1. Introduction
The increasing prevalence of antibiotic resistance coupled with the waning production of new drugs calls for the development of alternative treatments against bacterial pathogens [
1,
2,
3,
4,
5,
6,
7,
8,
9,
10]. One possibility is phage therapy, which uses virulent bacteriophages (phages) to treat bacterial infections. Soon after their discovery, phages were utilized as antibacterial treatments, especially in Eastern Europe [
11]. Although largely ignored by Western medicine following the discovery of broad-spectrum antibiotics, a recent resurgence of interest in phage therapy has sparked studies that successfully demonstrate the efficacy of phages for treating infections in animal models [
2,
3,
4,
5,
6,
7]. Phages isolated from natural microbial communities are effective in treating experimental bacterial infections, and some studies have evolved naturally isolated phages to improve their infectivity, thereby improving prospects for efficient use in phage therapy [
12,
13,
14,
15]. Here, we used short-term in vitro experimental evolution of phage PP01 to increase its virulence (killing efficiency) on the bacterial pathogen
Escherichia coli O157:H7.
E. coli O157:H7 is an important food-borne pathogen that causes hemorrhagic colitis, and in severe cases hemolytic-uric syndrome [
1]. Phage PP01 has a dsDNA genome that is ~140 kb in size, and is related to phages in the family
Myoviridae, for example T-even-like phages [
1,
9]. In 2002, phage PP01 was isolated alongside
E. coli O157:H7 from swine stool samples in Japan [
1]. Although phage PP01 growth potency on
E. coli O157:H7 can vary (presumably due to subtle lab effects [
15]), their co-isolation suggests that the bacteria constitute a natural host of the virus, whose binding specificity [
16] could be harnessed in phage therapy targeting this pathogen [
17,
18,
19,
20]. The current study shows that phage PP01 grows inefficiently on
E. coli O157:H7, affording the opportunity to harness short-term experimental evolution to study generalities of how the virus initially adapts to improve host binding as an important fitness component of lytic reproduction. We predicted that phage PP01 adsorption (attachment) would be a major initial target for selection because preliminary adsorption experiments revealed that PP01 poorly attached to
E. coli O157:H7. Improved adsorption should greatly increase PP01’s rate of infection events [
21], and thus we expected that mutations responsible for faster attachment should be strongly selected in evolving phage populations.
T-even phage adsorption is mediated by long tail fibers that recognize and bind host cellular receptors such as lipopolysaccharides (LPS) and outer membrane proteins [
22,
23,
24,
25,
26]. In phage T2, gene
gp37 encodes the distal tail fibers, which on their tips contain the homologous host recognition and binding protein, encoded by
gp38 [
22,
25]. In phage PP01, protein Gp38 reversibly binds to the outer membrane protein C (OmpC) on
E. coli O157:H7’s cell surface [
26]. This event prompts the short tail fibers, encoded by
gp12, to irreversibly bind to the LPS of
E. coli O157:H7, allowing PP01 to inject its DNA into the host and begin the lytic infection cycle [
8]. Thus, we predicted that the initial evolution of PP01 populations on
E. coli O157:H7 would select for mutations in genes
gp37,
gp38 and/or
gp12, allowing the phage to adsorb faster to the bacteria through improvements in the reversible and irreversible binding processes.
We allowed four lineages of phage PP01 to evolve independently on E. coli O157:H7 via serial transfer in a short-term experimental evolution study comprising 21 passages on this host. Consistent with our prediction, we observed that all four evolved virus lineages improved their adsorption ability on E. coli O157:H7, and adapted to kill host bacteria faster than the wildtype ancestor. Sequencing of candidate genes revealed that the phage populations evolutionarily converged, evidenced by shared point mutations in gp38. Furthermore, all of the evolved populations unexpectedly underwent sizeable deletions (579–686 bp) in the same region of gp37. Comparisons between phage clones drawn from the same evolved population strongly suggested that the deletion mutation in gp37 alone could result in improved host attachment. In contrast, no mutations were observed in gp12. Our study demonstrates how experimental evolution can be used to select for viral traits that improve phage attack of an important bacterial pathogen, and that the molecular targets of selection include loci contributing to cell attachment, and thereby impacting phage antibacterial virulence. Future studies investigating the therapeutic potential of naturally isolated phages should continue to capitalize on short-term experimental evolution designed to improve key viral traits useful for phage therapy.
3. Results
3.1. Increased Attachment in Evolved Phage Populations
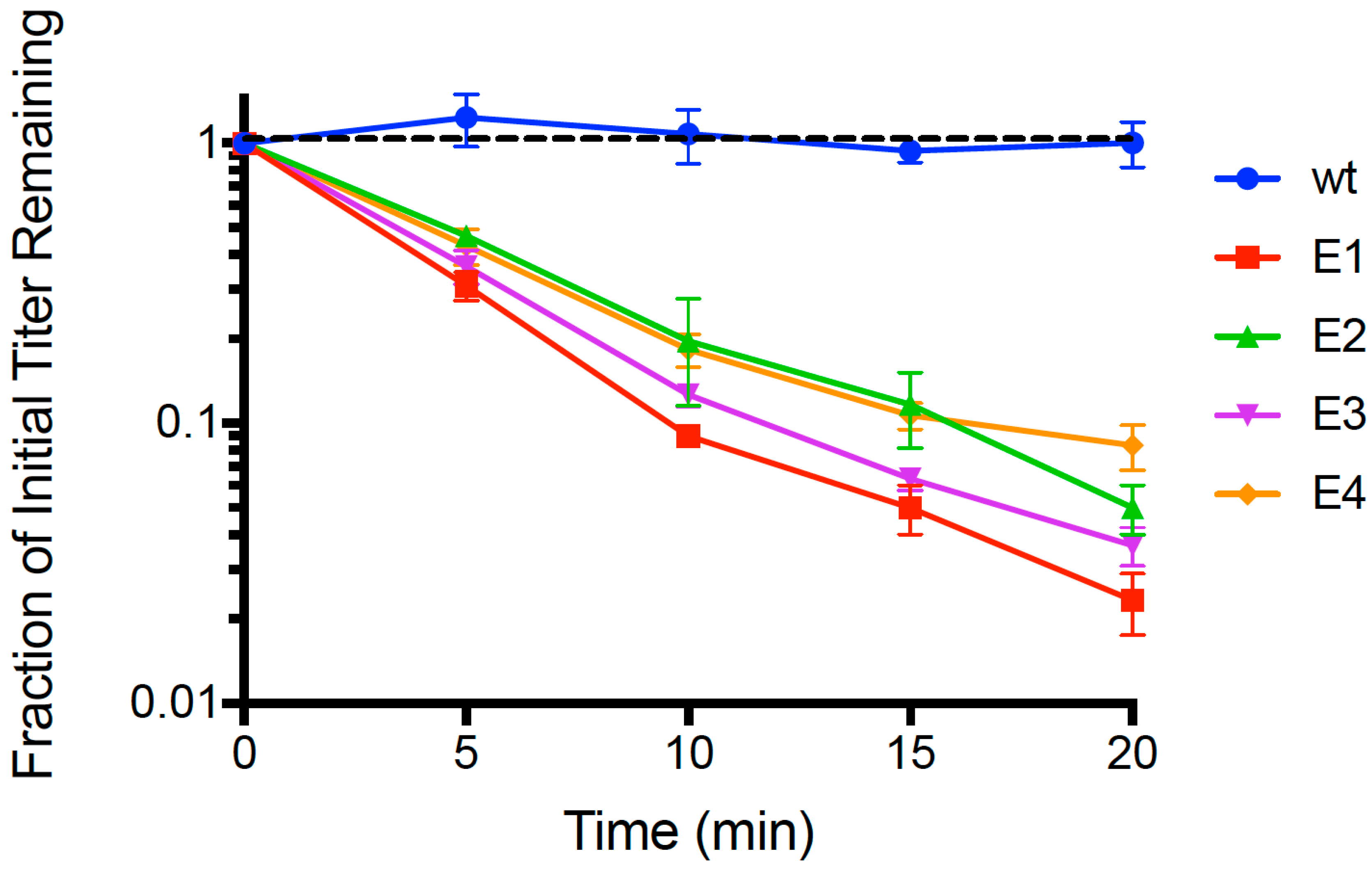
Following 21 passages of experimental evolution, we conducted repeated (
n = 3) dead-cell attachment assays (see Methods) that measured adsorption ability of evolved populations (E1, E2, E3, E4) and the wildtype (wt) ancestor. We then estimated the mean fraction of unattached particles relative to initial titer (pfu/mL) in the supernatant, for each test phage; results are shown in
Figure 1. After 20 min, we observed that the wt ancestor showed no measurable decrease in titer (−0.00241 ± 0.0027 SE;
p > 0.05), indicating that any cell attachment by the wt in the time allowed was below the level of detection. In contrast, all four evolved lineages showed ≥90% mean disappearance (cell attachment) in this same time period. We used linear regression to estimate the rate of disappearance (negative slope) in each of the replicated population assays, and the grand mean rate of decline for each population. For all of the evolved populations, the negative slope of the grand mean regression was statistically significantly different than zero (
p < 0.01). In addition, we used ANOVA to compare the attachment rates across all assays, and found that there was a significant difference in the grand mean regression slopes among the populations (
p = 0.005). This outcome was consistent with E1 showing the most rapid observed mean decline (−0.0811 ± 0.0042 SE), which was significantly faster (
p < 0.05) than the adsorption of E2 (−0.0646 ± 0.0038 SE), E3 (−0.0723 ± 0.0030 SE) and E4 (−0.0550 ± 0.0036 SE). Moreover, populations E2 and E3 did not significantly differ in grand mean slope (
p = 0.156), but attached significantly faster than E4 (
p < 0.05). In addition, we subjected the
Figure 1 dataset to an additional analysis using multiple-comparisons ANOVA that compared each evolved population’s ‘fraction of initial titer remaining’ to that of wt at each time point (
t = 5, 10, 15, 20 min); in all cases the evolved population titer was statistically significantly less than wt (
p < 0.0001). From these analyses, we concluded that short-term evolution of PP01 populations on
E. coli O157:H7 bacteria led to faster attachment of viruses to these host cells.
3.2. Improved Killing Efficiency of Evolved PP01 Populations on E. coli O157:H7
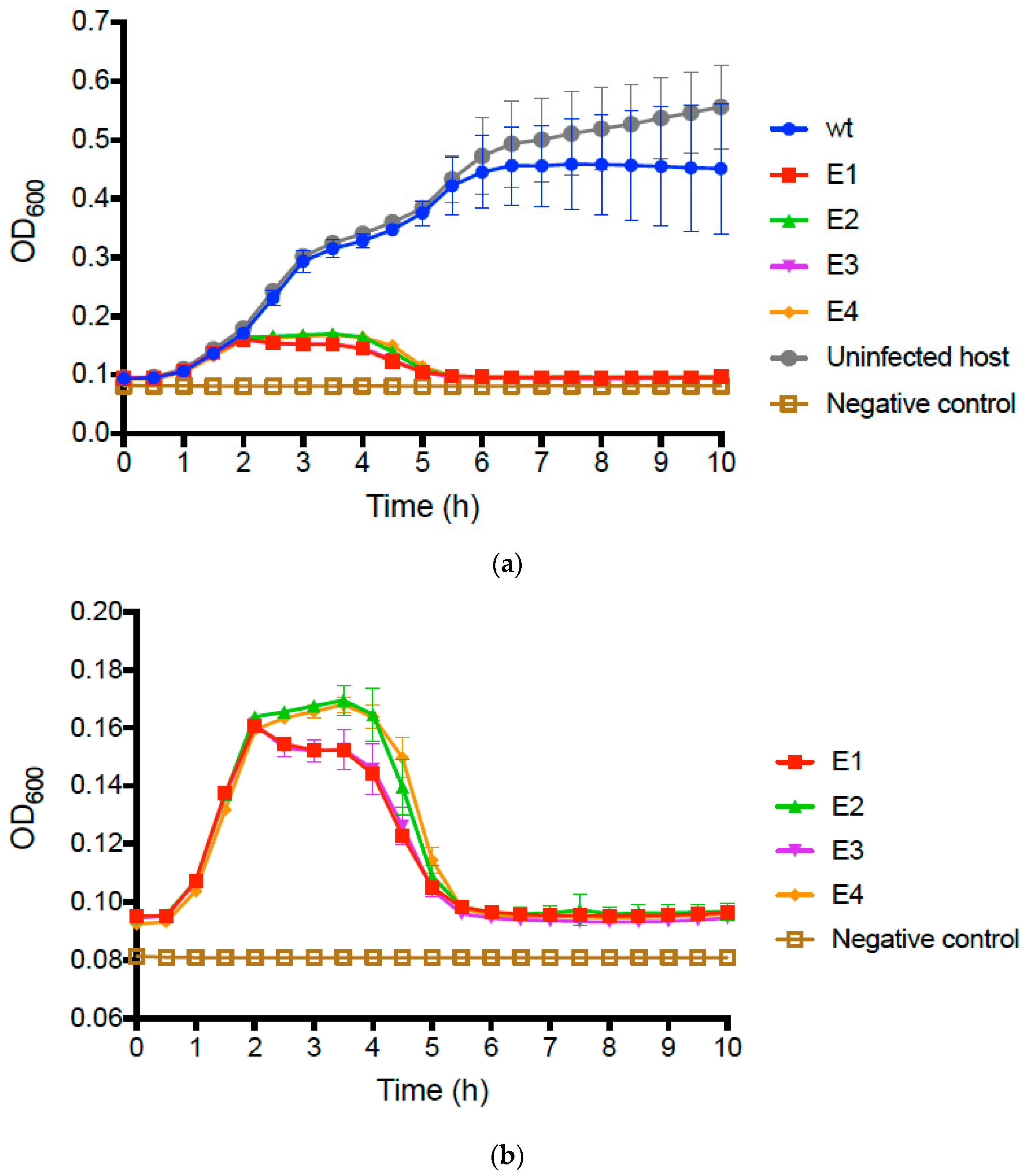
We compared the growth of uninfected
E. coli O157:H7 bacteria to that of bacteria infected with the wt PP01 ancestor by conducting repeated (
n = 3) assays (see Methods) that tracked changes in bacterial densities (OD
600 of growing cultures) over time. Results (
Figure 2a) showed that infected and uninfected bacteria initially grew similarly, presenting mean OD
600 ≈ 0.40 after 5 h and comparable growth trajectories. However, we then observed that growth of the wt PP01-infected bacteria plateaued at mean OD
600 ≈ 0.45, whereas the uninfected bacteria continued to grow and reached mean OD
600 ≈ 0.60 by 10 h (
Figure 2a).
We then conducted identical assays comparing growth of bacteria infected with wt PP01 relative to bacteria infected by each evolved population. The data for the evolved populations qualitatively matched growth curves in assays for the uninfected and wt-infected bacteria during the first 2 h (
Figure 2a); however, in the remaining 8 h of the assay, we observed dramatic differences between the assays containing evolved populations and those for the wt ancestor (
Figure 2b). In particular, growth of bacteria infected by each of the evolved populations was highly similar in the latter stages of the assay; cells only achieved a maximum of OD
600 ≈ 0.16, and after 5 h decreased to OD readings nearly as low as the negative controls containing no bacteria (
Figure 2b). We performed multiple-comparisons ANOVA to compare the OD
600 values of the test samples at 10 h. The differences between the wt, and each evolved population and the uninfected bacteria were statistically significant (in all cases,
p ≤ 0.0001). Moreover, we determined that 10 h OD
600 values did not statistically significantly differ for the evolved populations (
p ≥ 0.573). These results confirmed that following experimental evolution, the phage populations generally evolved to kill the host bacteria far more efficiently than their wt ancestor.
3.3. Sequence Analysis of PP01 Tail Fiber Genes
We sequenced three genes that encoded tail fiber proteins in phage PP01: gp12, gp37, and gp38. The short tail fiber is encoded by gp12 and the distal section of the long tail fiber is encoded by gp37. At the tip of the distal tail fiber is the host recognition protein encoded by gp38. Results of the dead-cell assays above suggested that evolved changes in phage populations included improvements in reversible and/or irreversible binding, indicating that mutations in either the short or long tail fibers could explain improved adsorption.
The NCBI consensus sequence (GenBank #AB180231) of gp12 encoding the short tail fibers in phage PP01 shows a G/C polymorphism at bp 1196. Our sequencing results for gp12 showed no differences between the ancestor and the evolved populations in this gene; all strains had a G at bp 1196 and no mutations at other sites in gp12 (GenBank accession numbers pending). The lack of observed mutations in the short tail fibers indicated that irreversible attachment alone was not the target for selection in our experiment. The faster attachment rate of the evolved populations must then be attributed to an improvement in the reversible binding process, (mediated by gp38), and/or increased efficiency of the conformational changes in the phage as it deploys its short tail fibers prior to irreversible binding.
We sequenced gp38 in the ancestor and evolved populations and compared these data to the NCBI consensus sequence (GenBank #AF349975). Results (GenBank accession numbers pending) revealed that the evolved populations all shared two non-synonymous polymorphisms: at bp 483 an A → C transversion led to a Q161H substitution, and at bp 620 a C → T transition caused a T207I substitution. In addition, each population showed a unique mutation in gp38. At bp 368, population E1 showed an A123E substitution, whereas population E4 showed an A123V substitution. Population E2 showed an N163H substitution, and population E3 had a Y159H substitution located nearby. The observed mutations in gp38 suggested that suboptimal reversible binding was a target for strong selection in the experiment. In addition, the two mutations Q161H and T207I likely played key roles in the improved attachment since all four independently-evolved populations experienced these same convergent mutations.
To preliminarily examine the role of individual allele substitutions in improved adsorption, we chose to study one representative population from the four evolved lineages; we sequenced gp38 in seven plaque-purified clones (GenBank accession numbers pending) randomly isolated from population E3, at the end of the study. These seven clones were abbreviated as E3a through E3g. The sequences of the seven E3 clones showed that the polymorphisms in the E3 population were due to the simultaneous presence of genotypes with the wt gp38 allele (E3b, E3c, E3d, E3e) and genotypes with all three novel mutations in gp38 (E3f, E3g). In addition, the sequencing revealed that clone E3a had the Q161H mutation and wildtype alleles at the other loci. The association of these mutations with improved binding is further examined below.
Lastly, we sequenced gp37, which encodes the distal long tail fiber. The wt ancestor’s gp37 sequence did not differ from the published sequence (GenBank #AF349974). In contrast, all four evolved populations had deletions in gp37 that varied in length from 579 bp to 686 bp, but were found in the same general region: 1980–2650 bp of gp37 (GenBank accession numbers pending). Populations E1 and E3 shared an identical 579 bp deletion in gp37 (2061–2640 bp). Population E2’s deletion was 620 bp (1987–2607 bp), and population E4 had a 686 bp deletion (1945–2631 bp). Gene gp37 is 3300 bp long; thus, the observed deletions comprised ~20% of the gene. PCR probes for the deleted sequence were unable to detect the presence of full-length gp37 in any of the evolved populations. We concluded that evolution on the E. coli O157:H7 host bacteria led to convergent evolution of deletions in gp37, and below we discuss how this outcome may relate to the evolution of improved attachment in the phage PP01 derivatives.
3.4. Mutations in gp37 and gp38 Associated with Adsorption Changes in Population E3
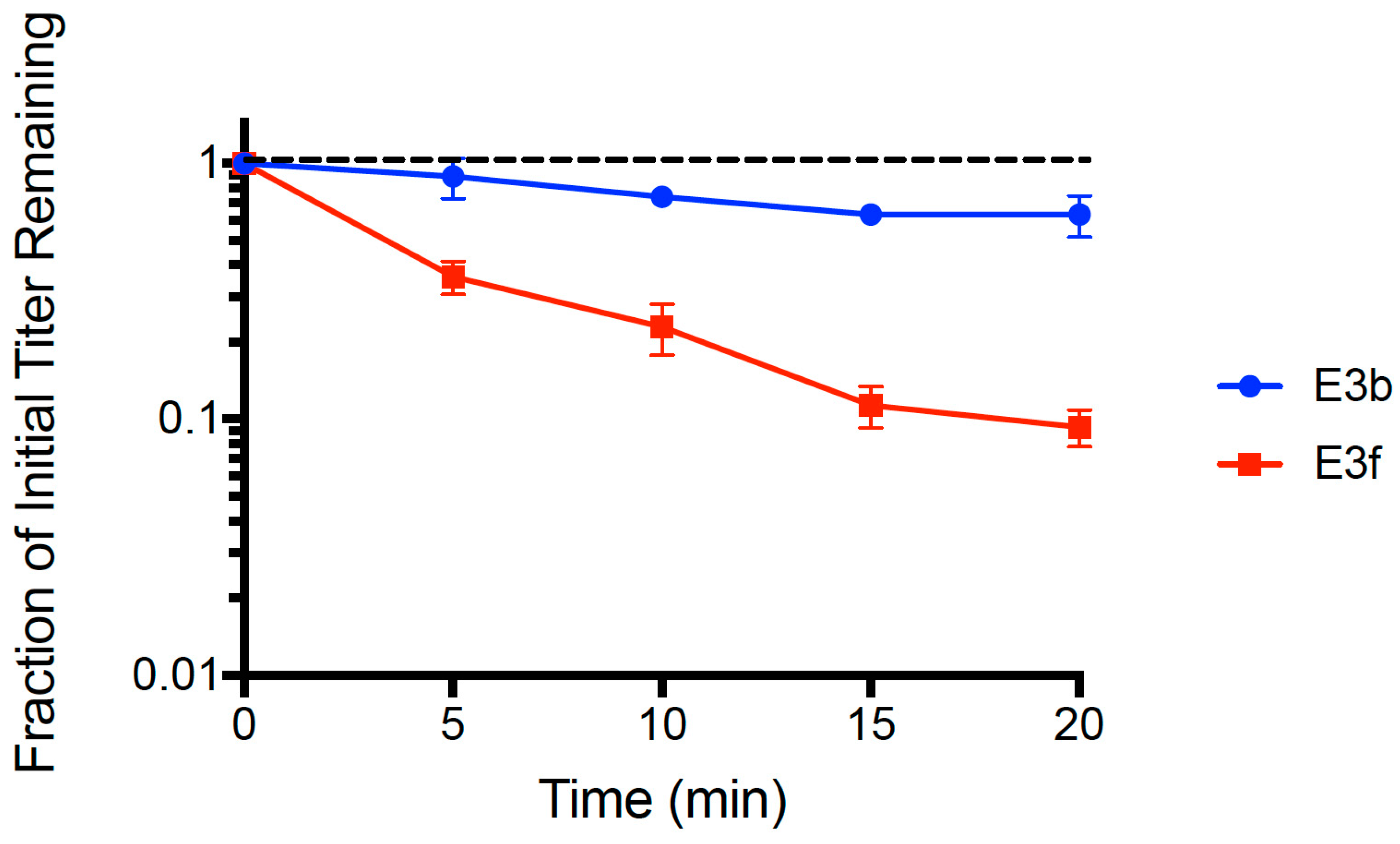
We sought preliminary evidence that allele changes in gp37 and/or gp38 played a role in improved attachment of phage PP01 on E. coli O157:H7. To do so, we focused on assays containing clones E3b and E3f that were isolated from evolved population E3. This comparison was meaningful because clone E3b contained a large deletion in gene gp37 and the wildtype sequence in gene gp38, whereas clone E3f contained the identical large deletion in gp37 and all three mutations in gp38.
We used the dead cell assay (see Methods) to compare binding abilities of clones E3b and E3f. Results (
Figure 3) showed that both strains adsorbed faster than the wt ancestor (see
Figure 1). A multiple comparisons ANOVA test was performed comparing the wt ancestor, E3b, and E3f for ‘fraction of initial titer remaining’ values at each time point (
t = 5, 10, 15, 20 min) in the assay. The test showed statistically significant differences in the adsorption of all three phage populations at every time point (
p < 0.005). The mean linear regression slopes for E3b (−0.0112 ± 0.0018 SE) and E3f (−0.0516 ± 0.0040 SE) were significantly less than the wt ancestor slope (−0.00241 ± 0.0027 SE,
p < 0.01). Clone E3f adsorbed faster than clone E3b (
p < 0.01), suggesting that one or more of the
gp38 mutations plays an important role in the improved binding. Importantly, clone E3b adsorbed faster (
p < 0.05) than the wt ancestor indicating that improved binding of evolved PP01 populations could solely be due to the observed deletion in
gp37. That is, because the E3b clone contained the deletion in
gp37 but was wildtype in
gp38, our data indicated that the
gp37 deletion alone could explain much of the improvement in binding by evolved phage PP01 strains.
3.5. Amino Acid Sequence Alignment of gp38
An alignment of the
gp38 protein sequence of PP01 and its closest relatives revealed the highly conserved 120 amino acid N-terminus and 25 amino acid C-terminus regions of
gp38 (
Figure 4) [
31,
32]. In
Figure 4, the un-shaded regions signify those that are highly conserved amongst the viruses, whereas the shaded regions are not conserved. The alignment also revealed the conserved glycine repeats that flank the shaded hyper-variable regions of
gp38, which serve as the protein’s binding domains. The mutations observed in the current study are circled in
Figure 4. Three of the six mutations (Y159H, Q161H, N163H) were in the 2nd binding domain and were within seven amino acids of each other. These three mutations encode a histidine (
Figure 4). Two mutations (A123E, A123V) were found in the 1st binding domain, and one mutation in the 4th domain (T207I). All of the observed mutations were located in Gp38’s binding domains, which corroborate the contribution of these mutations in increased adsorption of the evolved populations.
4. Discussion
We used short-term experimental evolution to select for improved binding performance of phage PP01 on
E. coli O157:H7; this approach allowed us to examine whether experimental evolution led to increased infectivity of a candidate virus potentially useful in phage therapy. Wildtype PP01 was used to found four lineages independently evolved on non-co-evolving
E. coli O157:H7 bacteria via 21 serial passages. We predicted that increased adsorption would be a major target for selection because viruses that more quickly bound to and infected host cells would on average produce progeny faster and maximize their fitness [
21]. A dead cell assay was used to measure PP01 adsorption, where test PP01 populations were mixed with dead
E. coli O157:H7 cells at an MOI of 0.01. The wt ancestor did not appreciably bind to the dead cells in the time allowed, whereas after 20 minutes the four evolved populations showed ~90% reduction in the supernatant (relatively increased binding) of the initial starting inoculum of virus particles. These results validated our hypothesis that cellular attachment would be a major target for selection in the short-term evolution experiment.
The dead cell assay measures irreversible binding, meaning that alterations in Gp12 and/or Gp38 could be responsible for an increase in adsorption. However, sequencing of gp12 revealed that the ancestor and the evolved populations were identical in this gene for the short tail fiber, indicating that selection pressure to increase irreversible attachment by improving the ability of Gp12 to bind with the cell surface was not very strong in the time duration of our experiment. Moreover, these data suggest that complete irreversible attachment to E. coli O157:H7’s LPS might be a highly optimized step in PP01 adsorption, which does not have a high capacity for adaptive improvement over relatively brief evolutionary time.
The lack of mutations in gp12 signified that the increase in attachment rate of the evolved populations likely involved improved reversible binding mediated by Gp38. Sequencing of gp38 revealed all four evolved populations shared two polymorphisms (Q161H and T207I). The convergence of these mutations indicates that they play key roles in attachment. Each population also had a single unique polymorphism. Our hypothesis that the mutations in gp38 should facilitate faster adsorption was supported by the dead cell assay performed with the E3b and E3f clones from population E3. Although both clones contained the gp37 deletion (further discussed below), E3f had the Y159H, Q161H, and T207I mutations fixed in gp38 while E3b had the wt gp38. Clone E3f attached faster to the dead host cells than E3b.
Further support that the
gp38 mutations function in enhancing adsorption stemmed from the locations of these mutations in
gp38’s binding domains. Analysis of the mutations in
gp38 suggests that charged and hydrophobic amino acids allow the protein to bind to
E. coli O157:H7 faster. Three of the six mutations in this study are to histidine (N163H in E2, Y159H in E3, and convergent mutation Q161H in all lineages), which has a positively charged side chain. Population E1’s A123E mutation encodes for glutamate, a negatively charged amino acid. These four mutations may allow Gp38 to better bind
E. coli O157:H7 in a charge dependent manner. Furthermore, the three histidine mutations are in close proximity to one another in the 2nd binding domain, which suggests that this domain plays a key role in host recognition and may benefit from a more positively-charged binding site. In a past study, Morita et al. isolated PP01 host range mutants capable of infecting cells lacking OmpC. The host range mutants had mutations in
gp38 that were to positively charged amino acids arginine and histidine (Q161H, Q161R, W189R) [
27]. This result suggests that the selection of positively charged amino acids in this region, especially at Q161, may increase adsorption by a mechanism other than direct binding to OmpC. Two of the polymorphisms in the evolved populations were to amino acids with hydrophobic side chains: A123V in population E3 and convergent mutation T207I. The A123V and T207I mutations suggest that Gp38 may better bind
E. coli O157:H7 via hydrophobic interactions. The A123V mutation is particularly illuminating of the potential role of hydrophobic residues in
gp38 binding. Valine with its methyl group is marginally more hydrophobic than alanine, yet the A123V mutation was targeted by selection in population E4. Furthermore, the convergent T207I mutation converts tyrosine, a polar uncharged amino acid, to isoleucine, which has an uncharged hydrophobic side chain. Detailed knowledge of the specific residues that regulate attachment in
gp38’s binding domains allows for the possibility of enhancing phage adsorption and altering host ranges in phage therapy candidates [
33]. Current experiments in our lab concern the utility of genetic engineering of phages to increase their host attachment and specificity via
gp38 alterations.
At the onset of the experiment, we anticipated that the evolved populations would harbor mutations in
gp12 and
gp38 that facilitated faster attachment to the host. Surprisingly,
gp37 deletions in the 1980–2650 bp region arose early, (2nd or 3rd passage; data not shown) in all of the evolved populations and persisted until the 21st endpoint passage. The deletion mutation was unexpected because
gp37 encodes the distal long tail fibers and is not directly involved with adsorption to the host [
34]. Nevertheless, the orientation of the proximal long tail fibers (gene
gp34) to the baseplate determines short tail fiber triggering. It is possible that the shortened distal tail fiber causes more downward force to be applied at the baseplate by each bound long tail fiber. This could result in improved phage adsorption through a faster transition from reversible to irreversible binding [
24]. T-even phages with faster contracting tail fibers are described as “trigger happy” and have been isolated in past experiments [
22,
23,
25]. Further experiments are needed to confirm that our observations are explained by the “trigger happy” phenotype evolving in association with a mutation in the
gp37 distal tail fiber rather than in the baseplate genes [
35]. Because phage PP01 seems to naturally infect
E. coli O157:H7, one might ask: why has the phage not previously evolved this mechanism in the wild? To examine this question, we conducted a preliminary analysis using publicly available sequence data in GenBank.
Figure 5 shows genetic data for
gp37 distal tail fiber genes in four coliphages with high sequence similarity to phage PP01, in comparison to phages in the current study. This comparison indicated that at least two other viruses (phages HY01 and UFV-AREG1) that could infect
E. coli O157:H7 also possessed similar deletions as those observed when phage PP01 was subjected to experimental evolution; in contrast, phages AR1 and Ac3 do not show evidence of the
gp37 deletion. One possibility is that being “trigger happy” is not advantageous under some natural conditions, because this committed mechanism could lessen (or altogether prevent) phage detachment from sub-optimal host types, which are likely to be encountered in species-diverse natural communities of bacteria. In contrast, constant selection on a single host type in the laboratory may constitute a cost-free environment for evolving the “trigger happy” phenotype. This idea is speculative and warrants further investigation.
The
gp37 deletion evolutionarily preceded the mutations in
gp38 and was presumably the primary target of selection. The
gp38 mutations arose later in the experiment and were polymorphic in the final populations. Several factors could explain why the mutant alleles did not all fix in the final passages. First, the
gp38 mutations may have recently appeared and are in the process of fixing. Second, the large population size within each bottleneck (~10
7 viruses) of the serial passage might have slowed the rate of fixation of beneficial mutations due to clonal interference, including mutations in
gp38 [
36]. Lastly, phages with wt
gp38 may have beneficial mutations in other loci that confer an equal or greater fitness advantage than viruses with mutated
gp38. Overall, we note that other loci may function in the adaptive changes allowing the PP01 derivatives in our study to better kill
E. coli O157:H7, which can be addressed in future work involving single-step growth experiments and whole-genomics of the evolved populations.
In addition to faster host adsorption, the evolved populations were also more virulent against E. coli O157:H7 than the wt ancestor. E. coli O157:H7 infected with wt PP01 was similar to the growth profile of the uninfected host and did not show a characteristic decline in density due to phage lysis over the time course of our growth assays. In contrast, the host infected with the evolved populations by 5h had OD600 measurements that nearly matched the negative control wells containing only growth medium. These results strongly suggest that the evolved PP01 populations would make better phage therapy candidates than wt PP01 because they not only attach to E. coli O157:H7 faster, but also are more virulent and exhibit higher bactericidal activity. Thus, after 21 passages of phage experimental evolution, we obtained evolved PP01 variants with greater therapeutic potential, which could be confirmed through additional studies, especially experimental mouse infections.
In the current study, we passaged phage PP01 populations on ‘static’ (non-coevolving)
E. coli O157:H7 cultures, to rigorously investigate how the phage generally adapts to improve its infectivity on host bacteria. In the context of phage therapy, however, the host bacteria may not be static, and could instead co-evolve in response to the phage. Phage can exert selection pressure on the population of the target bacterial pathogen to evolve greater ability to resist phage attack; in response, the phage population can be selected to better overcome bacterial resistance mechanisms. We note that this co-evolutionary dynamic is possible, but not inevitable; rather, it depends on whether spontaneous mutations occurring in both the bacterial and phage populations provide the useful genetic variation needed to drive reciprocal evolutionary change. Another possibility is that phage evolution outpaces bacterial evolution (or vice versa), such that one interactor drives the other to extinction. Nevertheless, phage-bacteria co-evolutionary arms races have been documented previously through in vitro studies [
15,
37,
38]; these co-evolutionary dynamics between phage and bacteria can be complex, with the possibility that populations of both interactors can evolve innovative defense strategies [
39]. While the current study focused on evolved strategies that phages employ to better infect static host-bacteria targets, additional research could explore whether phage-bacteria coevolution leads to different phage adaptations than documented here.
Previous studies have examined the effectiveness of phage-therapy candidates—or of phage approved for emergency treatment—by relying on the spontaneous generation of highly virulent phage mutants or on isolates of virulent phage from natural sources [
9,
40,
41,
42]. In contrast, the current study suggests that experimental evolution may be an effective method to increase the killing potential of naturally isolated viruses targeted for phage therapy use, where parallel evolution among phage populations [
43,
44] can illuminate how phage-therapy candidates mechanistically improve killing ability on target bacterial pathogens. Future studies investigating the therapeutic potential of naturally isolated phage could benefit from using experimental evolution to improve key traits such as adsorption, virulence, and burst size before using phage to treat in vitro and in vivo experimental infections.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}




