Genomic Phylogeography of Gymnocarpos przewalskii (Caryophyllaceae): Insights into Habitat Fragmentation in Arid Northwestern China



Abstract
1. Introduction
2. Materials and Methods
2.1. Plant Collection and DNA Extraction
2.2. GBS Sequencing and SNP Calling
2.3. Genetic Structure Analyses
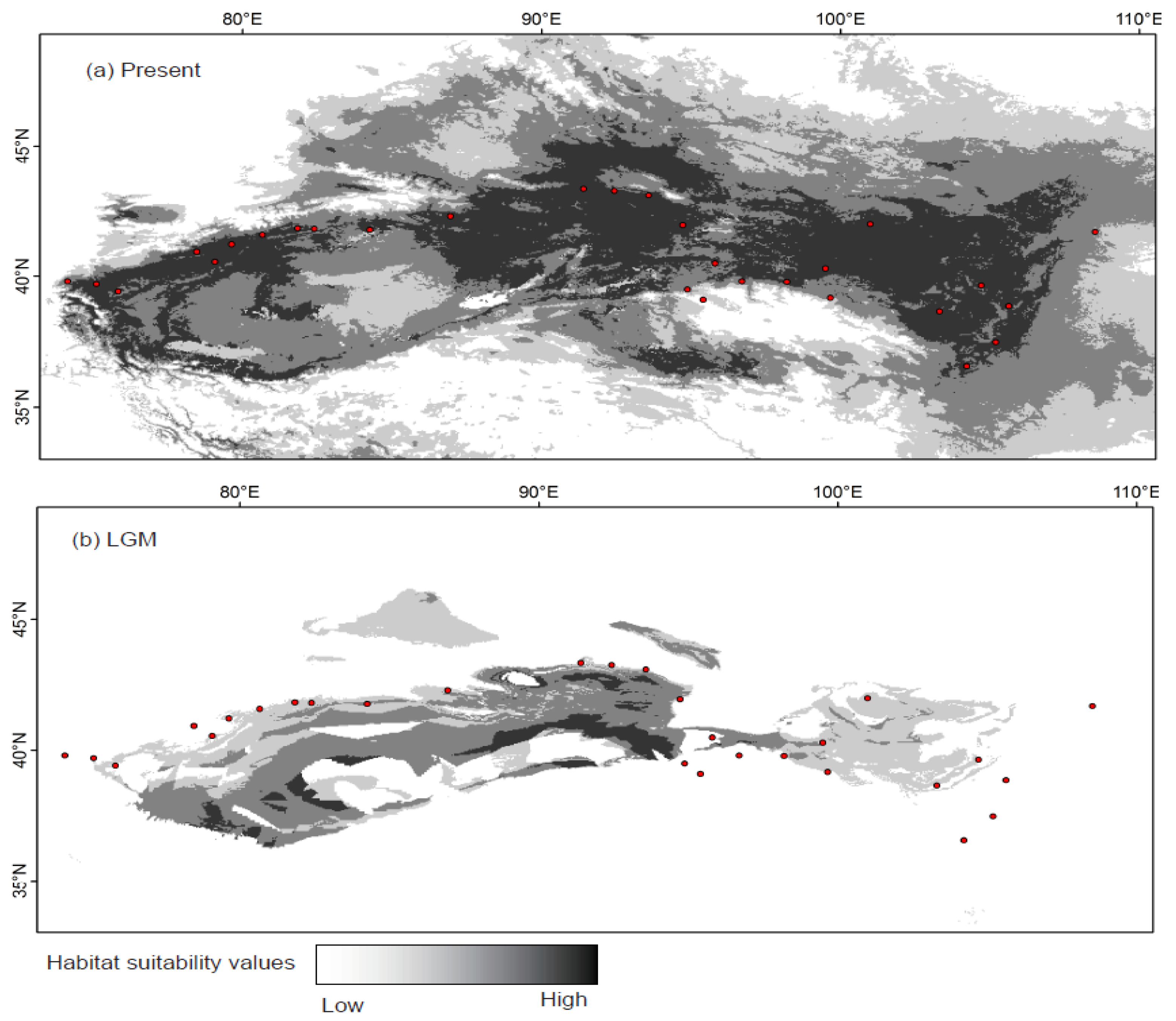
2.4. Species Distribution Model
2.5. Landscape Genetic Analysis
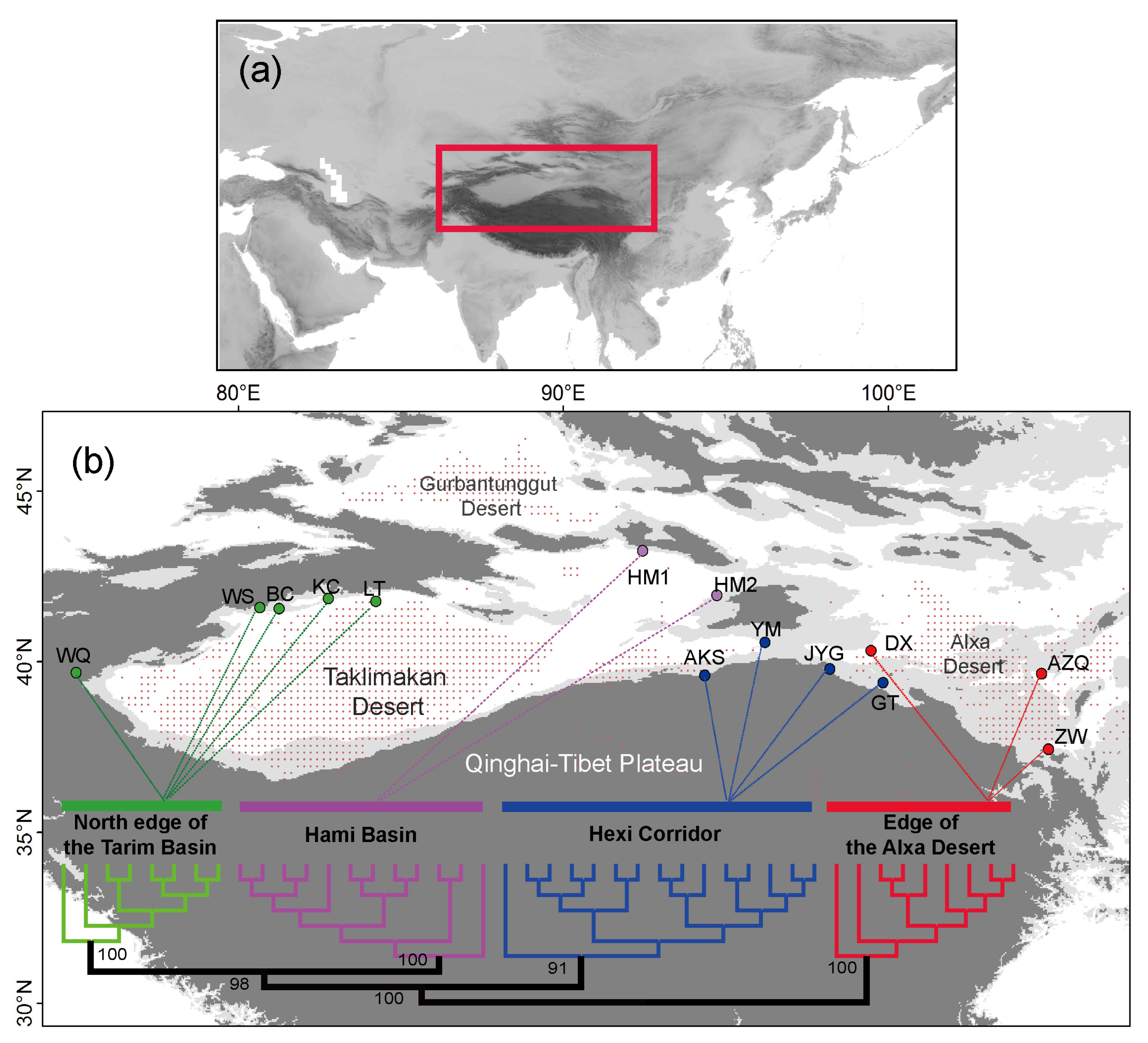
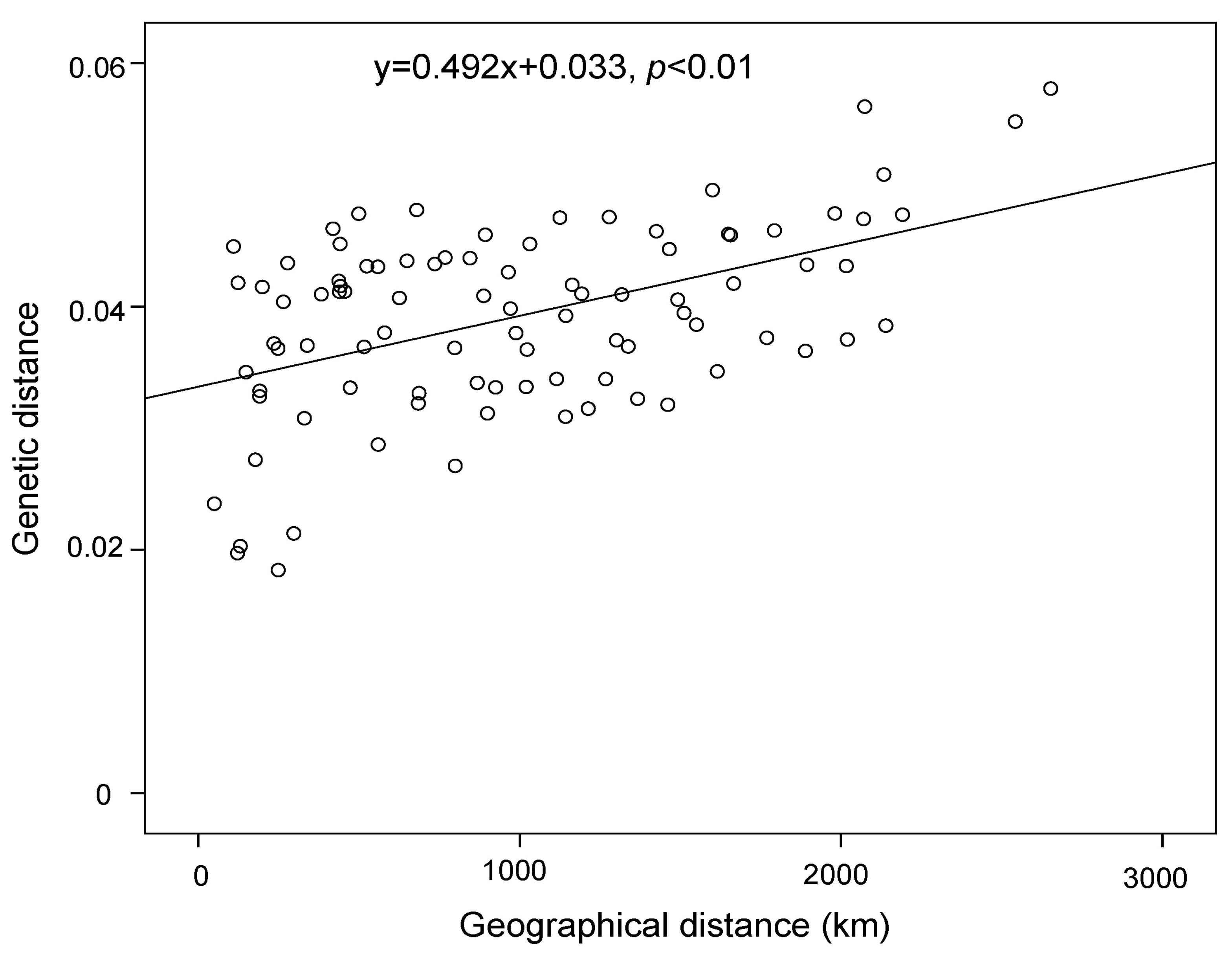
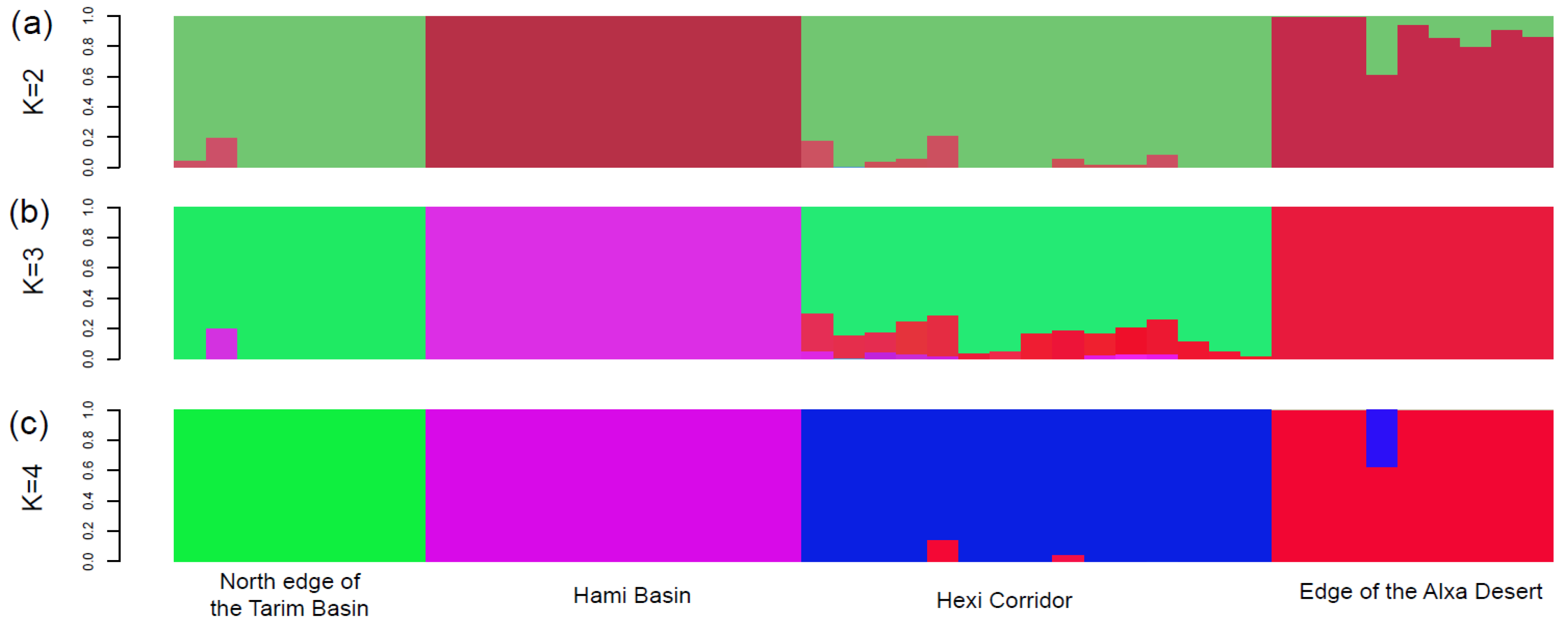
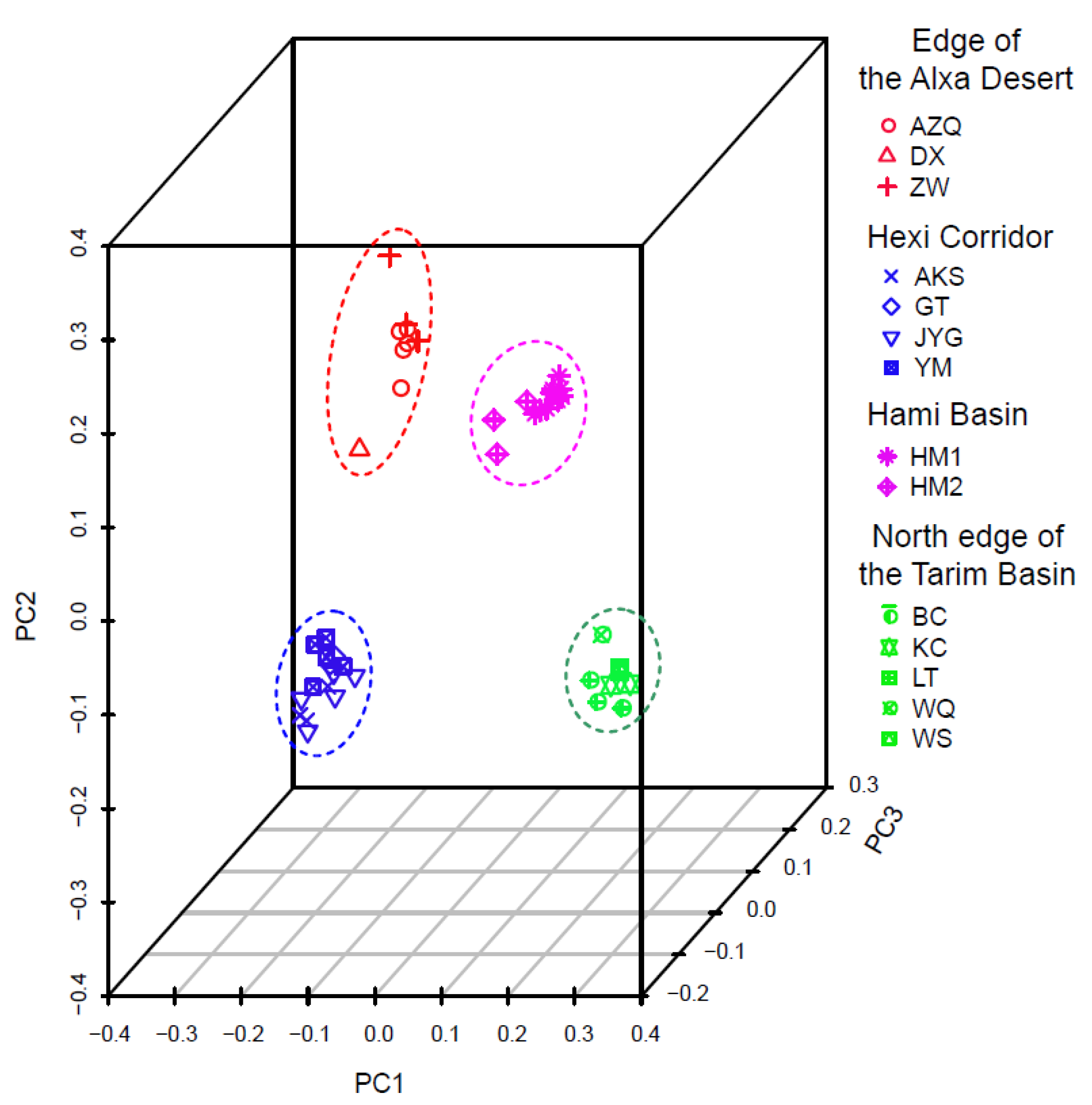
3. Results
4. Discussion
4.1. Clarifying Population Structure of G. przewalskii from the Genome-Wide SNPs
4.2. Effect of Historical and Current Environmental Conditions on the Spatial Genetic Pattern of G. przewalskii
4.3. Conservation Implications
5. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Haddad, N.M.; Brudvig, L.A.; Clobert, J.; Davies, K.F.; Gonzalez, A.; Holt, R.D.; Lovejoy, T.E.; Sexton, J.O.; Austin, M.P.; Collins, C.D.; et al. Habitat fragmentation and its lasting impact on Earth’s ecosystems. Sci. Adv. 2015, 1, e1500052. [Google Scholar] [CrossRef] [PubMed]
- Perrino, E.V.; Tomaselli, V.; Costa, R.; Pavone, P. Conservation status of habitats (Directive 92/43 EEC) of coastal and low hill belts in a Mediterranean biodiversity hot spot (Gargano-Italy). Plant Biosyst. 2013, 147, 1006–1028. [Google Scholar] [CrossRef]
- Yuan, N.; Comes, H.P.; Cao, Y.N.; Guo, R.; Zhang, Y.H.; Qiu, Y.X. A comparative study on genetic effects of artificial and natural habitat fragmentation on Loropetalum chinense (Hamamelidaceae) in Southeast China. Heredity 2015, 114, 544–551. [Google Scholar] [CrossRef][Green Version]
- Halbritter, D.A.; Storer, C.G.; Kawahara, A.Y.; Daniels, J.C. Phylogeography and population genetics of pine butterflies: Sky islands increase genetic divergence. Ecol. Evol. 2019, 9, 13389–13401. [Google Scholar] [CrossRef]
- Chen, J.H.; Huang, Y.; Brachi, B.; Yun, Q.Z.; Zhang, W.; Lu, W.; Li, H.N.; Li, W.Q.; Sun, X.D.; Wang, G.Y.; et al. Genome-wide analysis of Cushion willow provides insights into alpine plant divergence in a biodiversity hotspot. Nat. Commun. 2019, 10, 5230. [Google Scholar] [CrossRef]
- Edwards, C.E.; Albrecht, M.A.; Bassuener, B.; Yatskievych, G.A. Population genetic analysis reveals a predominantly selfing mating system and strong genetic structuring in a naturally fragmented, threatened plant. Conserv. Genet. 2019, 20, 1437–1448. [Google Scholar] [CrossRef]
- Yang, X.; Scuderi, L.; Paillou, P.; Liu, Z.; Li, H.; Ren, X. Quaternary environmental changes in the drylands of China—A critical review. Quat. Sci. Rev. 2011, 30, 3219–3233. [Google Scholar] [CrossRef]
- Guan, Q.; Pan, B.; Li, N.; Zhang, J.; Xue, L. Timing and significance of the initiation of present day deserts in the northeastern Hexi Corridor, China. Palaeogeogr. Palaeoclimatol. Palaeoecol. 2011, 306, 70–74. [Google Scholar]
- Xu, Z.; Zhang, M.L.; Cohen, J.I. Phylogeographic History of Atraphaxis Plants in Arid Northern China and the Origin of A. bracteata in the Loess Plateau. PLoS ONE 2016, 11, e0163243. [Google Scholar] [CrossRef]
- Li, Y.; Song, F.; Zhang, X.N.; Lv, G.H. Phylogeography suggest the Yili Valley being the glacial refuge of the genus Ixiolirion (Amaryllidaceae) in China. Syst. Biodivers. 2019, 17, 385–401. [Google Scholar] [CrossRef]
- Su, Z.; Lu, W.; Zhang, M. Phylogeographical patterns of two closely related desert shrubs, Nitraria roborowskii and N. sphaerocarpa (Nitrariaceae), from arid north-western China. Bot. J. Linn. Soc. 2016, 180, 334–347. [Google Scholar] [CrossRef]
- Ma, S.M.; Nie, Y.B.; Jiang, X.L.; Xu, Z.; Ji, W.Q. Genetic structure of the endangered, relict shrub Amygdalus mongolica (Rosaceae) in arid northwest China. Aust. J. Bot. 2019, 67, 128–139. [Google Scholar] [CrossRef]
- Zeng, Y.F.; Zhang, J.G.; Abuduhamiti, B.; Wang, W.T.; Jia, Z.Q. Phylogeographic patterns of the desert poplar in Northwest China shaped by both geology and climatic oscillations. BMC Evol. Biol. 2018, 18, 75. [Google Scholar] [CrossRef] [PubMed]
- Shi, X.J.; Zhang, M.L. Phylogeographical structure inferred from cpDNA sequence variation of Zygophyllum xanthoxylon across north-west China. J. Plant Res. 2015, 128, 269–282. [Google Scholar] [CrossRef]
- Zhang, H.X.; Zhang, M.L.; Sanderson, S.C. Spatial genetic structure of forest and xerophytic plant species in arid Eastern Central Asia: Insights from comparative phylogeography and ecological niche modelling. Biol. J. Linn. Soc. 2017, 120, 612–625. [Google Scholar] [CrossRef]
- Fu, L.G. China Plant Red Data Book; Science Press: Beijing, China, 1992. [Google Scholar]
- Jia, S.W.; Zhang, M.L. Pleistocene climate change and phylogeographic structure of the Gymnocarpos przewalskii (Caryophyllaceae) in the northwest China: Evidence from plastid DNA, ITS sequences, and Microsatellite. Ecol. Evol. 2019, 9, 5219–5235. [Google Scholar] [CrossRef]
- Cutter, A.D. Integrating phylogenetics, phylogeography and population genetics through genomes and evolutionary theory. Mol. Phylogenet. Evol. 2013, 69, 1172–1185. [Google Scholar] [CrossRef]
- Emerson, K.J.; Merz, C.R.; Catchen, J.M.; Hohenlohe, P.A.; Cresko, W.A.; Bradshaw, W.E.; Holzapfel, C.M. Resolving postglacial phylogeography using high-throughput sequencing. Proc. Natl. Acad. Sci. USA 2010, 107, 16196–16200. [Google Scholar] [CrossRef]
- Zhang, H.X.; Li, H.Y.; Li, Y.X. Identifying evolutionarily significant units for conservation of the endangered Malus sieversii using genome-wide RADseq data. Nord. J. Bot. 2018, 36, e01733. [Google Scholar] [CrossRef]
- Davey, J.L.; Blaxter, M.W. RADSeq: Next-generation population genetics. Brief. Funct. Genom. 2010, 9, 416–423. [Google Scholar] [CrossRef]
- Wallace, J.G.; Mitchell, S.E. Genotyping-by-Sequencing. Curr. Protoc. Plant Biol. 2017, 2, 64–77. [Google Scholar] [CrossRef] [PubMed]
- Sun, X.; Liu, D.; Zhang, X.; Li, W.; Liu, H.; Hong, W.; Jiang, C.; Guan, N.; Ma, C.; Zeng, H.; et al. SLAF-seq: An Efficient Method of Large-Scale De Novo SNP Discovery and Genotyping Using High-Throughput Sequencing. PLoS ONE 2013, 8, e58700. [Google Scholar] [CrossRef] [PubMed]
- Elshire, R.J.; Glaubitz, J.C.; Sun, Q.; Poland, J.A.; Kawamoto, K.; Buckler, E.S.; Mitchell, S.E. A Robust, Simple Genotyping-by-Sequencing (GBS) Approach for High Diversity Species. PLoS ONE 2011, 6, e19379. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Durbin, R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 2009, 25, 1754–1760. [Google Scholar] [CrossRef]
- Li, H.; Handsaker, B.; Wysoker, A.; Fennell, T.; Ruan, J.; Homer, N.; Marth, G.; Abecasis, G.; Durbin, R.; Genome Project Data Processing Subgroup. The Sequence Alignment/Map format and SAMtools. Bioinformatics 2009, 25, 2078–2079. [Google Scholar] [CrossRef]
- Sudhir, K.; Glen, S.; Li, M.; Christina, K.; Koichiro, T. MEGA X: Molecular Evolutionary Genetics Analysis across computing platforms. Mol. Biol. Evol. 2018, 35, 1547–1549. [Google Scholar]
- Alexander, D.H.; Novembre, J.; Lange, K. Fast model-based estimation of ancestry in unrelated individuals. Genome Res. 2009, 19, 1655–1664. [Google Scholar] [CrossRef]
- Yang, J.; Lee, S.H.; Goddard, M.E.; Visscher, P.M. GCTA: A Tool for Genome-wide Complex Trait Analysis. Am. J. Hum. Genet. 2011, 88, 76–82. [Google Scholar] [CrossRef]
- Excoffier, L.; Lischer, H.E.L. Arlequin suite ver 3.5: A new series of programs to perform population genetics analyses under Linux and Windows. Mol. Ecol. Resour. 2010, 10, 564–567. [Google Scholar] [CrossRef]
- Hijmans, R.J.; Cameron, S.E.; Parra, J.L.; Jones, P.G.; Jarvis, A. Very high resolution interpolated climate surfaces for global land areas. Int. J. Climatol. 2005, 25, 1965–1978. [Google Scholar] [CrossRef]
- Phillips, S.J.; Anderson, R.P.; Schapire, R.E. Maximum entropy modeling of species geographic distributions. Ecol. Model. 2006, 190, 231–259. [Google Scholar] [CrossRef]
- Jiang, X.L.; Deng, M.; Li, Y. Evolutionary history of subtropical evergreen broad-leaved forest in Yunnan Plateau and adjacent areas: An insight from Quercus schottkyana (Fagaceae). Tree Genet. Genomes 2016, 12, 104. [Google Scholar] [CrossRef]
- Jiang, X.L.; An, M.; Zheng, S.S.; Deng, M.; Su, Z.H. Geographical isolation and environmental heterogeneity contribute to the spatial genetic patterns of Quercus kerrii (Fagaceae). Heredity 2018, 120, 219–233. [Google Scholar] [CrossRef] [PubMed]
- Edwards, S.V.; Shultz, A.J.; Campbell-Staton, S.C. Next-generation sequencing and the expanding domain of phylogeography. Folia Zool. 2015, 64, 187–206. [Google Scholar] [CrossRef]
- Reitzel, A.M.; Herrera, S.; Layden, M.J.; Martindale, M.Q.; Shank, T.M. Going where traditional markers have not gone before: Utility of and promise for RAD sequencing in marine invertebrate phylogeography and population genomics. Mol. Ecol. 2013, 22, 2953–2970. [Google Scholar] [CrossRef]
- Bougeois, L.; Dupont-Nivet, G.; de Rafelis, M.; Tindall, J.C.; Proust, J.N.; Reichart, G.J.; de Nooijer, L.J.; Guo, Z.; Ormukov, C. Asian monsoons and aridification response to Paleogene sea retreat and Neogene westerly shielding indicated by seasonality in Paratethys oysters. Earth Planet. Sci. Lett. 2018, 485, 99–110. [Google Scholar] [CrossRef]
- Sun, J.; Gong, Z.; Tian, Z.; Jia, Y.; Windley, B. Late Miocene stepwise aridification in the Asian interior and the interplay between tectonics and climate. Palaeogeogr. Palaeoclimatol. Palaeoecol. 2015, 421, 48–59. [Google Scholar] [CrossRef]
- Roberts, D.R.; Hamann, A. Glacial refugia and modern genetic diversity of 22 western North American tree species. Proc. R. Soc. B Biol. Sci. 2015, 282, 20142903. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Region | Location | Longitude/Latitude | Nind | π (SD) | Ho (SD) | He (SD) |
|---|---|---|---|---|---|---|
| Edge of the Alxa Desert | 9 | 0.2181 (0.1087) | 0.3825 (0.2348) | 0.3251 (0.1441) | ||
| AZQ | 104.71/39.65 | 5 | ||||
| ZW | 104.92/37.43 | 3 | ||||
| DX | 99.46/40.32 | 1 | ||||
| Hexi Corridor | 15 | 0.2370 (0.1155) | 0.3279 (0.2202) | 0.2753 (0.1384) | ||
| GT | 99.83/39.38 | 2 | ||||
| JYG | 98.19/39.78 | 5 | ||||
| YM | 96.20/40.56 | 5 | ||||
| AKS | 94.34/39.59 | 3 | ||||
| Hami Basin | 12 | 0.2269 (0.1115) | 0.3422 (0.2196) | 0.2947 (0.1409) | ||
| HM1 | 92.43/43.25 | 9 | ||||
| HM2 | 94.72/41.95 | 3 | ||||
| North edge of the Tarim Basin | 8 | 0.2223 (0.1116) | 0.4196 (0.2524) | 0.3442 (0.1491) | ||
| LT | 84.23/41.77 | 1 | ||||
| KC | 82.77/41.85 | 2 | ||||
| BC | 81.25/41.55 | 3 | ||||
| WS | 80.66/41.59 | 1 | ||||
| WQ | 75.01/39.68 | 1 |
| Allelic Richness (Ar) | Expected Heterozygosity (He) | |||||||
|---|---|---|---|---|---|---|---|---|
| Est | SE | t | p | Est | SE | t | p | |
| (Intercept) | −5.846 × 108 | 2.347 × 108 | −2.490 | 0.03 | −8.255 × 107 | 3.943 × 107 | −2.094 | 0.06 |
| Longitude | - | - | - | - | 2.481 × 10−3 | 1.806 × 10−3 | 1.373 | 0.19 |
| Epre | - | - | - | - | 1.467 × 10−2 | 9.424 × 10-3 | 1.557 | 0.15 |
| ELGM | 5.911 × 10−2 | 4.090 × 10−2 | 1.445 | 0.17 | - | - | - | - |
| Npre | 5.846 × 108 | 2.347 × 108 | 2.490 | 0.03 | 8.255 × 107 | 3.943 × 107 | 2.094 | 0.06 |
| NLGM | −5.846 × 108 | 2.347 × 108 | −2.490 | 0.03 | −8.255 × 107 | 3.943 × 107 | −2.094 | 0.06 |
| Nstab | 5.846 × 108 | 2.347 × 108 | 2.490 | 0.03 | 8.255 × 107 | 3.943 × 107 | 2.094 | 0.06 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhang, H.-X.; Wang, Q.; Jia, S.-W. Genomic Phylogeography of Gymnocarpos przewalskii (Caryophyllaceae): Insights into Habitat Fragmentation in Arid Northwestern China. Diversity 2020, 12, 335. https://doi.org/10.3390/d12090335
Zhang H-X, Wang Q, Jia S-W. Genomic Phylogeography of Gymnocarpos przewalskii (Caryophyllaceae): Insights into Habitat Fragmentation in Arid Northwestern China. Diversity. 2020; 12(9):335. https://doi.org/10.3390/d12090335
Chicago/Turabian StyleZhang, Hong-Xiang, Qian Wang, and Su-Wen Jia. 2020. "Genomic Phylogeography of Gymnocarpos przewalskii (Caryophyllaceae): Insights into Habitat Fragmentation in Arid Northwestern China" Diversity 12, no. 9: 335. https://doi.org/10.3390/d12090335
APA StyleZhang, H.-X., Wang, Q., & Jia, S.-W. (2020). Genomic Phylogeography of Gymnocarpos przewalskii (Caryophyllaceae): Insights into Habitat Fragmentation in Arid Northwestern China. Diversity, 12(9), 335. https://doi.org/10.3390/d12090335