disease Gene Prioritization. BioData Min. 2011, 4, 19. [Google Scholar] [CrossRef] [PubMed]
- Guney, E.; Oliva, B. Analysis of the Robustness of Network-Based Disease-Gene Prioritization Methods Reveals Redundancy in the Human Interactome and Functional Diversity of Disease-Genes. PLoS ONE 2014, 9, e94686. [Google Scholar] [CrossRef] [PubMed]
- Guney, E. Investigating Side Effect Modules in the Interactome and Their Use in Drug Adverse Effect Discovery. In Complex Networks VIII; Springer: Cham, Switzerland, 2017; pp. 239–250. [Google Scholar] [CrossRef]
- Guney, E.; Oliva, B. Exploiting Protein-Protein Interaction Networks for Genome-Wide Disease-Gene Prioritization. PLoS ONE 2012, 7, e43557. [Google Scholar] [CrossRef] [PubMed]
- Piñero, J.; Bravo, A.; Queralt-Rosinach, N.; Gutiérrez-Sacristán, A.; Deu-Pons, J.; Centeno, E.; García-García, J.; Sanz, F.; Furlong, L.I. DisGeNET: A comprehensive platform integrating information on human disease-associated genes and variants. Nucleic Acids Res. 2017, 45, D833–D839. [Google Scholar] [CrossRef] [PubMed]
- UniProt Consortium. UniProt: A hub for protein information. Nucleic Acids Res. 2015, 43, D204–D212. [Google Scholar] [CrossRef]
- Landrum, M.J.; Lee, J.M.; Benson, M.; Brown, G.; Chao, C.; Chitipiralla, S.; Gu, B.; Hart, J.; Hoffman, D.; Hoover, J.; et al. ClinVar: Public archive of interpretations of clinically relevant variants. Nucleic Acids Res. 2016, 44, D862–D868. [Google Scholar] [CrossRef] [PubMed]
- Rath, A.; Olry, A.; Dhombres, F.; Brandt, M.M.; Urbero, B.; Ayme, S. Representation of rare diseases in health information systems: The Orphanet approach to serve a wide range of end users. Hum. Mutat. 2012, 33, 803–808. [Google Scholar] [CrossRef] [PubMed]
- Welter, D.; MacArthur, J.; Morales, J.; Burdett, T.; Hall, P.; Junkins, H.; Klemm, A.; Flicek, P.; Manolio, T.; Hindorff, L.; et al. The NHGRI GWAS Catalog, a curated resource of SNP-trait associations. Nucleic Acids Res. 2014, 42, D1001–D1006. [Google Scholar] [CrossRef] [PubMed]
- Davis, A.P.; Grondin, C.J.; Lennon-Hopkins, K.; Saraceni-Richards, C.; Sciaky, D.; King, B.L.; Wiegers, T.C.; Mattingly, C.J. The Comparative Toxicogenomics Database’s 10th year anniversary: Update 2015. Nucleic Acids Res. 2015, 43, D914–D920. [Google Scholar] [CrossRef] [PubMed]
- Wishart, D.S.; Feunang, Y.D.; Guo, A.C.; Lo, E.J.; Marcu, A.; Grant, J.R.; Sajed, T.; Johnson, D.; Li, C.; Sayeeda, Z.; et al. DrugBank 5.0: A major update to the DrugBank database for 2018. Nucleic Acids Res. 2018, 46, D1074–D1082. [Google Scholar] [CrossRef] [PubMed]
- Liberzon, A.; Subramanian, A.; Pinchback, R.; Thorvaldsdóttir, H.; Tamayo, P.; Mesirov, J.P. Molecular signatures database (MSigDB) 3.0. Bioinformatics 2011, 27, 1739–1740. [Google Scholar] [CrossRef] [PubMed]
- Croft, D.; Mundo, A.F.; Haw, R.; Milacic, M.; Weiser, J.; Wu, G.; Caudy, M.; Garapati, P.; Gillespie, M.; Kamdar, M.R.; et al. The Reactome pathway knowledgebase. Nucleic Acids Res. 2014, 42, D472–D477. [Google Scholar] [CrossRef] [PubMed]
- Zhou, X.; Menche, J.; Barabási, A.L.; Sharma, A. Human symptoms—Disease network. Nat. Commun. 2014, 5, 4212. [Google Scholar] [CrossRef] [PubMed]
- Hidalgo, C.A.; Blumm, N.; Barabási, A.L.; Christakis, N.A. A Dynamic Network Approach for the Study of Human Phenotypes. PLoS Comput. Biol. 2009, 5, e1000353. [Google Scholar] [CrossRef] [PubMed]
- Kibbe, W.A.; Arze, C.; Felix, V.; Mitraka, E.; Bolton, E.; Fu, G.; Mungall, C.J.; Binder, J.X.; Malone, J.; Vasant, D.; et al. Disease Ontology 2015 update: An expanded and updated database of human diseases for linking biomedical knowledge through disease data. Nucleic Acids Res. 2015, 43, D1071–D1078. [Google Scholar] [CrossRef] [PubMed]
- Clark, N.R.; Ma’ayan, A. Introduction to statistical methods for analyzing large data sets: Gene-set enrichment analysis. Sci. Signal. 2011, 4, tr4. [Google Scholar] [CrossRef] [PubMed]
- Benjamini, Y.; Hochberg, Y. Controlling the False Discovery Rate: A Practical and Powerful Approach to Multiple Testing. J. R. Stat. Soc. Ser. B (Methodol.) 1995, 57, 289–300. [Google Scholar]
- Shannon, P.; Markiel, A.; Ozier, O.; Baliga, N.S.; Wang, J.T.; Ramage, D.; Amin, N.; Schwikowski, B.; Ideker, T. Cytoscape: A software environment for integrated models of biomolecular interaction networks. Genome Res. 2003, 13, 2498–2504. [Google Scholar] [CrossRef] [PubMed]
- VanderPlas, J. Python Data Science Handbook; O’Reilly Media, Inc.: Sebastopol, CA, USA, 2018. [Google Scholar]
- Wickham, H. ggplot2: Elegant Graphics for Data Analysis (Use R!); Springer: New York, NY, USA, 2009. [Google Scholar]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Disease | # of Pathways | |
|---|---|---|
| Overlap | Proximity | |
| celiac disease | 7 | 143 |
| Crohn’s disease | 5 | 116 |
| diabetes mellitus, insulin-dependent | 16 | 121 |
| Graves’ disease | 3 | 92 |
| lupus erythematosus, systemic | 17 | 98 |
| multiple sclerosis | 12 | 138 |
| psoriasis | 5 | 50 |
| rheumatoid arthritis | 55 | 17 |
| ulcerative colitis | 6 | 138 |
| Pathway | # of Shared Diseases | |
|---|---|---|
| Overlap | Proximity | |
| interferon gamma signaling | 5 | 8 |
| costimulation by the CD28 family | 5 | 7 |
| cytokine signaling in immune system | 5 | 7 |
| translocation of ZAP-70 to immunological synapse | 5 | 6 |
| phosphorylation of CD3 and TCR zeta chains | 5 | 6 |
| PD1 signaling | 5 | 4 |
| IL-6 signaling | 4 | 8 |
| generation of second messenger molecules | 4 | 6 |
| TCR signaling | 4 | 6 |
| signaling by ILs | 3 | 9 |
| immune system | 3 | 7 |
| downstream TCR signaling | 3 | 7 |
| interferon signaling | 3 | 7 |
| adaptive immune system | 3 | 3 |
| regulation of KIT signaling | 2 | 7 |
| IL-7 signaling | 2 | 6 |
| CTLA4 inhibitory signaling | 2 | 5 |
| chemokine receptors bind chemokines | 2 | 3 |
| extrinsic pathway for apoptosis | 2 | 3 |
| MHC class II antigen presentation | 2 | 2 |
| IL receptor SHC signaling | - | 9 |
| IL-3, 5 and GM CSF signaling | - | 9 |
| signaling by the B cell receptor BCR | - | 8 |
| regulation of IFNG signaling | - | 8 |
| growth hormone receptor signaling | - | 8 |
| IL-2 signaling | - | 8 |
| regulation of signaling by CBL | - | 8 |
| Drug | ATC | Hetionet Indication | DrugBank Indication | PxEA Score | Adjusted p-Value |
|---|---|---|---|---|---|
| orlistat | A08 | obesity, type 2 diabetes | obesity | 94.07 | |
| obeticholic acid, chenodeoxycholic acid | A05 | primary biliary cirrhosis (C) | liver disease (O), primary biliary cholangitis (O), gallbladders (C) | 74.06 | <0.0001 |
| esmolol, practolol | C07 | hypertension (E) | atrial fibrillation (E), noncompensatory sinus tachycardia (E), cardiac arrhythmias (P) | 70.55 | <0.0001 |
| clenbuterol | R03 | - | asthma | 70.44 | <0.0001 |
| erythrityl tetranitrate | C01 | - | angina | 70.32 | <0.0001 |
| fenoterol, arbutamine, bupranolol | R03 (F), G02 (F) C01 (A), C07 (B) | - | asthma (F), coronary artery disease (A), hypertension (B), tachycardia (B), glaucoma (B) | 68.97 | <0.0001 |
| dalfampridine | N07 | multiple sclerosis | multiple sclerosis | 68.44 | <0.0001 |
| magnesium sulfate | D11, V04, A06, B05, A12 | - | eclampsia, acute nephritis, acute hypomagnesemia, uterine tetany | 68.27 | <0.0001 |
| roflumilast, crisaborole | R03 (R) | chronic obstructive pulmonary disease (R) | chronic obstructive pulmonary disease (R), dermatitis (C), psoriasis (C) | 66.33 | <0.0001 |
| montelukast | R03 | chronic obstructive pulmonary disease, asthma, allergic rhinitis | asthma | 65.94 | <0.0001 |
| Disease | # of Genes | Genes |
|---|---|---|
| celiac disease | 11 | IL21 CCR4 HLA-DQA1 BACH2 RUNX3 ICOSLG SH2B3 CTLA4 MYO9B ZMIZ1 ETS1 |
| Crohn’s disease | 19 | DNMT3A IL12B IRGM IL10 CCL2 FUT2 SMAD3 TYK2 ATG16L1 BACH2 |
| IL2RA NKX2-3 PTPN2 NOD2 TAGAP MST1 DENND1B IL23R ERAP2 | ||
| diabetes mellitus, insulin-dependent | 18 | IL10 GLIS3 HLA-DQA1 HLA-DRB1 PTPN22 SLC29A3 INS BACH2 CLEC16A |
| PAX4 HLA-DQB1 IL2RA CD69 IL27 HNF1A CTSH SH2B3 C1QTNF6 | ||
| Graves’ disease | 4 | RNASET2 CTLA4 FCRL3 TSHR |
| lupus erythematosus, systemic | 29 | IKZF1 CFB RASGRP3 PDCD1 RASGRP1 DNASE1 HLA-DRB1 PTPN22 ETS1 TNIP1 |
| FCGR2B TNFSF4 IRF5 C2 PRDM1 PXK TLR5 TREX1 TNFAIP3 SLC15A4 PHRF1 | ||
| HLA-DQA1 STAT4 ITGAX ITGAM BLK C4A BANK1 CR2 | ||
| multiple sclerosis | 15 | CD58 CD6 IRF8 HLA-DQB1 CBLB HLA-DRA KIF1B IL2RA |
| TNFSF14 VCAM1 IL7R HLA-DRB1 CD24 TNFRSF1A PTPRC | ||
| psoriasis | 15 | IL12B TNIP1 LCE3D IL13 IL23R TYK2 HLA-DQB1 HLA-C FBXL19 |
| ERAP1 TRAF3IP2 TNFAIP3 TNF REL NOS2 | ||
| rheumatoid arthritis | 23 | MIF CD40 ANKRD55 HLA-DRB1 PTPN22 RBPJ IL2RA AFF3 CCL21 REL SLC22A4 CCR6 |
| IRF5 SPRED2 CTLA4 PADI4 TNFAIP3 NFKBIL1 HLA-DQA2 STAT4 IL6 BLK TRAF1 | ||
| ulcerative colitis | 24 | IL12B JAK2 ICOSLG IL1R2 LSP1 CXCR2 IL10 IL7R CXCR1 DAP NKX2-3 CARD9 GNA12 |
| IRF5 PRDM1 HNF4A CCNY SLC26A3 FCGR2A IL23R IL17REL MST1 TNFSF15 CDH3 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Aguirre-Plans, J.; Piñero, J.; Menche, J.; Sanz, F.; Furlong, L.I.; Schmidt, H.H.H.W.; Oliva, B.; Guney, E. Proximal Pathway Enrichment Analysis for Targeting Comorbid Diseases via Network Endopharmacology. Pharmaceuticals 2018, 11, 61. https://doi.org/10.3390/ph11030061
Aguirre-Plans J, Piñero J, Menche J, Sanz F, Furlong LI, Schmidt HHHW, Oliva B, Guney E. Proximal Pathway Enrichment Analysis for Targeting Comorbid Diseases via Network Endopharmacology. Pharmaceuticals. 2018; 11(3):61. https://doi.org/10.3390/ph11030061
Chicago/Turabian StyleAguirre-Plans, Joaquim, Janet Piñero, Jörg Menche, Ferran Sanz, Laura I. Furlong, Harald H. H. W. Schmidt, Baldo Oliva, and Emre Guney. 2018. "Proximal Pathway Enrichment Analysis for Targeting Comorbid Diseases via Network Endopharmacology" Pharmaceuticals 11, no. 3: 61. https://doi.org/10.3390/ph11030061
APA StyleAguirre-Plans, J., Piñero, J., Menche, J., Sanz, F., Furlong, L. I., Schmidt, H. H. H. W., Oliva, B., & Guney, E. (2018). Proximal Pathway Enrichment Analysis for Targeting Comorbid Diseases via Network Endopharmacology. Pharmaceuticals, 11(3), 61. https://doi.org/10.3390/ph11030061