
A Calibration-Free pH Sensor Using an In-Situ Modified Ir Electrode for Bespoke Application in Seawater



Abstract
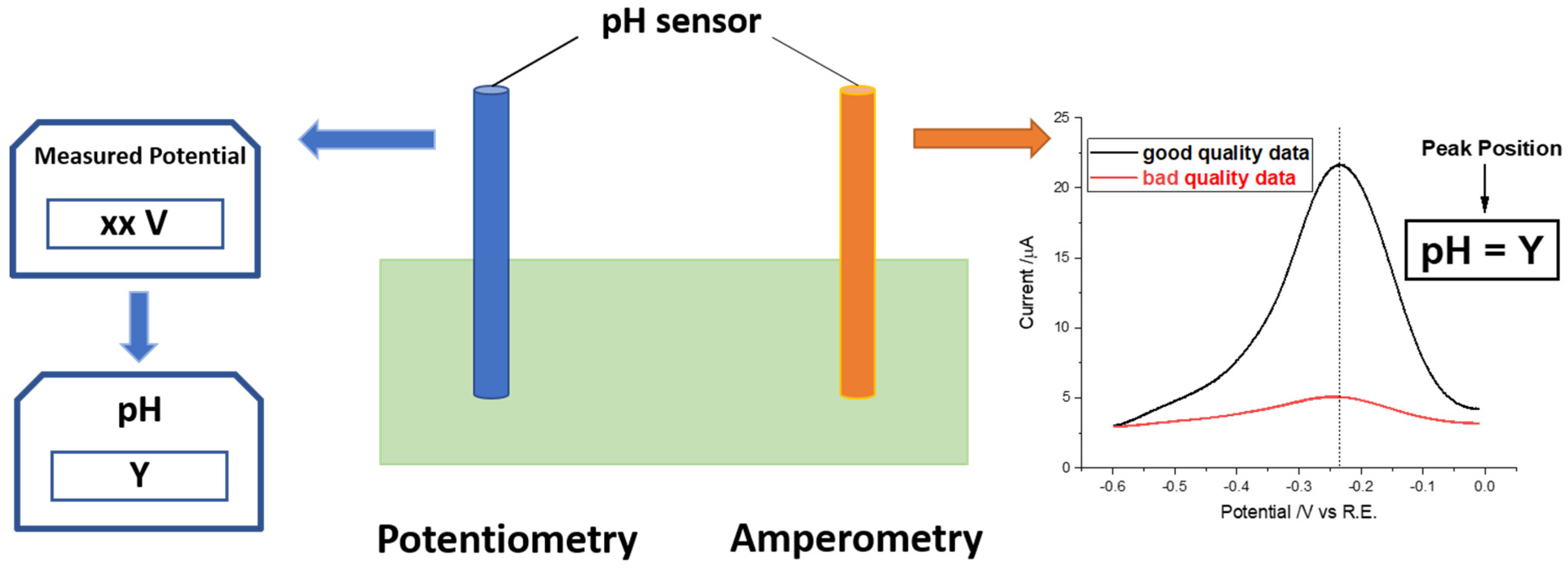
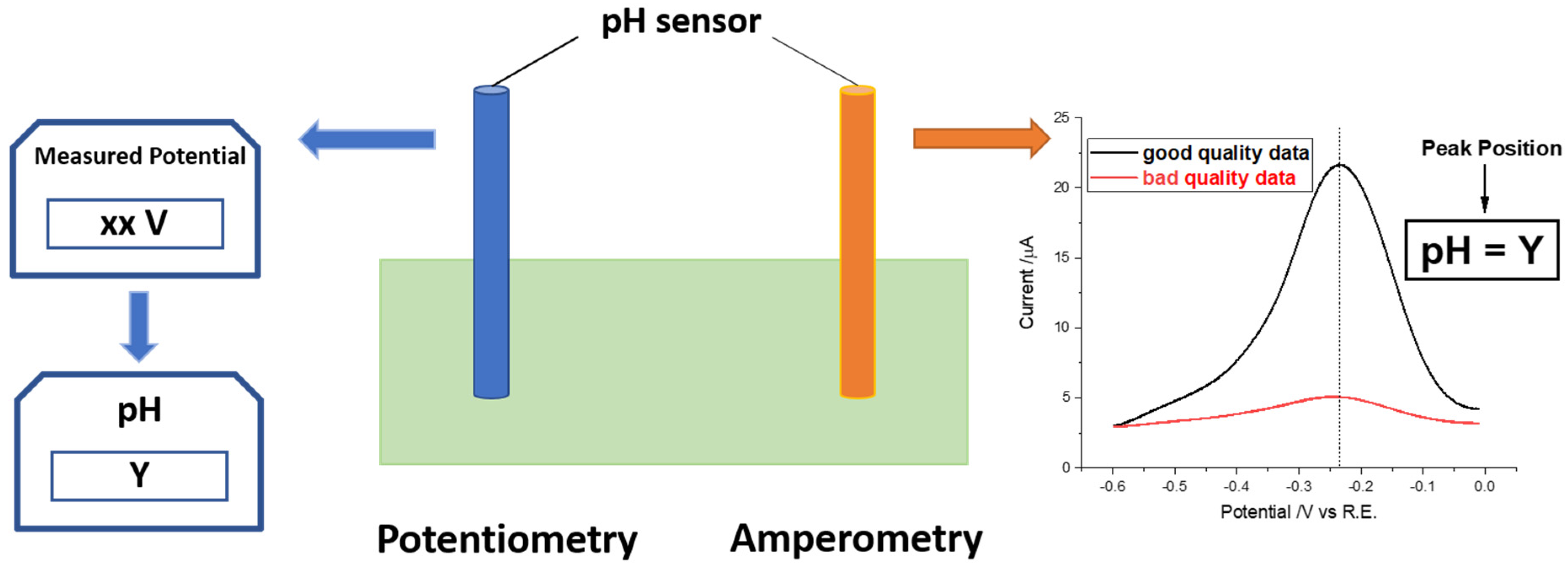
:1. Introduction
2. Experimental Section
3. Results and Discussion
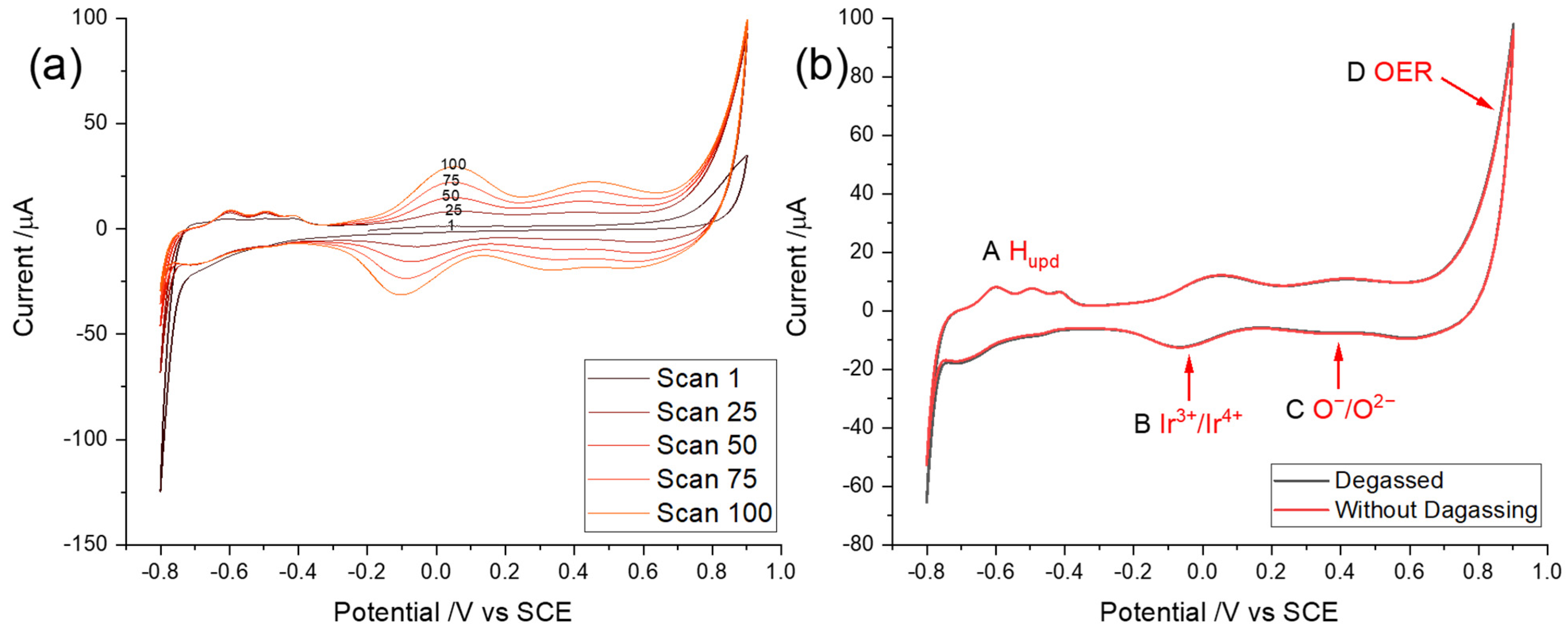
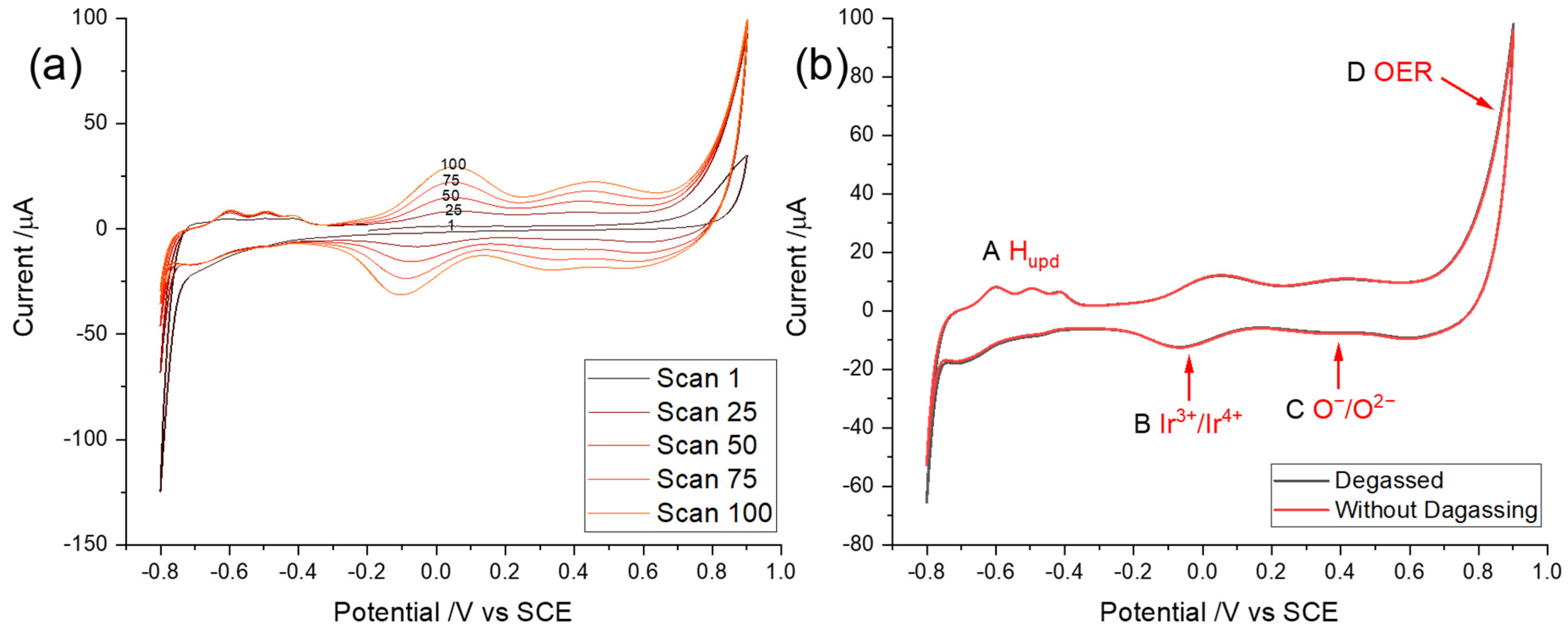
3.1. Cyclic Voltammetry of Iridium and Iridium Oxides
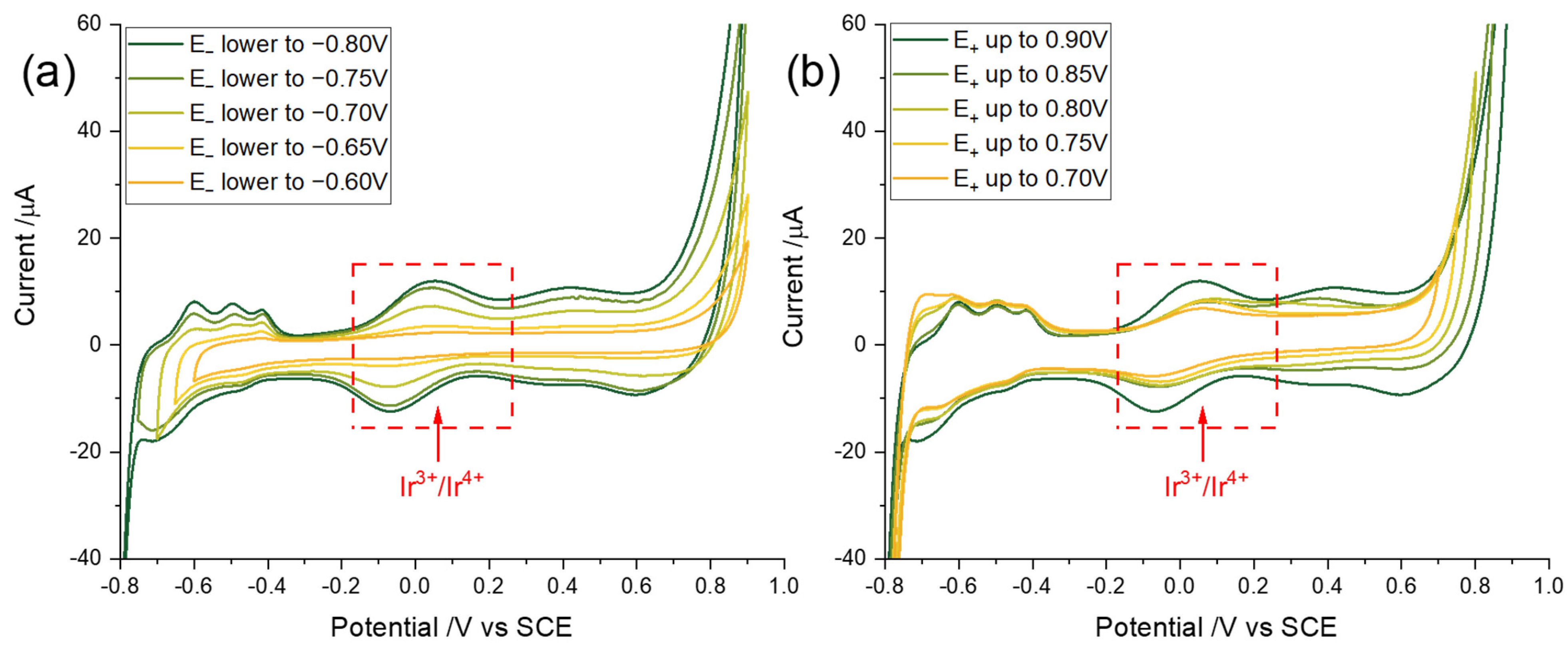
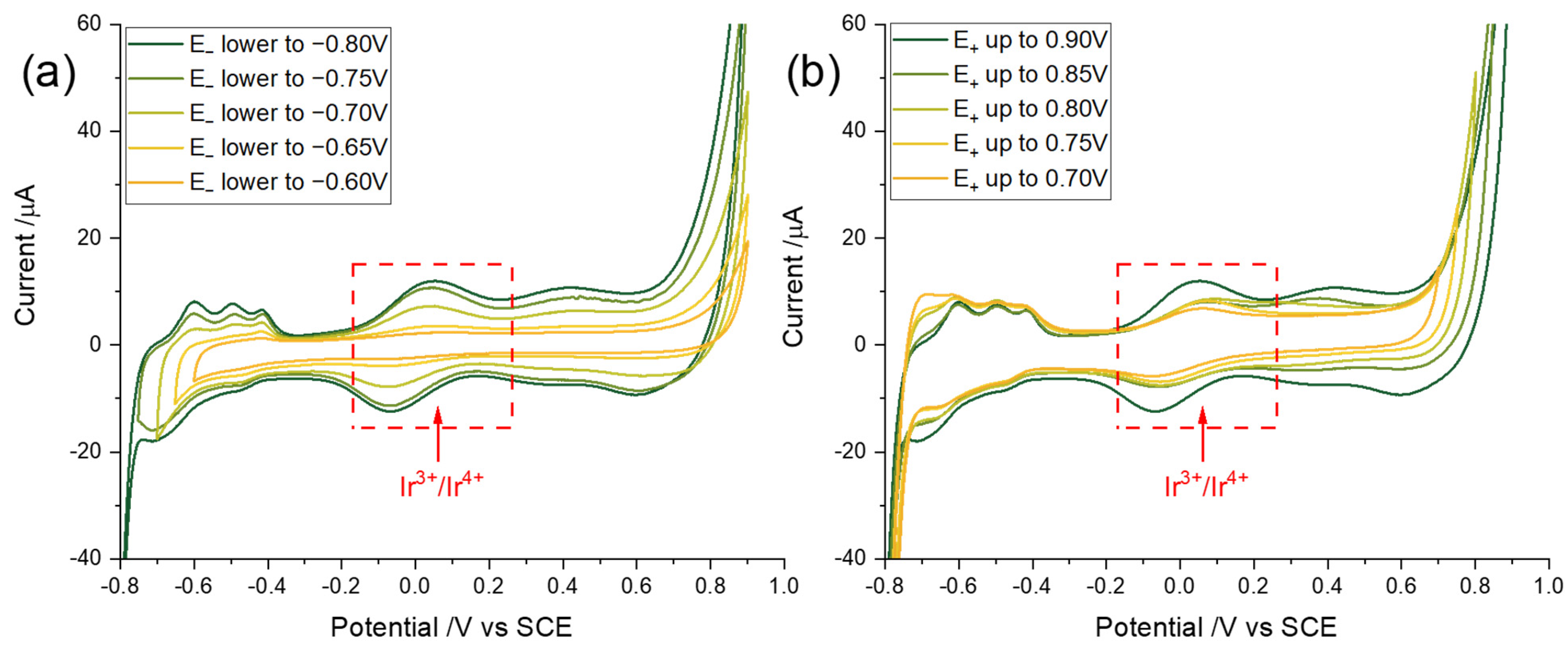
3.2. Optimization of Potential Cycling Activation
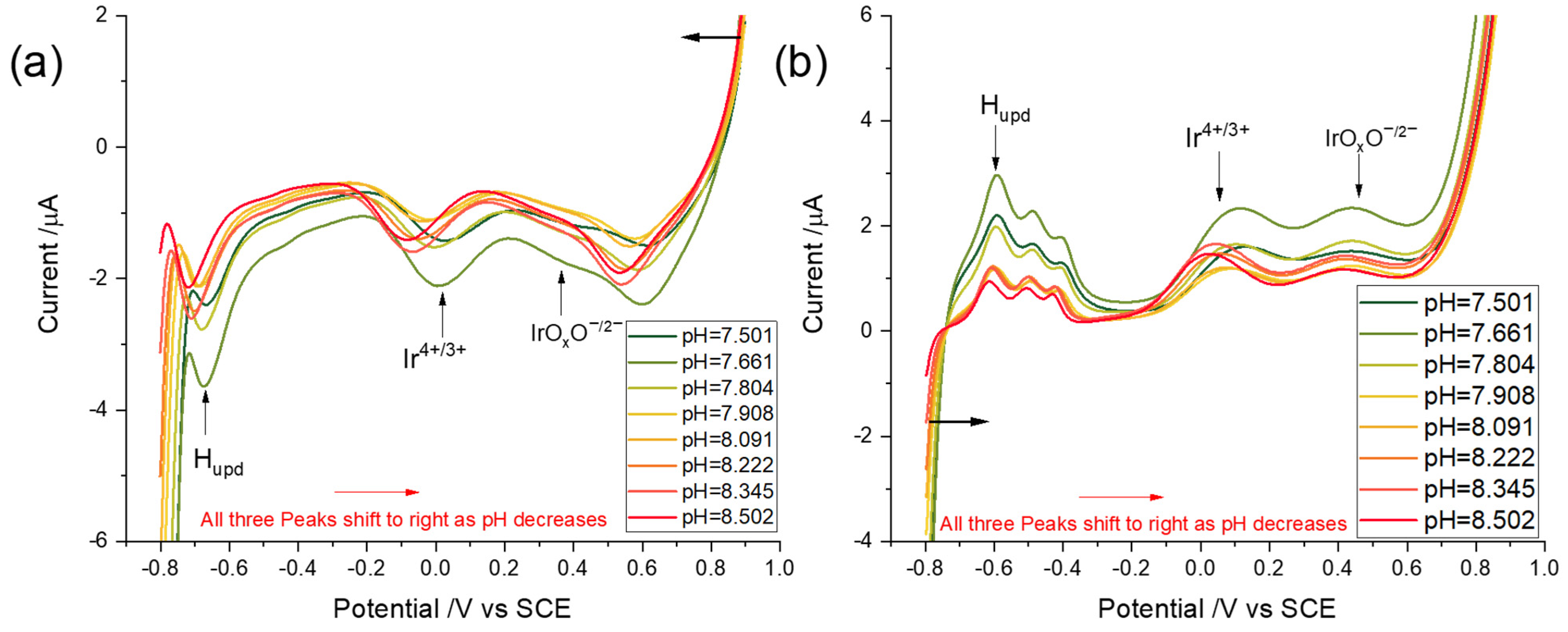
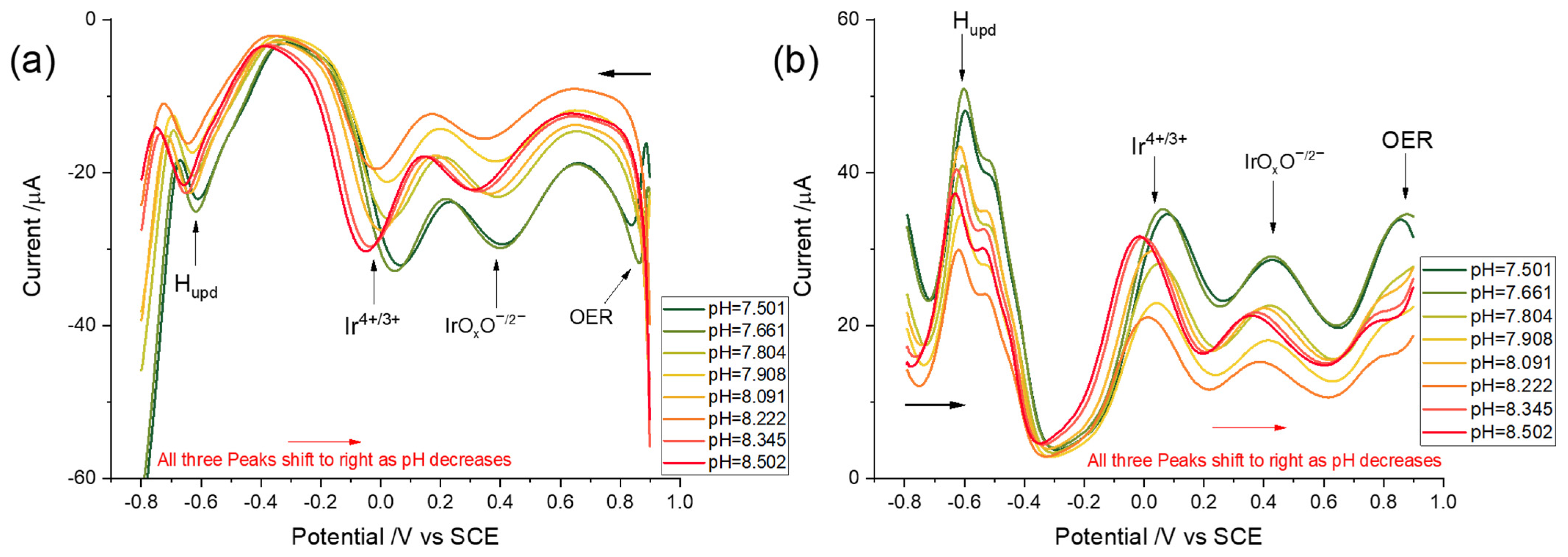
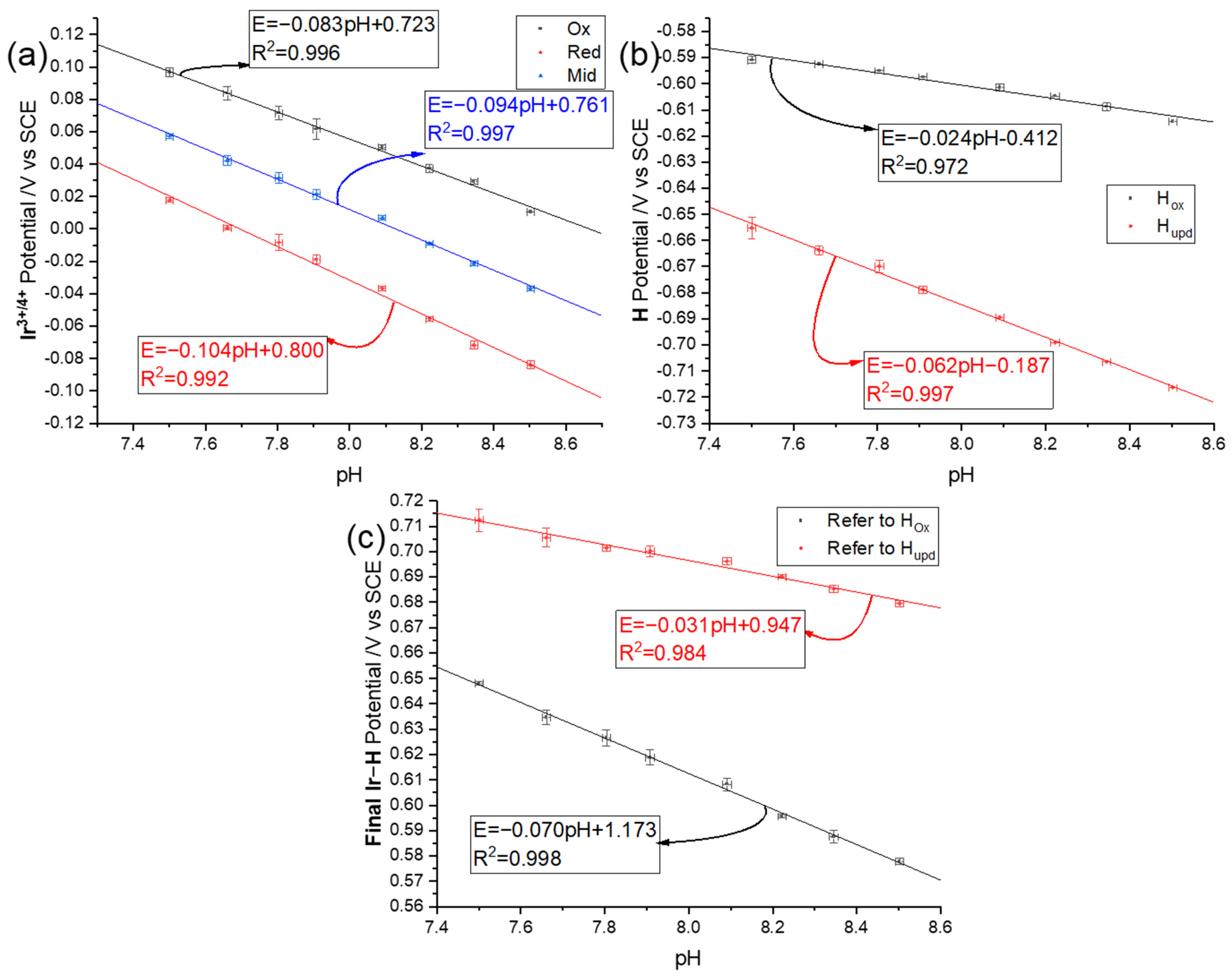
3.3. pH Dependency of the Voltammetric Responses of the Iridium Hydrous Oxide Layers
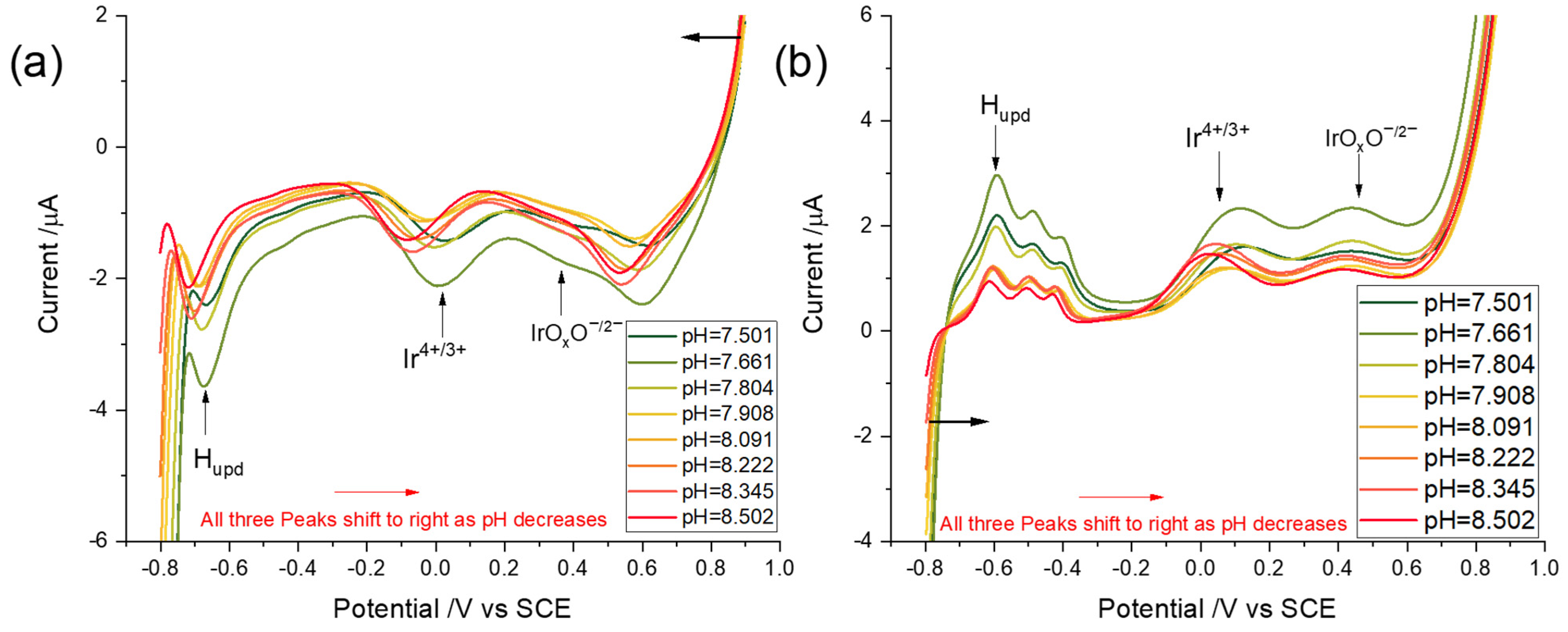
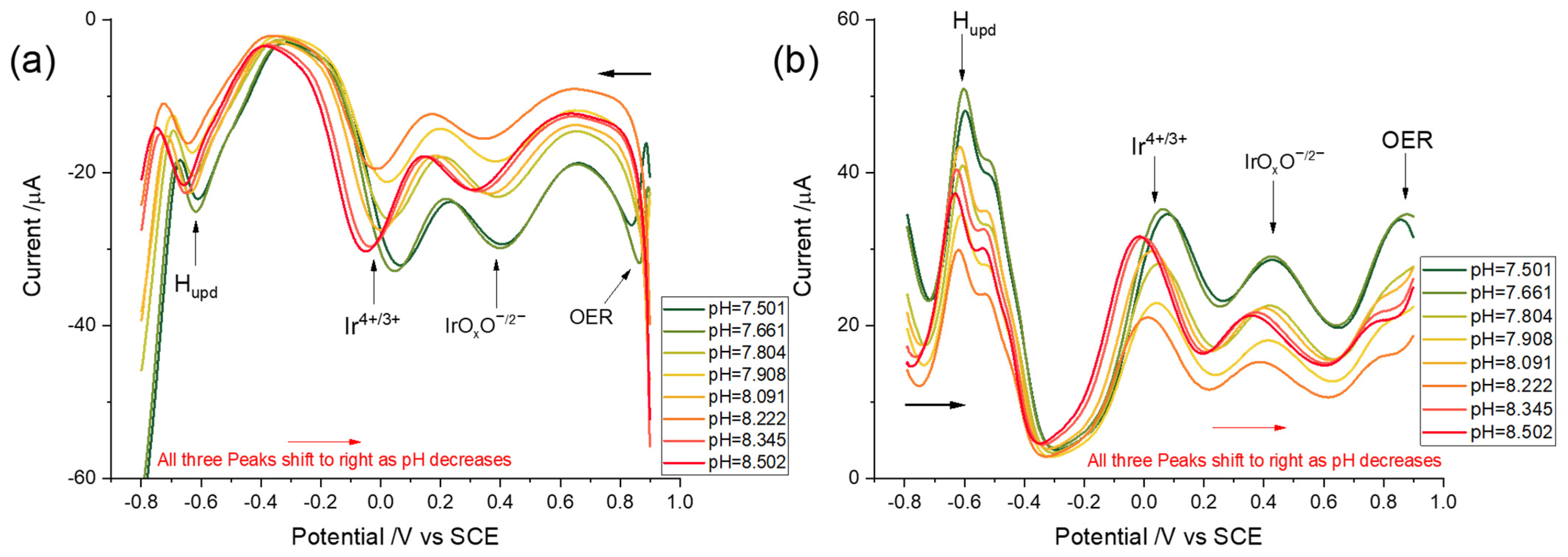
3.3.1. Linear Sweep Voltammetry
3.3.2. Square Wave Voltammetry
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Mills, I. Quantities, Units and Symbols in Physical Chemistry; Blackwell Science: Oxford, UK; Boston, MA, USA; CRC Press: Boca Raton, FL, USA, 1993. [Google Scholar]
- Buck, R.; Rondinini, S.; Covington, A.; Baucke, F.; Brett, C.M.; Camoes, M.; Milton, M.; Mussini, T.; Naumann, R.; Pratt, K. Measurement of pH. Definition, standards, and procedures (IUPAC Recommendations 2002). Pure Appl. Chem. 2002, 74, 2169–2200. [Google Scholar] [CrossRef] [Green Version]
- Hückel, E.; Debye, P. The theory of electrolytes: I. lowering of freezing point and related phenomena. Phys. Z 1923, 24, 1. [Google Scholar]
- Bates, R.G.; Vijh, A.K. Determination of pH: Theory and practice. J. Electrochem. Soc. 1973, 120, 263C. [Google Scholar] [CrossRef]
- Hunter, Y.R.; Kuwabara, J.S. Ionic strength and DOC determinations from various freshwater sources to the San Francisco Bay. Bull. Environ. Contam. Toxicol. 1994, 52, 311–318. [Google Scholar] [CrossRef] [PubMed]
- Guo, Y.; Compton, R.G. A bespoke chloride sensor for seawater: Simple and fast with a silver electrode. Talanta 2021, 232, 122502. [Google Scholar] [CrossRef] [PubMed]
- Zang, C.; Huang, S.; Wu, M.; Du, S.; Scholz, M.; Gao, F.; Lin, C.; Guo, Y.; Dong, Y. Comparison of Relationships Between pH, Dissolved Oxygen and Chlorophyll a for Aquaculture and Non-aquaculture Waters. Water Air Soil Pollut. 2011, 219, 157–174. [Google Scholar] [CrossRef]
- Scholz, M. Wetland Systems to Control Urban Runoff; Elsevier: Amsterdam, The Netherlands, 2015. [Google Scholar]
- U.S. Environmental Protection Agency. Available online: https://www.epa.gov/ (accessed on 6 December 2021).
- Bowmer, K.H.; Muirhead, W.A. Inhibition of algal photosynthesis to control pH and reduce ammonia volatilization from rice floodwater. Fertil. Res. 1987, 13, 13–29. [Google Scholar] [CrossRef]
- Jin, L.; Wang, Z.; Zhu, X.; Zhang, Y.; Sheng, L. Environmental Ecology; HigherEducation: Beijing, China, 1992. [Google Scholar]
- Fondriest Environmental Learning Centre. Algae, Phytoplankton and Chlorophyll. Available online: https://www.fondriest.com/environmental-measurements/parameters/water-quality/algae-phytoplankton-and-chlorophyll (accessed on 12 December 2021).
- Yang, M.; Batchelor-Mcauley, C.; Chen, L.; Guo, Y.; Zhang, Q.; Rickaby, R.E.M.; Bouman, H.A.; Compton, R.G. Fluoro-electrochemical microscopy reveals group specific differential susceptibility of phytoplankton towards oxidative damage. Chem. Sci. 2019, 10, 7988–7993. [Google Scholar] [CrossRef] [Green Version]
- Halevy, I.; Bachan, A. The geologic history of seawater pH. Science 2017, 355, 1069–1071. [Google Scholar] [CrossRef] [PubMed]
- Chuan, M.C.; Shu, G.Y.; Liu, J.C. Solubility of heavy metals in a contaminated soil: Effects of redox potential and pH. Water Air Soil Pollut. 1996, 90, 543–556. [Google Scholar] [CrossRef]
- Bourg, A.C.M.; Loch, J.P.G. Mobilization of Heavy Metals as Affected by pH and Redox Conditions; Springer: Berlin/Heidelberg, Germany, 1995; pp. 87–102. [Google Scholar]
- Sörensen, S.; Palitzsch, S. Sur le mesurage de la concentration en ions hydrogene de l’eau de mer. C. R. 1910, 9, 8. [Google Scholar]
- Millero, F.J.; Zhang, J.-Z.; Fiol, S.; Sotolongo, S.; Roy, R.N.; Lee, K.; Mane, S. The use of buffers to measure the pH of seawater. Mar. Chem. 1993, 44, 143–152. [Google Scholar] [CrossRef]
- Dickson, A. pH scales and proton-transfer reactions in saline media such as sea water. Geochim. Cosmochim. Acta 1984, 48, 2299–2308. [Google Scholar] [CrossRef]
- Culberson, C. Direct potentiometry. In Marine Electrochemistry; Whitfield, M., Jagner, D., Eds.; Wiley: Chichester, UK, 1981; pp. 187–261. [Google Scholar]
- Millero, F.J. The pH of estuarine waters. Limnol. Oceanogr. 1986, 31, 839–847. [Google Scholar] [CrossRef] [Green Version]
- Whitfield, M.; Butler, R.; Covington, A. The determination of ph in estuarine waters. 1. Definition of ph scales and the selection of buffers. Oceanol. Acta 1985, 8, 423–432. [Google Scholar]
- Ramette, R.W.; Culberson, C.H.; Bates, R.G. Acid-base properties of tris (hydroxymethyl) aminomethane (Tris) buffers in sea water from 5 to 40 degree C. Anal. Chem. 1977, 49, 867–870. [Google Scholar] [CrossRef]
- Bates, R.; Culberson, C. Hydrogen ions and the thermodynamic state of marine systems. In The Fate of Fossil Fuel CO2 in the Oceans; Anderson, N.R., Malahoff, A., Eds.; Plenum Press: New York, NY, USA, 1977; pp. 45–61. [Google Scholar]
- Khoo, K.; Ramette, R.W.; Culberson, C.H.; Bates, R.G. Determination of hydrogen ion concentrations in seawater from 5 to 40 degree C: Standard potentials at salinities from 20 to 45%. Anal. Chem. 1977, 49, 29–34. [Google Scholar] [CrossRef]
- Hansson, I. A new set of pH-scales and standard buffers for sea water. Deep Sea Res. Oceanogr. Abstr. 1973, 20, 479–491. [Google Scholar] [CrossRef]
- Dickson, A.; Riley, J. The estimation of acid dissociation constants in seawater media from potentionmetric titrations with strong base. I. The ionic product of water—Kw. Mar. Chem. 1979, 7, 89–99. [Google Scholar] [CrossRef]
- Dickson, A.G. Thermodynamics of the dissociation of boric acid in synthetic seawater from 273.15 to 318.15 K. Deep Sea Res. Part A Oceanogr. Res. Pap. 1990, 37, 755–766. [Google Scholar] [CrossRef]
- Dickson, A.; Millero, F.J. A comparison of the equilibrium constants for the dissociation of carbonic acid in seawater media. Deep Sea Res. Part A Oceanogr. Res. Pap. 1987, 34, 1733–1743. [Google Scholar] [CrossRef]
- Dickson, A.G. pH buffers for sea water media based on the total hydrogen ion concentration scale. Deep Sea Res. Part I Oceanogr. Res. Pap. 1993, 40, 107–118. [Google Scholar] [CrossRef]
- Bates, R.G.; Calais, J.G. Thermodynamics of the dissociation of Bis H+ in seawater from 5 to 40 °C. J. Solut. Chem. 1981, 10, 269–279. [Google Scholar] [CrossRef]
- Bates, R.G.; Erickson, W.P. Thermodynamics of the dissociation of 2-aminopyridinium ion in synthetic seawater and a standard for pH in marine systems. J. Solut. Chem. 1986, 15, 891–901. [Google Scholar] [CrossRef]
- Czerminski, J.B.; Dickson, A.G.; Bates, R.G. Thermodynamics of the dissociation of morpholinium ion in seawater from 5 to 40 °C. J. Solut. Chem. 1982, 11, 79–89. [Google Scholar] [CrossRef]
- Min, R.; Liu, Z.; Pereira, L.; Yang, C.; Sui, Q.; Marques, C. Optical fiber sensing for marine environment and marine structural health monitoring: A review. Opt. Laser Technol. 2021, 140, 107082. [Google Scholar] [CrossRef]
- Byrne, R.H.; Breland, J.A. High precision multiwavelength pH determinations in seawater using cresol red. Deep Sea Res. Part A Oceanogr. Res. Pap. 1989, 36, 803–810. [Google Scholar] [CrossRef]
- Clayton, T.D.; Byrne, R.H. Spectrophotometric seawater pH measurements: Total hydrogen ion concentration scale calibration of m-cresol purple and at-sea results. Deep Sea Res. Part I Oceanogr. Res. Pap. 1993, 40, 2115–2129. [Google Scholar] [CrossRef]
- Hudson-Heck, E.; Byrne, R.H. Purification and characterization of thymol blue for spectrophotometric pH measurements in rivers, estuaries, and oceans. Anal. Chim. Acta 2019, 1090, 91–99. [Google Scholar] [CrossRef] [PubMed]
- Byrne, R.; Robert-Baldo, G.; Thompson, S.; Chen, C. Seawater pH measurements: An at-sea comparison of spectrophotometric and potentiometric methods. Deep Sea Res. Part A Oceanogr. Res. Pap. 1988, 35, 1405–1410. [Google Scholar] [CrossRef]
- King, D.W.; Kester, D.R. Determination of seawater pH from 1.5 to 8.5 using colorimetric indicators. Mar. Chem. 1989, 26, 5–20. [Google Scholar] [CrossRef]
- Breland, J.; Byrne, R. Spectrophotometric determination of the total alkalinity of sea water using bromocresol green. Deep-Sea Res. 1993, 40, 629–641. [Google Scholar] [CrossRef]
- Byrne, R.H. Standardization of standard buffers by visible spectrometry. Anal. Chem. 1987, 59, 1479–1481. [Google Scholar] [CrossRef]
- Takahashi, T.; Broecker, W.S.; Bainbridge, A.E.; Weiss, R.F. Carbonate chemistry of the Atlantic, Pacific and Indian Oceans: The Results of the Geosecs Expeditions, 1972–1978; Lamont-Doherty Geological Observatory: Palisades, NY, USA, 1980. [Google Scholar]
- Dickson, A.G. The measurement of sea water pH. Mar. Chem. 1993, 44, 131–142. [Google Scholar] [CrossRef]
- Cuartero, M.; Pankratova, N.; Cherubini, T.; Crespo, G.A.; Massa, F.; Confalonieri, F.; Bakker, E. In Situ Detection of Species Relevant to the Carbon Cycle in Seawater with Submersible Potentiometric Probes. Environ. Sci. Technol. Lett. 2017, 4, 410–415. [Google Scholar] [CrossRef]
- Compton, R.G.; Batchelor-Mcauley, C.; Chaisiwamongkhol, K.; Vatish, M.; Kennedy, S. Method for Determining Human Blood pH. Patent Number WO/2020/217056, 29 October 2020. [Google Scholar]
- Chaisiwamongkhol, K.; Batchelor-Mcauley, C.; Compton, R.G. Amperometric micro pH measurements in oxygenated saliva. Analyst 2017, 142, 2828–2835. [Google Scholar] [CrossRef]
- Chaisiwamongkhol, K.; Batchelor-Mcauley, C.; Compton, R.G. Optimising amperometric pH sensing in blood samples: An iridium oxide electrode for blood pH sensing. Analyst 2019, 144, 1386–1393. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Li, X.; Li, D.; Batchelor-McAuley, C.; Compton, R.G. A simplified methodology: pH sensing using an in situ fabricated Ir electrode under neutral conditions. J. Solid State Electrochem. 2021, 25, 2821–2833. [Google Scholar] [CrossRef]
- Lawrence, N.S.; Pagels, M.; Hackett, S.F.; McCormack, S.; Meredith, A.; Jones, T.G.; Wildgoose, G.G.; Compton, R.G.; Jiang, L. Triple component carbon epoxy pH probe. Electroanal. Int. J. Devoted Fundam. Pract. Asp. Electroanal. 2007, 19, 424–428. [Google Scholar] [CrossRef]
- Streeter, I.; Leventis, H.C.; Wildgoose, G.G.; Pandurangappa, M.; Lawrence, N.S.; Jiang, L.; Jones, T.G.; Compton, R.G. A sensitive reagentless pH probe with a ca. 120 mV/pH unit response. J. Solid State Electrochem. 2004, 8, 718–721. [Google Scholar] [CrossRef]
- Wildgoose, G.G.; Pandurangappa, M.; Lawrence, N.S.; Jiang, L.; Jones, T.G.J.; Compton, R.G. Anthraquinone-derivatised carbon powder: Reagentless voltammetric pH electrodes. Talanta 2003, 60, 887–893. [Google Scholar] [CrossRef]
- Leventis, H.C.; Streeter, I.; Wildgoose, G.G.; Lawrence, N.S.; Jiang, L.; Jones, T.G.J.; Compton, R.G. Derivatised carbon powder electrodes: Reagentless pH sensors. Talanta 2004, 63, 1039–1051. [Google Scholar] [CrossRef] [PubMed]
- Wildgoose, G.G.; Leventis, H.C.; Streeter, I.; Lawrence, N.S.; Wilkins, S.J.; Jiang, L.; Jones, T.G.; Compton, R.G. Abrasively immobilised multiwalled carbon nanotube agglomerates: A novel electrode material approach for the analytical sensing of pH. ChemPhysChem 2004, 5, 669–677. [Google Scholar] [CrossRef] [PubMed]
- Phair, J.; Newton, L.; McCormac, C.; Cardosi, M.F.; Leslie, R.; Davis, J. A disposable sensor for point of care wound pH monitoring. Analyst 2011, 136, 4692. [Google Scholar] [CrossRef] [PubMed]
- Dai, C.; Chan, C.-W.I.; Barrow, W.; Smith, A.; Song, P.; Potier, F.; Wadhawan, J.D.; Fisher, A.C.; Lawrence, N.S. A Route to Unbuffered pH Monitoring: A Novel Electrochemical Approach. Electrochim. Acta 2016, 190, 879–886. [Google Scholar] [CrossRef]
- Sisodia, N.; Miranda, M.; McGuinness, K.L.; Wadhawan, J.D.; Lawrence, N.S. Intra- and Inter-molecular Sulf- hydryl Hydrogen Bonding: Facilitating Proton Transfer Events for Determination of pH in Sea Water. Electroanalysis 2020, 33, 559–562. [Google Scholar] [CrossRef]
- Nguyen, C.; Rao, S.; Yang, X.; Dubey, S.; Mays, J.; Cao, H.; Chiao, J.-C. Sol-Gel Deposition of Iridium Oxide for Biomedical Micro-Devices. Sensors 2015, 15, 4212–4228. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, W.-D.; Cao, H.; Deb, S.; Chiao, M.; Chiao, J.C. A flexible pH sensor based on the iridium oxide sensing film. Sens. Actuators A Phys. 2011, 169, 1–11. [Google Scholar] [CrossRef]
- Kuo, L.-M.; Chou, Y.-C.; Chen, K.-N.; Lu, C.-C.; Chao, S. A precise pH microsensor using RF-sputtering IrO2 and Ta2O5 films on Pt-electrode. Sens. Actuators B Chem. 2014, 193, 687–691. [Google Scholar] [CrossRef]
- Kreider, K.G.; Tarlov, M.J.; Cline, J.P. Sputtered thin-film pH electrodes of platinum, palladium, ruthenium, and iridium oxides. Sens. Actuators B Chem. 1995, 28, 167–172. [Google Scholar] [CrossRef]
- Ardizzone, S.; Carugati, A.; Trasatti, S. Properties of thermally prepared iridium dioxide electrodes. J. Electroanal. Chem. Interfacial Electrochem. 1981, 126, 287–292. [Google Scholar] [CrossRef]
- Hitchman, M.L.; Ramanathan, S. A field-induced poising technique for promoting convergence of standard electrode potential values of thermally oxidized iridium pH sensors. Talanta 1992, 39, 137–144. [Google Scholar] [CrossRef]
- Kinoshita, E.; Ingman, F.; Edwall, G.; Thulin, S.; Gła̧b, S. Polycrystalline and monocrystalline antimony, iridium and palladium as electrode material for pH-sensing electrodes. Talanta 1986, 33, 125–134. [Google Scholar] [CrossRef]
- Hitchman, M.L.; Ramanathan, S. Evaluation of iridium oxide electrodes formed by potential cycling as pH probes. Analyst 1988, 113, 35–39. [Google Scholar] [CrossRef] [PubMed]
- Olthuis, W.; Robben, M.A.M.; Bergveld, P.; Bos, M.; Van Der Linden, W.E. pH sensor properties of electrochemically grown iridium oxide. Sens. Actuators B Chem. 1990, 2, 247–256. [Google Scholar] [CrossRef] [Green Version]
- Compton, R.G.; Banks, C.E. Understanding Voltammetry; World Scientific: Singapore, 2018. [Google Scholar]
- Xiong, L.; Batchelor-McAuley, C.; Compton, R.G. Calibrationless pH sensors based on nitrosophenyl and ferrocenyl co-modified screen printed electrodes. Sens. Actuators B Chem. 2011, 159, 251–255. [Google Scholar] [CrossRef]
- Jiang, L.I.; Jones Timothy Gareth, J.; Compton, R.; Lawrence, N.; Wildgoose Gregory, G.; Pandurangappa, M.; Mullins Oliver, C.; Meredith, A. Electro-Chemical pH Sensor. European Patent EP 1702208 B1, 22 December 2004. [Google Scholar]
- Pickup, P.G.; Birss, V.I. The electrochemistry of iridium oxide films in some nonaqueous solvents. J. Electrochem. Soc. 1988, 135, 41. [Google Scholar] [CrossRef]
- Pfeifer, V.; Jones, T.E.; Velasco Velez, J.J.; Arrigo, R.; Piccinin, S.; Havecker, M.; Knop-Gericke, A.; Schlogl, R. In situ observation of reactive oxygen species forming on oxygen-evolving iridium surfaces. Chem. Sci. 2017, 8, 2143–2149. [Google Scholar] [CrossRef] [Green Version]
- Pfeifer, V.; Jones, T.E.; Velasco Velez, J.J.; Massue, C.; Greiner, M.T.; Arrigo, R.; Teschner, D.; Girgsdies, F.; Scherzer, M.; Allan, J.; et al. The electronic structure of iridium oxide electrodes active in water splitting. Phys. Chem. Chem. Phys. 2016, 18, 2292–2296. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dubouis, N.; Grimaud, A. The hydrogen evolution reaction: From material to interfacial descriptors. Chem. Sci. 2019, 10, 9165–9181. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jerkiewicz, G. Electrochemical Hydrogen Adsorption and Absorption. Part 1: Under-potential Deposition of Hydrogen. Electrocatalysis 2010, 1, 179–199. [Google Scholar] [CrossRef]
- Yang, X.; Nash, J.; Oliveira, N.; Yan, Y.; Xu, B. Understanding the pH Dependence of Underpotential Deposited Hydrogen on Platinum. Angew. Chem. 2019, 131, 17882–17887. [Google Scholar] [CrossRef]
- Zheng, J.; Sheng, W.; Zhuang, Z.; Xu, B.; Yan, Y. Universal dependence of hydrogen oxidation and evolution reaction activity of platinum-group metals on pH and hydrogen binding energy. Sci. Adv. 2016, 2, e1501602. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pfeifer, V.; Jones, T.E.; Velasco Vélez, J.J.; Massué, C.; Arrigo, R.; Teschner, D.; Girgsdies, F.; Scherzer, M.; Greiner, M.T.; Allan, J.; et al. The electronic structure of iridium and its oxides. Surf. Interface Anal. 2016, 48, 261–273. [Google Scholar] [CrossRef] [Green Version]
- Burke, L.D.; Lyons, M.E.; O’Sullivan, E.J.M.; Whelan, D.P. Influence of hydrolysis on the redox behaviour of hydrous oxide films. J. Electroanal. Chem. Interfacial Electrochem. 1981, 122, 403–407. [Google Scholar] [CrossRef]
- Carroll, S.; Baldwin, R.P. Self-calibrating microfabricated iridium oxide pH electrode array for remote monitoring. Anal. Chem. 2010, 82, 878–885. [Google Scholar] [CrossRef]
- Burke, L.D.; Mulcahy, J.K.; Whelan, D.P. Preparation of an oxidized iridium electrode and the variation of its potential with pH. J. Electroanal. Chem. Interfacial Electrochem. 1984, 163, 117–128. [Google Scholar] [CrossRef]
- Pickup, P.G.; Birss, V.I. A model for anodic hydrous oxide growth at iridium. J. Electroanal. Chem. Interfacial Electrochem. 1987, 220, 83–100. [Google Scholar] [CrossRef] [Green Version]
- Kasian, O.; Grote, J.-P.; Geiger, S.; Cherevko, S.; Mayrhofer, K.J.J. The Common Intermediates of Oxygen Evolution and Dissolution Reactions during Water Electrolysis on Iridium. Angew. Chem. Int. Ed. 2018, 57, 2488–2491. [Google Scholar] [CrossRef] [PubMed]
- Angerstein-Kozlowska, H.; Conway, B.E.; Sharp, W.B.A. The real condition of electrochemically oxidized platinum surfaces: Part I. Resolution of component processes. J. Electroanal. Chem. Interfacial Electrochem. 1973, 43, 9–36. [Google Scholar] [CrossRef]
- Kluge, R.M.; Haid, R.W.; Bandarenka, A.S. Assessment of active areas for the oxygen evolution reaction on an amorphous iridium oxide surface. J. Catal. 2021, 396, 14–22. [Google Scholar] [CrossRef]
- Sanchez Casalongue, H.G.; Ng, M.L.; Kaya, S.; Friebel, D.; Ogasawara, H.; Nilsson, A. In Situ Observation of Surface Species on Iridium Oxide Nanoparticles during the Oxygen Evolution Reaction. Angew. Chem. 2014, 126, 7297–7300. [Google Scholar] [CrossRef] [Green Version]
- Baur, J.E.; Spaine, T.W. Electrochemical deposition of iridium (IV) oxide from alkaline solutions of iridium (III) oxide. J. Electroanal. Chem. 1998, 443, 208–216. [Google Scholar] [CrossRef]
- Salimi, A.; Hyde, M.E.; Banks, C.E.; Compton, R.G. Boron doped diamond electrode modified with iridium oxide for amperometic detection of ultra trace amounts of arsenic (III). Analyst 2004, 129, 9–14. [Google Scholar] [CrossRef]
- Tarlov, M.J.; Semancik, S.; Kreider, K.G. Mechanistic and response studies of iridium oxide pH sensors. Sens. Actuators B Chem. 1990, 1, 293–297. [Google Scholar] [CrossRef]
- Wang, M.; Yao, S.; Madou, M. A long-term stable iridium oxide pH electrode. Sens. Actuators B Chem. 2002, 81, 313–315. [Google Scholar] [CrossRef]
- Głab, S.; Hulanicki, A.; Edwall, G.; Ingman, F. Metal-Metal Oxide and Metal Oxide Electrodes as pH Sensors. Crit. Rev. Anal. Chem. 1989, 21, 29–47. [Google Scholar] [CrossRef]
- Yao, S.; Wang, M.; Madou, M. A pH electrode based on melt-oxidized iridium oxide. J. Electrochem. Soc. 2001, 148, H29. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Constituent | Moles | Weight/g in 0.5 L | Final pH | |
|---|---|---|---|---|
| Synthetic Seawater | NaCl | 0.388 | 11.32 | N/A |
| KCl | 0.011 | 0.39 | ||
| MgCl2 | 0.055 | 2.61 | ||
| CaCl2 | 0.011 | 0.60 | ||
| Na2SO4 | 0.029 | 2.08 | ||
| HCl | 0.04 | 1.73 (mL in vol.) | ||
| One of | 2-Aminopyrine | 0.08 | 3.76 | 6.77 |
| Tris | 0.08 | 4.84 | 8.07 | |
| Bis | 0.08 | 4.20 | 8.81 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chen, Y.; Compton, R. A Calibration-Free pH Sensor Using an In-Situ Modified Ir Electrode for Bespoke Application in Seawater. Sensors 2022, 22, 3286. https://doi.org/10.3390/s22093286
Chen Y, Compton R. A Calibration-Free pH Sensor Using an In-Situ Modified Ir Electrode for Bespoke Application in Seawater. Sensors. 2022; 22(9):3286. https://doi.org/10.3390/s22093286
Chicago/Turabian StyleChen, Yuqi, and Richard Compton. 2022. "A Calibration-Free pH Sensor Using an In-Situ Modified Ir Electrode for Bespoke Application in Seawater" Sensors 22, no. 9: 3286. https://doi.org/10.3390/s22093286
APA StyleChen, Y., & Compton, R. (2022). A Calibration-Free pH Sensor Using an In-Situ Modified Ir Electrode for Bespoke Application in Seawater. Sensors, 22(9), 3286. https://doi.org/10.3390/s22093286