
Formation of a β-(3-Chlorobenzoyloxy)-α-hydroxyketone from a TBS-Protected Chalcone upon Oxidation with m-Chloroperbenzoic Acid

 , , , and
, , , and
Abstract
{kind=link}
{kind=link}
1. Introduction
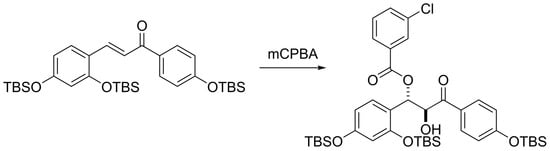
2. Results and Discussion
3. Materials and Methods
3.1. (E)-3-(2,4-Bis((tert-butyldimethylsilyl)oxy)phenyl)1-(4-((tert-butyldimethylsilyl)oxy) phenyl)prop-2-en-1-one (3)
3.2. 1-(2,4-Bis((tert-butyldimethylsilyl)oxy)phenyl)-3-(4-((tert-butyldimethylsilyl)oxy) phenyl)-2-hydroxy-3-oxopropyl 3-chlorobenzoate of (6)
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Zhuang, C.; Zhang, W.; Sheng, C.; Zhang, W.; Xing, C.; Miao, Z. Chalcone: A Privileged Structure in Medicinal Chemistry. Chem. Rev. 2017, 117, 7762–7810. [Google Scholar] [CrossRef] [PubMed]
- Elkanzi, N.A.A.; Hrichi, H.; Alolayan, R.A.; Derafa, W.; Zahou, F.M.; Bakr, R.B. Synthesis of Chalcones Derivatives and Their Biological Activities: A Review. ACS Omega 2022, 7, 27769–27786. [Google Scholar] [CrossRef] [PubMed]
- Zhai, J.; Sun, B.; Sang, F. Progress of isolation, chemical synthesis and biological activities of natural chalcones bearing 2-hydroxy-3-methyl-3-butenyl group. Front. Chem. 2022, 10, 964089. [Google Scholar] [CrossRef] [PubMed]
- Gaonkar, S.L.; Vignesh, U.N. Synthesis and pharmacological properties of chalcones: A review. Res. Chem. Intermed. 2017, 43, 6043–6077. [Google Scholar] [CrossRef]
- Donaire-Arias, A.; Poulsen, M.L.; Ramón-Costa, J.; Montagut, A.M.; Estrada-Tejedor, R.; Borrell, J.I. Synthesis of Chalcones: An Improved High-Yield and Substituent-Independent Protocol for an Old Structure. Molecules 2023, 28, 7576. [Google Scholar] [CrossRef] [PubMed]
- Holt, H., Jr.; LeBlanc, R.; Dickson, J.; Brown, T.; Maddox, J.R.; Lee, M. Reaction of Chalcones with Basic Hydrogen Peroxide: A Structure and Reactivity Study. Heterocycl. Commun. 2005, 11, 465–470. [Google Scholar] [CrossRef]
- Mathew, S.P.; Gunathilagan, S.; Roberts, S.M.; Blackmond, D.G. Mechanistic Insights from Reaction Progress Kinetic Analysis of the Polypeptide-Catalyzed Epoxidation of Chalcone. Org. Lett. 2005, 7, 4847–4850. [Google Scholar] [CrossRef] [PubMed]
- Osorio-Olivares, M.E.; Vásquez-Martínez, Y.; Díaz, K.; Canelo, J.; Taborga, L.; Espinoza-Catalán, L. Antibacterial and Antioxidant Activity of Synthetic Polyoxygenated Flavonoids. Int. J. Mol. Sci. 2024, 25, 5999. [Google Scholar] [CrossRef] [PubMed]
- Wuts, P.G.M. Greene’s Protective Groups in Organic Synthesis; John Wiley & Sons, Inc.: Hoboken, NJ, USA, 2025. [Google Scholar]
- Lauret, C. Epoxy ketones as versatile building blocks in organic synthesis. Tetrahedron Asymmetry 2001, 12, 2359–2383. [Google Scholar] [CrossRef]
- Majdecki, M.; Tyszka-Gumkowska, A.; Jurczak, J. Highly Enantioselective Epoxidation of α,β-Unsaturated Ketones Using Amide-Based Cinchona Alkaloids as Hybrid Phase-Transfer Catalysts. Org. Lett. 2020, 22, 8687–8691. [Google Scholar] [CrossRef] [PubMed]
- House, H.O. The Rearrangement of α,β-Epoxy Ketones. I. Chalcone Oxides. J. Am. Chem. Soc. 1954, 76, 1235–1237. [Google Scholar] [CrossRef]
- Berenguel Gómez, S.; Moreno-Gutiérrez, I.; Muñoz-Dorado, M.; Álvarez-Corral, M.; Rodríguez-García, I. (E)-4,2′,4′-Trimethoxychalcone (Z)-N-Tosyl Hydrazone. Molbank 2025, 2025, M1997. [Google Scholar] [CrossRef]
- Wang, Y.; Shou, X.; Xu, Y.; Zhou, X. Versatile C─H Alkylation and Alkylidenation via Catalytic Alkylidene Transfer of Enones. Angew. Chem. Int. Ed. 2025, 64, e202502619. [Google Scholar] [CrossRef] [PubMed]
- Aslam, S.N.; Stevenson, P.C.; Phythian, S.J.; Veitch, N.C.; Hall, D.R. Synthesis of cicerfuran, an antifungal benzofuran, and some related analogues. Tetrahedron 2006, 62, 4214–4226. [Google Scholar] [CrossRef]
- Almtorp, G.T.; Bachmann, T.L.; Torssell, K.B.G. Syntheses of flavonoids via the isoxazoline route. Acta Chem. Scand. 1991, 45, 212. [Google Scholar] [CrossRef]

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2026 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license.
Share and Cite
Berenguel-Gómez, S.; Moreno-Gutiérrez, I.; Acien-García, J.; Muñoz-Dorado, M.; Álvarez-Corral, M.; Rodríguez-García, I. Formation of a β-(3-Chlorobenzoyloxy)-α-hydroxyketone from a TBS-Protected Chalcone upon Oxidation with m-Chloroperbenzoic Acid. Molbank 2026, 2026, M2175. https://doi.org/10.3390/M2175
Berenguel-Gómez S, Moreno-Gutiérrez I, Acien-García J, Muñoz-Dorado M, Álvarez-Corral M, Rodríguez-García I. Formation of a β-(3-Chlorobenzoyloxy)-α-hydroxyketone from a TBS-Protected Chalcone upon Oxidation with m-Chloroperbenzoic Acid. Molbank. 2026; 2026(3):M2175. https://doi.org/10.3390/M2175
Chicago/Turabian StyleBerenguel-Gómez, Sonia, Irene Moreno-Gutiérrez, Jenifer Acien-García, Manuel Muñoz-Dorado, Míriam Álvarez-Corral, and Ignacio Rodríguez-García. 2026. "Formation of a β-(3-Chlorobenzoyloxy)-α-hydroxyketone from a TBS-Protected Chalcone upon Oxidation with m-Chloroperbenzoic Acid" Molbank 2026, no. 3: M2175. https://doi.org/10.3390/M2175
APA StyleBerenguel-Gómez, S., Moreno-Gutiérrez, I., Acien-García, J., Muñoz-Dorado, M., Álvarez-Corral, M., & Rodríguez-García, I. (2026). Formation of a β-(3-Chlorobenzoyloxy)-α-hydroxyketone from a TBS-Protected Chalcone upon Oxidation with m-Chloroperbenzoic Acid. Molbank, 2026(3), M2175. https://doi.org/10.3390/M2175