Perceptions of Similarity Can Mislead Provenancing Strategies—An Example from Five Co-Distributed Acacia Species

 ,
, 
Abstract
1. Introduction
2. Materials and Methods
2.1. Study Species: Acacia linifolia, A. longifolia, A. suaveolens, A. terminalis and A. ulicifolia
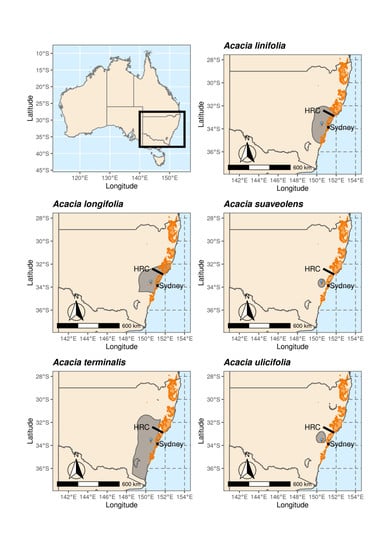
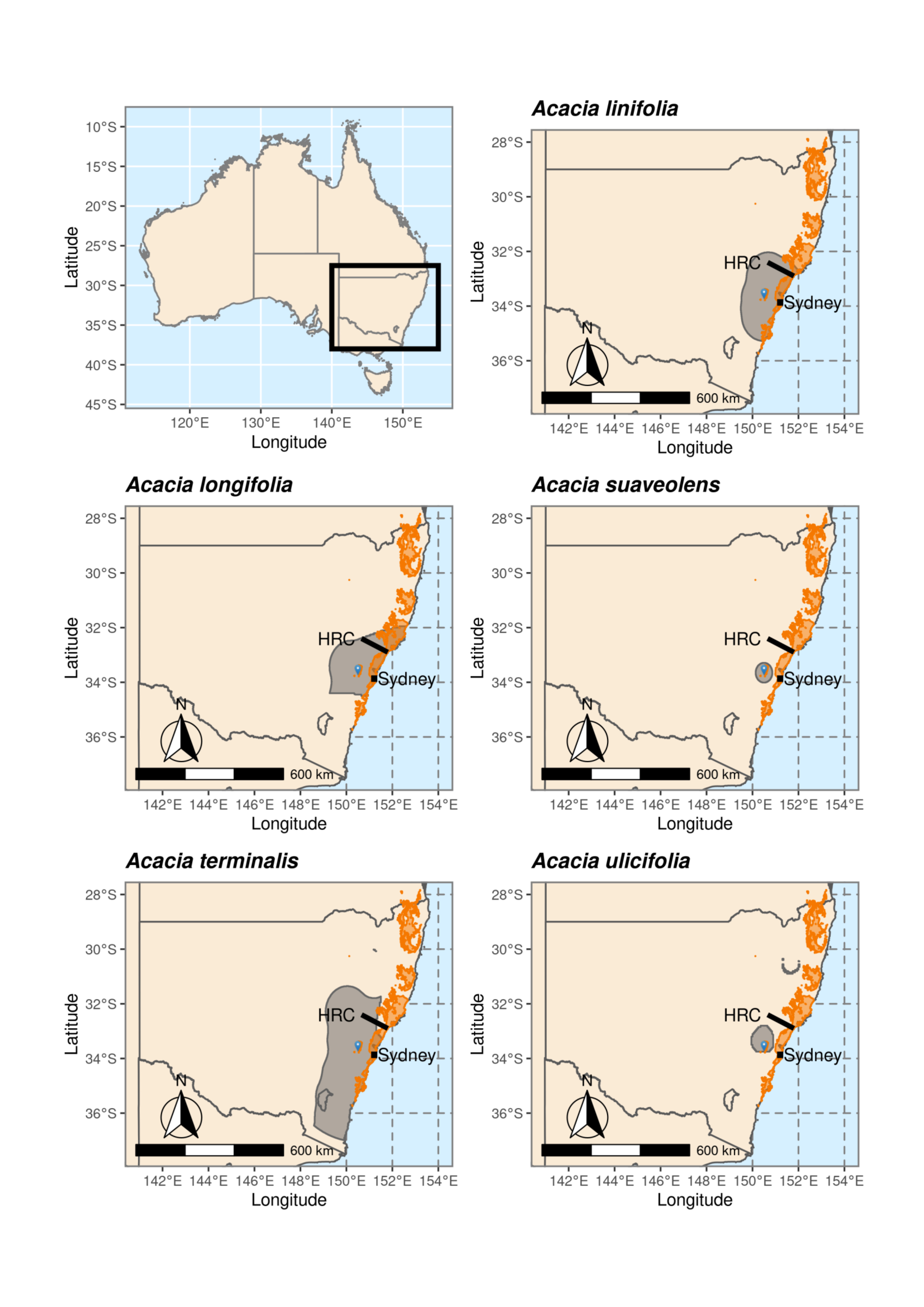
2.2. Species Distributions and Environmental Niche Models—Comparing Niche Overlap and Future Expectations
2.3. A New Site Matching Tool
2.4. Sampling Strategy and SNP Datasets
2.5. Comparing Landscap Genomics among Five Co-Distributed Acacia Species
2.6. Comparing Provenance Delineation among Five Co-Distributed Acacia Species
3. Results
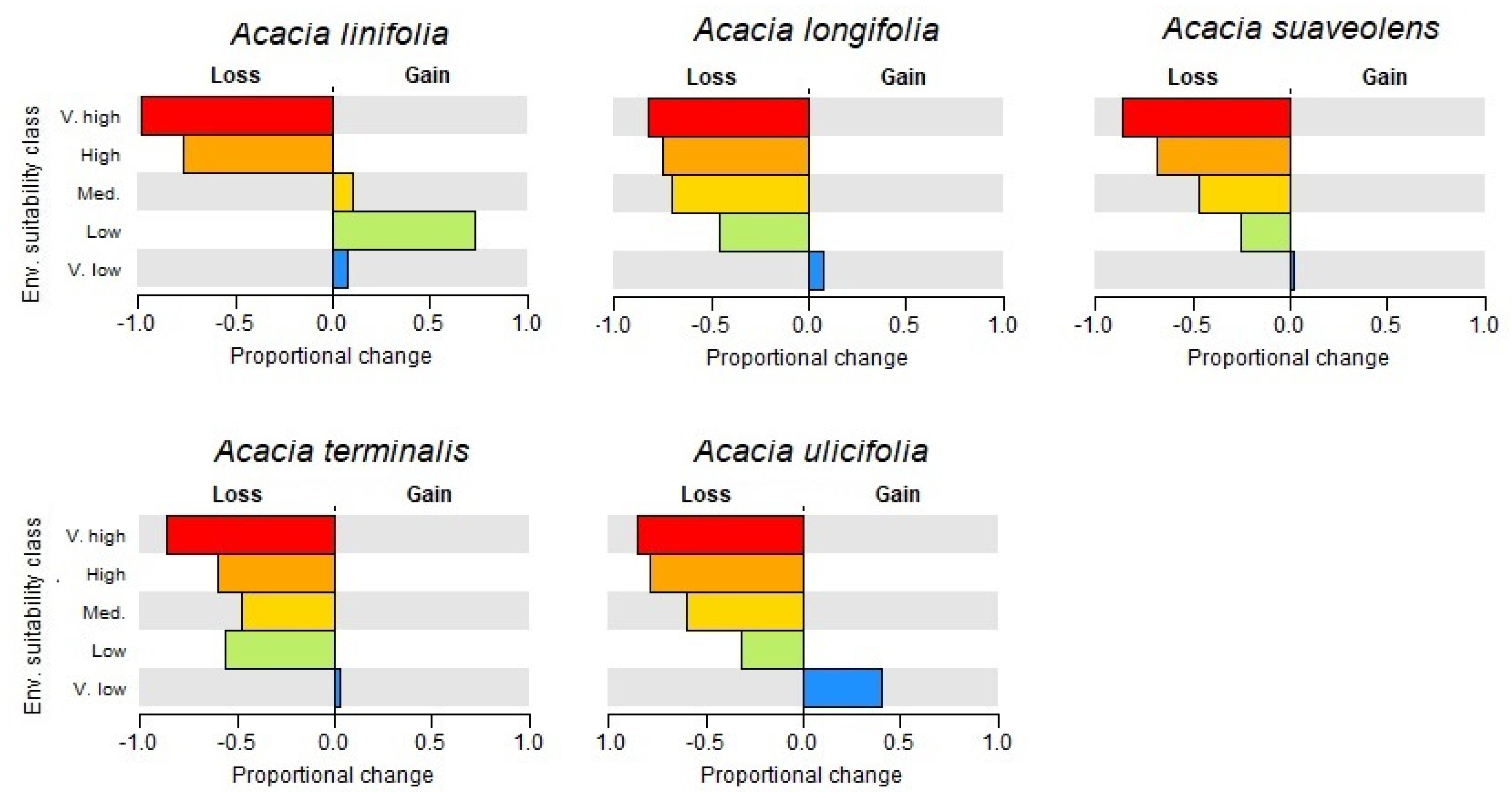
3.1. Species Distributions and Environmental Niche Models—Comparing Niche Overlap and Future Expectations
3.2. Comparing Knowledge-Based Provenance Delineation and Landscape-Genomics
4. Discussion
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Clewell, A.F.; Aronson, J. Motivations for the restoration of ecosystems. Conserv. Biol. 2006, 20, 420–428. [Google Scholar] [CrossRef] [PubMed]
- Gann, G.D.; McDonald, T.; Walder, B.; Aronson, J.; Nelson, C.R.; Jonson, J.; Hua, F. International principles and standards for the practice of ecological restoration. Restor. Ecol. 2019, 27, S1–S46. [Google Scholar] [CrossRef]
- Jones, T.A. Ecosystem restoration: Recent advances in theory and practice. Rangel. J. 2018, 39, 417–430. [Google Scholar] [CrossRef]
- Breed, M.F.; Harrison, P.A.; Blyth, C.; Byrne, M.; Gaget, V.; Gellie, N.J.; Steane, D.A. The potential of genomics for restoring ecosystems and biodiversity. Nat. Rev. Genet. 2019, 20, 615–628. [Google Scholar] [CrossRef]
- Broadhurst, L.M.; Lowe, A.; Coates, D.J.; Cunningham, S.A.; McDonald, M.; Vesk, P.A.; Yates, C. Seed supply for broadscale restoration: Maximizing evolutionary potential. Evol. Appl. 2008, 1, 587–597. [Google Scholar] [CrossRef]
- St. Clair, A.B.; Dunwiddie, P.W.; Fant, J.B.; Kaye, T.N.; Kramer, A.T. Mixing source populations increases genetic diversity of restored rare plant populations. Restor. Ecol. 2020, 28, 583–593. [Google Scholar] [CrossRef]
- Rossetto, M.; Bragg, J.; Kilian, A.; McPherson, H.; van der Merwe, M.; Wilson, P.D. Restore and Renew: A genomics-era framework for species provenance delimitation. Restor. Ecol. 2019, 27, 538–548. [Google Scholar] [CrossRef]
- Hancock, N.; Hughes, L. How far is it to your local? A survey on local provenance use in New South Wales. Ecol. Manag. Restor. 2012, 13, 259–266. [Google Scholar] [CrossRef]
- Hoban, S.; Callicrate, T.; Clark, J.; Deans, S.; Dosmann, M.; Fant, J.; Gailing, O.; Havens, K.; Hipp, A.L.; Kadav, P.; et al. Taxonomic similarity does not predict necessary sample size for ex situ conservation: A comparison among five genera. Proc. R. Soc. B 2020, 287, 20200102. [Google Scholar] [CrossRef]
- Kettenring, K.M.; Mercer, K.L.; Reinhardt Adams, C.; Hines, J. Application of genetic diversity–ecosystem function research to ecological restoration. J. Appl. Ecol. 2014, 51, 339–348. [Google Scholar] [CrossRef]
- Lesica, P.; Allendorf, F.W. Ecological genetics and the restoration of plant communities: Mix or match? Restor. Ecol. 1999, 7, 42–50. [Google Scholar] [CrossRef]
- Stockwell, C.A.; Kinnison, M.T.; Hendry, A.P.; Hamilton, J.A. Evolutionary restoration ecology. In Foundations of Restoration Ecology; Island Press: Washington, DC, USA, 2016; pp. 427–454. [Google Scholar]
- Hancock, N.; Gibson-Roy, P.; Driver, M.; Broadhurst, L. The Australian Native Seed Sector Survey Report; Australian Network for Plant Conservation: Canberra, Australia, 2020; Available online: https://www.anpc.asn.au/wp-content/uploads/2020/03/ANPC_NativeSeedSurveyReport_WEB.pdf (accessed on 1 February 2020).
- McKay, J.K.; Christian, C.E.; Harrison, S.; Rice, K.J. “How local is local?”—A review of practical and conceptual issues in the genetics of restoration. Restor. Ecol. 2005, 13, 432–440. [Google Scholar] [CrossRef]
- Breed, M.F.; Stead, M.G.; Ottewell, K.M.; Gardner, M.G.; Lowe, A.J. Which provenance and where? Seed sourcing strategies for revegetation in a changing environment. Conserv. Genet. 2013, 14, 1–10. [Google Scholar] [CrossRef]
- Prober, S.M.; Byrne, M.; McLean, E.H.; Steane, D.A.; Potts, B.M.; Vaillancourt, R.E.; Stock, W.D. Climate-adjusted provenancing: A strategy for climate-resilient ecological restoration. Front. Ecol. Evol. 2015, 3, 65. [Google Scholar] [CrossRef]
- Massatti, R.; Shriver, R.K.; Winkler, D.E.; Richardson, B.A.; Bradford, J.B. Assessment of population genetics and climatic variability can refine climate-informed seed transfer guidelines. Restor. Ecol. 2020, 28, 485–493. [Google Scholar] [CrossRef]
- Broadhurst, L.; Breed, M.; Lowe, A.; Bragg, J.; Catullo, R.; Coates, D.; Potts, B. Genetic diversity and structure of the Australian flora. Divers. Distrib. 2017, 23, 41–52. [Google Scholar] [CrossRef]
- Thiel-Egenter, C.; Gugerli, F.; Alvarez, N.; Brodbeck, S.; Cieślak, E.; Colli, L.; Negrini, R. Effects of species traits on the genetic diversity of high-mountain plants: A multi-species study across the Alps and the Carpathians. Glob. Ecol. Biogeogr. 2009, 18, 78–87. [Google Scholar] [CrossRef]
- Broadhurst, L.M.; Young, A.G.; Forrester, R. Genetic and demographic responses of fragmented Acacia dealbata (Mimosaceae) populations in southeastern Australia. Biol. Conserv. 2008, 141, 2843–2856. [Google Scholar] [CrossRef]
- FloraBank. Guidelines 5 Seed Collection from Woody Plants for Local Revegetation; FloraBank: Canberra, Australia, 1999; Available online: http://www.florabank.org.au/ (accessed on 1 February 2020).
- Gunn, B. Australian Tree Seed Centre Operations Manua; CSIRO Forestry and Forest Products: Canberra, Australia, 2001. [Google Scholar]
- Bernhardt, P. The floral ecology of Australian Acacia. Adv. Legum. Boil. Monogr. Syst. Bot. Mo. Bot. Gard. 1989, 29, 263–282. [Google Scholar]
- Kenrick, J.; Knox, R.B. Quantitative Analysis of Self-Incompatibility in Trees of Seven Species of Acacia. J. Hered. 1989, 80, 240–245. [Google Scholar] [CrossRef]
- Auld, T.D.; O’Connell, M.A. Predicting patterns of post-fire germination in 35 eastern Australian Fabaceae. Aust. J. Ecol. 1991, 16, 53–70. [Google Scholar] [CrossRef]
- Liyanage, G.S.; Ooi, M.K.J. Intra-population level variation in thresholds for physical dormancy-breaking temperature. Ann. Bot. 2015, 116, 123–131. [Google Scholar] [CrossRef] [PubMed]
- Aiello-Lammens, M.; Boria, R.; Radosavljevic, A.; Vilela, B.; Anderson, R. spThin: An R package for spatial thinning of species occurrence records for use in ecological niche models. Ecography 2015, 38, 541–545. [Google Scholar] [CrossRef]
- Xu, T.; Hutchinson, M.F. New developments and applications in the ANUCLIM spatial climatic and bioclimatic modelling package. Environ. Model. Softw. 2013, 40, 267–279. [Google Scholar] [CrossRef]
- Powney, G.D.; Roy, D.B.; Chapman, D.; Brereton, T.; Oliver, T.H. Measuring functional connectivity using long-term monitoring data. Methods Ecol. Evol. 2011, 2, 527–533. [Google Scholar] [CrossRef]
- Naimi, B. usdm: Uncertainty analysis for species distribution models. R Package Version 2013, 1, 1–12. [Google Scholar]
- Phillips, S.J.; Anderson, R.P.; Schapire, R.E. Maximum entropy modeling of species geographic distributions. Ecol. Model. 2006, 190, 231–259. [Google Scholar] [CrossRef]
- Phillips, S.J.; Dudík, M. Modeling of species distributions with Maxent: New extensions and a comprehensive evaluation. Ecography 2008, 31, 161–175. [Google Scholar] [CrossRef]
- Muscarella, R.; Galante, P.J.; Soley-Guardia, M.; Boria, R.A.; Kass, J.M.; Uriarte, M.; Anderson, R.P. ENM eval: An R package for conducting spatially independent evaluations and estimating optimal model complexity for Maxent ecological niche models. Methods Ecol. Evol. 2014, 5, 1198–1205. [Google Scholar] [CrossRef]
- Whetton, P.; Ekström, M.; Gerbing, C.; Grose, M.; Bhend, J.; Webb, L.; Colman, R. Climate Change in Australia. Information for Australia’s Natural Resource Management Regions: Technical Report; 2015. Available online: http://ccia2007.climatechangeinaustralia.gov.au/technical_report.php (accessed on 1 February 2020).
- Wilson, P.D. Binned Relative Environmental Change Indicator (BRECI): A tool to communicate the nature of differences between environmental niche model outputs. bioRxiv 2019, 672618. [Google Scholar] [CrossRef]
- Junker, R.R.; Kuppler, J.; Bathke, A.C.; Schreyer, M.L.; Trutschnig, W. Dynamic range boxes—A robust nonparametric approach to quantify size and overlap of n-dimensional hypervolumes. Methods Ecol. Evol. 2016, 7, 1503–1513. [Google Scholar] [CrossRef]
- Prunier, J.G.; Kaufmann, B.; Fenet, S.; Picard, D.; Pompanon, F.; Joly, P.; Lena, J.P. Optimizing the trade-off between spatial and genetic sampling efforts in patchy populations: Towards a better assessment of functional connectivity using an individual-based sampling scheme. Mol. Ecol. 2013, 22, 5516–5530. [Google Scholar] [CrossRef] [PubMed]
- Santos, A.S.; Gaiotto, F.A. Knowledge status and sampling strategies to maximize cost-benefit ratio of studies in landscape genomics of wild plants. Sci. Rep. 2020, 10, 3706. [Google Scholar] [CrossRef] [PubMed]
- Bragg, J.G.; Cuneo, P.; Sherieff, A.; Rossetto, M. Optimizing the genetic composition of a translocation population: Incorporating constraints and conflicting objectives. Mol. Ecol. Resour. 2020, 20, 54–65. [Google Scholar] [CrossRef]
- Bragg, J.G.; Supple, M.A.; Andrew, R.L.; Borevitz, J.O. Genomic variation across landscapes: Insights and applications. New Phytol. 2015, 207, 953–967. [Google Scholar] [CrossRef] [PubMed]
- Sansaloni, C.; Petroli, C.; Jaccoud, D.; Carling, J.; Detering, F.; Grattapaglia, D.; Kilian, A. Diversity Arrays Technology (DArT) and next-generation sequencing combined: Genome-wide, high throughput, highly informative genotyping for molecular breeding of Eucalyptus. In BMC Proceedings; BioMed Central: London, UK, 2011; Volume 5, p. 54. [Google Scholar]
- Kilian, A.; Wenzl, P.; Huttner, E.; Carling, J.; Xia, L.; Blois, H.; Aschenbrenner-Kilian, M. Diversity arrays technology: A generic genome profiling technology on open platforms. In Data Production and Analysis in Population Genomics; Humana Press: Totowa, NJ, USA, 2012; pp. 67–89. [Google Scholar]
- Rutherford, S.; Rossetto, M.; Bragg, J.G.; McPherson, H.; Benson, D.; Bonser, S.P.; Wilson, P.G. Speciation in the presence of gene flow: Population genomics of closely related and diverging Eucalyptus species. Heredity 2018, 121, 126–141. [Google Scholar] [CrossRef] [PubMed]
- Fahey, M.; Rossetto, M.; Wilson, P.D.; Ho, S.Y. Habitat preference differentiates the Holocene range dynamics but not barrier effects on two sympatric, congeneric trees (Tristaniopsis, Myrtaceae). Heredity 2019, 123, 532–548. [Google Scholar] [CrossRef]
- Huson, D.H.; Bryant, D. Application of phylogenetic networks in evolutionary studies. Mol. Biol. Evol. 2006, 23, 254–267. [Google Scholar] [CrossRef]
- Keenan, K.; McGinnity, P.; Cross, T.F.; Crozier, W.W.; Prodöhl, P.A. diveRsity: An R package for the estimation and exploration of population genetics parameters and their associated errors. Methods Ecol. Evol. 2013, 4, 782–788. [Google Scholar] [CrossRef]
- Kamvar, Z.N.; Tabima, J.F.; Grünwald, N.J. Poppr: An R package for genetic analysis of populations with clonal, partially clonal, and/or sexual reproduction. PeerJ 2014, 2, e281. [Google Scholar] [CrossRef]
- Weir, B.S.; Hill, W.G. Estimating F-statistics. Annu. Rev. Genet. 2002, 36, 721–750. [Google Scholar] [CrossRef] [PubMed]
- Zheng, X.; Zheng, M.X. Package ‘SNPRelate’. A package for Parallel Computing Toolset for Relatedness and Principal Component Analysis of SNP Data. 2013. Available online: http://github.com/zhengxwen/SNPRelate (accessed on 1 February 2020).
- Mantel, N. The detection of disease clustering and a generalized regression approach. Cancer Res. 1967, 27, 209–220. [Google Scholar] [PubMed]
- Oksanen, J.; Blanchet, F.G.; Kindt, R.; Legendre, P.; Minchin, P.R.; O’hara, R.B.; Oksanen, M.J. Package ‘vegan’. Community Ecol. Package Version 2013, 2, 1–295. [Google Scholar]
- Frichot, E.; François, O. LEA: An R package for landscape and ecological association studies. Methods Ecol. Evol. 2015, 6, 925–929. [Google Scholar] [CrossRef]
- Manion, G.; Lisk, M.; Ferrier, S.; Lugilde, K.M.; Fitzpatrick, M.C.; Fitzpatrick, M.M.C.; Rcpp, I. Package ‘gdm’. A Toolkit with Functions to Fit, Plot, and Summarize Generalized Dissimilarity Models: CRAN Repository, R. 2017. Available online: https://CRAN.R-project.org/package=gdm (accessed on 1 February 2020).
- Shryock, D.F.; Havrilla, C.A.; DeFalco, L.A.; Esque, T.C.; Custer, N.A.; Wood, T.E. Landscape genetic approaches to guide native plant restoration in the Mojave Desert. Ecol. Appl. 2017, 27, 429–445. [Google Scholar] [CrossRef]
- Supple, M.A.; Bragg, J.G.; Broadhurst, L.M.; Nicotra, A.B.; Byrne, M.; Andrew, R.L.; Borevitz, J.O. Landscape genomic prediction for restoration of a Eucalyptus foundation species under climate change. eLIFE 2018, 7, e31835. [Google Scholar] [CrossRef]
- Rue, H.; Martino, S.; Chopin, N. Approximate Bayesian inference for latent Gaussian models using inte-grated nested Laplace approximations (with discussion). J. R. Stat. Soc. Ser. B 2009, 71, 319–392. [Google Scholar] [CrossRef]
- Bivand, R.; Rundel, C. RGEOS: Interface to Geometry Engine—Open Source (‘GEOS’). R Package Version 0.5-2. 2019. Available online: https://CRAN.R-project.org/package=rgeos (accessed on 1 February 2020).
- van den Boogaart, K.G.; Tolosana, R.; Bren, M. Compositions: Compositional Data Analysis. R Package Version 1.40-1. 2014. Available online: https://CRAN.R-project.org/package=compositions (accessed on 1 February 2020).
- Booth, T.H. Identifying particular areas for potential seed collections for restoration plantings under climate change. Ecol. Manag. Restor. 2016, 17, 228–234. [Google Scholar] [CrossRef]
- Milner, M.L.; Rossetto, M.; Crisp, M.D.; Weston, P.H. The impact of multiple biogeographic barriers and hybridization on species-level differentiation. Am. J. Bot. 2012, 99, 2045–2057. [Google Scholar] [CrossRef]
- Bond, W.J.; Midgley, J.J. Ecology of sprouting in woody plants: The persistence niche. Trends Ecol. Evol. 2001, 16, 45–51. [Google Scholar] [CrossRef]
- Parmesan, C. Ecological and evolutionary responses to recent climate change. Annu. Rev. Ecol. Evol. Syst. 2006, 37, 637–669. [Google Scholar] [CrossRef]
- Williams, A.V.; Nevill, P.G.; Krauss, S.L. Next generation restoration genetics: Applications and opportunities. Trends Plant Sci. 2014, 19, 529–537. [Google Scholar] [CrossRef] [PubMed]
- Broadhurst, L.; Waters, C.; Coates, D. Native seed for restoration: A discussion of key issues using examples from the flora of southern Australia. Rangel. J. 2018, 39, 487–498. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Species | N Samples | N Sites | N Raw Loci | N Loci |
|---|---|---|---|---|
| Acacia linifolia | 168 | 29 | 43,111 | 15,274 |
| Acacia longifolia | 166 | 28 | 36,787 | 21,509 |
| Acacia suaveolens | 166 | 29 | 75,908 | 14,370 |
| Acacia terminalis | 256 | 44 | 62,423 | 24,182 |
| Acacia ulicifolia | 159 | 26 | 135,357 | 25,421 |
| Taxon | Provenance Coverage (Percent) | 20 km Radius Coverage (Percent) |
|---|---|---|
| Acacia linifolia | 100.00 | 8.18 |
| Acacia longifolia | 90.16 | 5.73 |
| Acacia suaveolens | 0.23 | 0.23 |
| Acacia terminalis | 62.72 | 1.32 |
| Acacia ulicifolia | 36.44 | 0.12 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rossetto, M.; Wilson, P.D.; Bragg, J.; Cohen, J.; Fahey, M.; Yap, J.-Y.S.; van der Merwe, M. Perceptions of Similarity Can Mislead Provenancing Strategies—An Example from Five Co-Distributed Acacia Species. Diversity 2020, 12, 306. https://doi.org/10.3390/d12080306
Rossetto M, Wilson PD, Bragg J, Cohen J, Fahey M, Yap J-YS, van der Merwe M. Perceptions of Similarity Can Mislead Provenancing Strategies—An Example from Five Co-Distributed Acacia Species. Diversity. 2020; 12(8):306. https://doi.org/10.3390/d12080306
Chicago/Turabian StyleRossetto, Maurizio, Peter D. Wilson, Jason Bragg, Joel Cohen, Monica Fahey, Jia-Yee Samantha Yap, and Marlien van der Merwe. 2020. "Perceptions of Similarity Can Mislead Provenancing Strategies—An Example from Five Co-Distributed Acacia Species" Diversity 12, no. 8: 306. https://doi.org/10.3390/d12080306
APA StyleRossetto, M., Wilson, P. D., Bragg, J., Cohen, J., Fahey, M., Yap, J.-Y. S., & van der Merwe, M. (2020). Perceptions of Similarity Can Mislead Provenancing Strategies—An Example from Five Co-Distributed Acacia Species. Diversity, 12(8), 306. https://doi.org/10.3390/d12080306