
Synthesis and Characterization of Amide-Based Cyclotriphosphazene Derivatives with Alkoxy Terminal Groups



Abstract
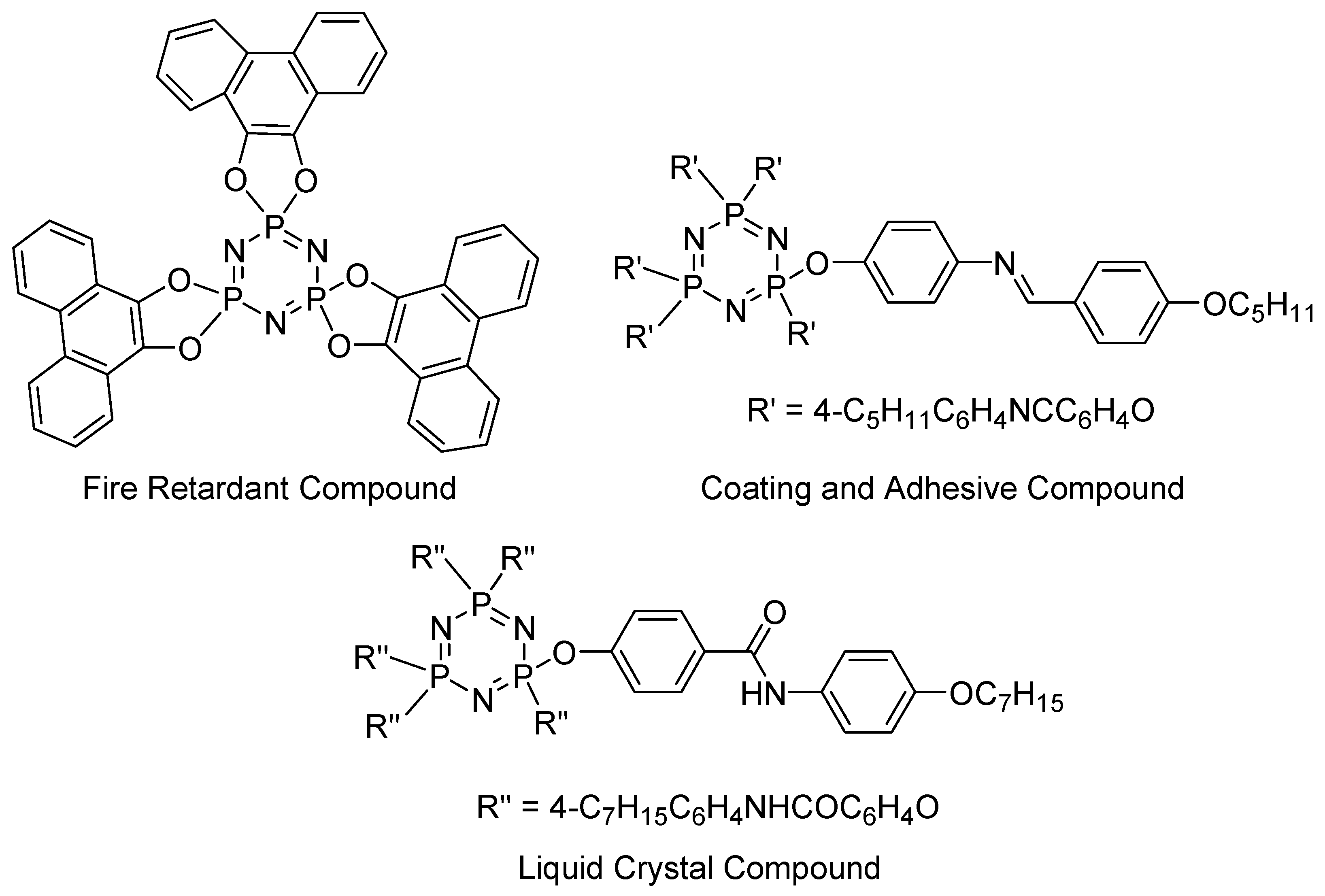
1. Introduction
2. Results and Discussion
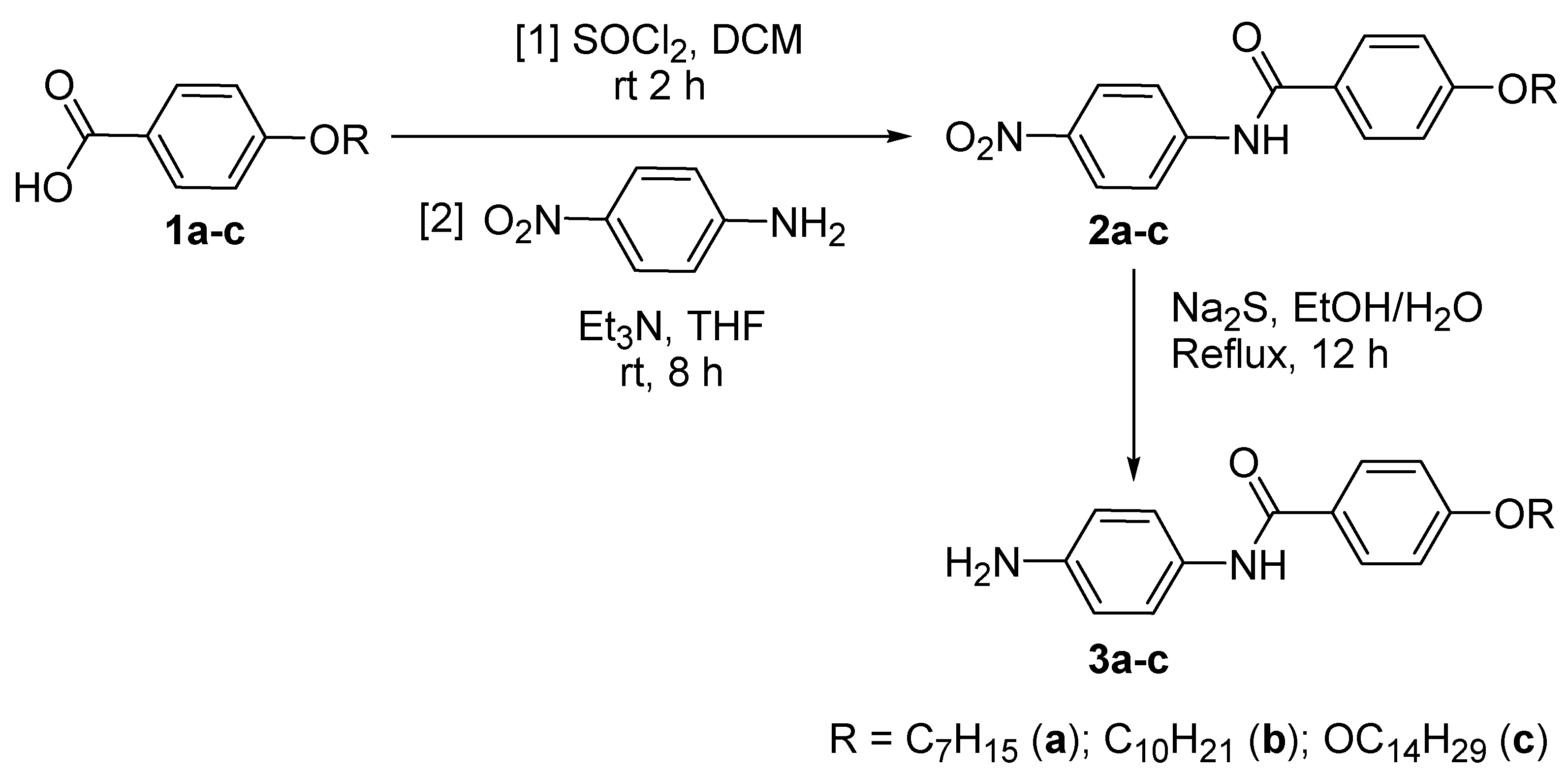
2.1. Synthesis Flow
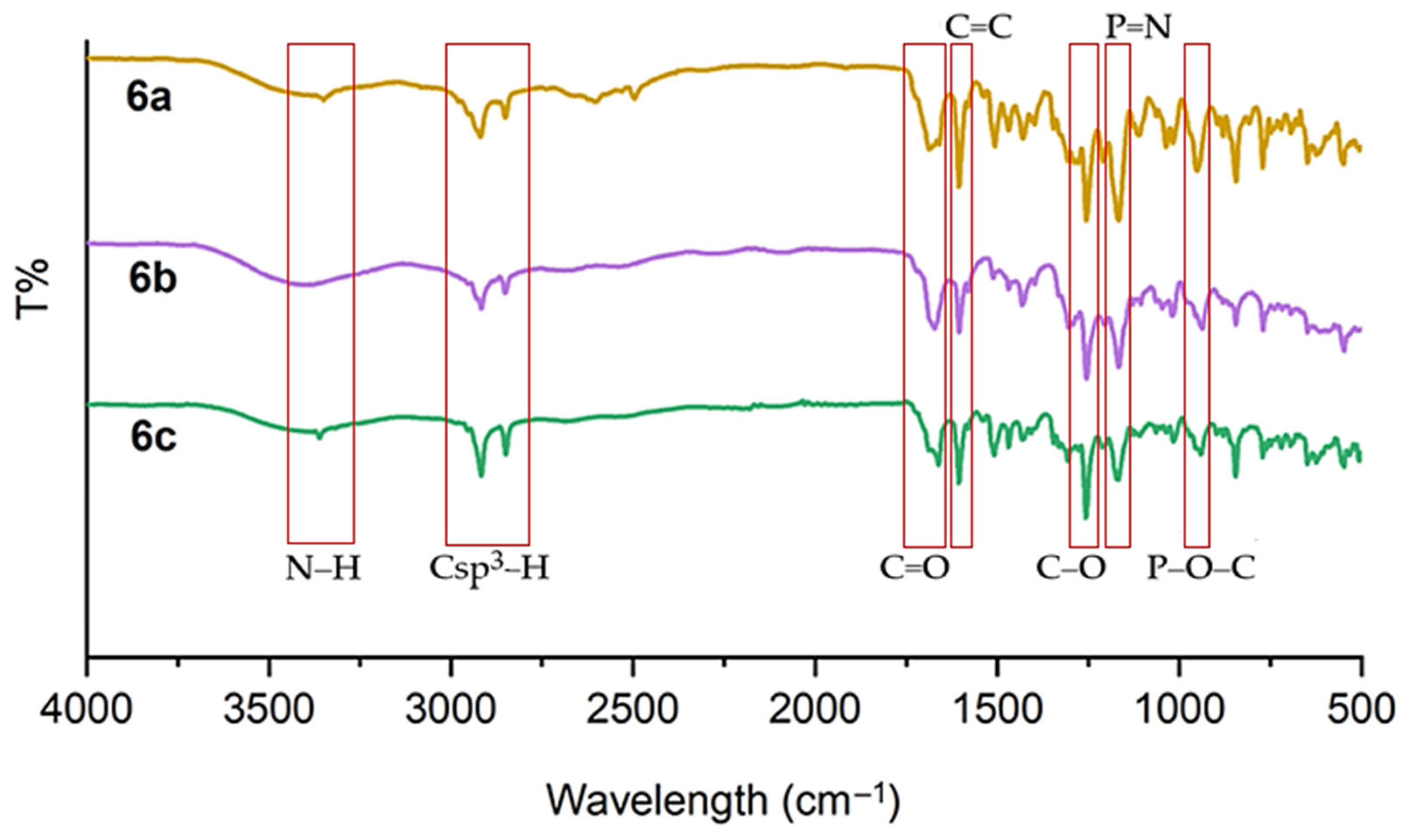
2.2. FTIR Spectral Discussion
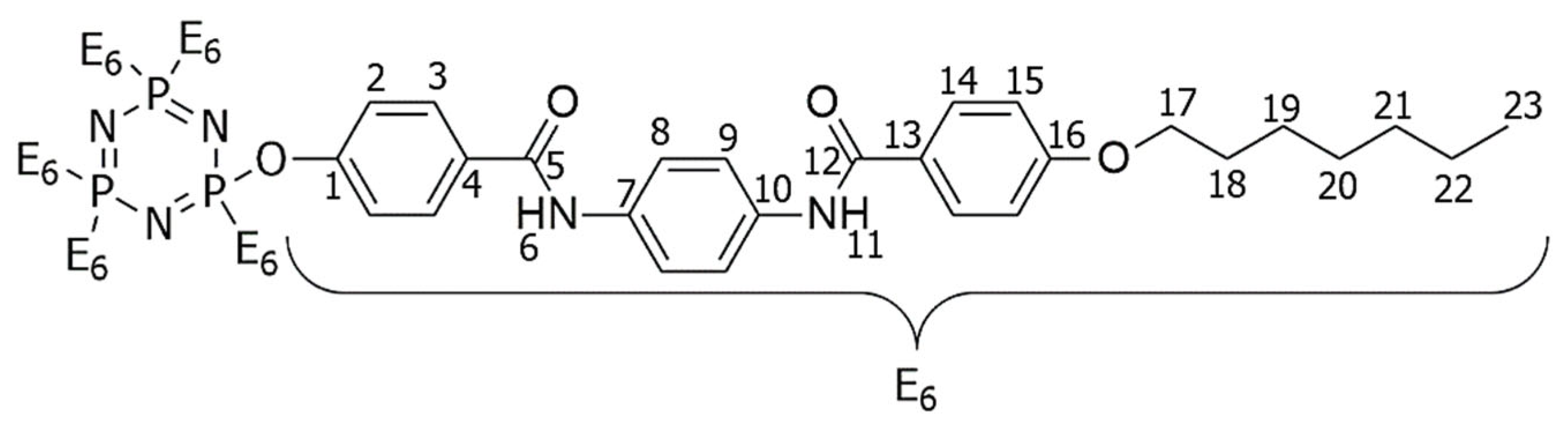
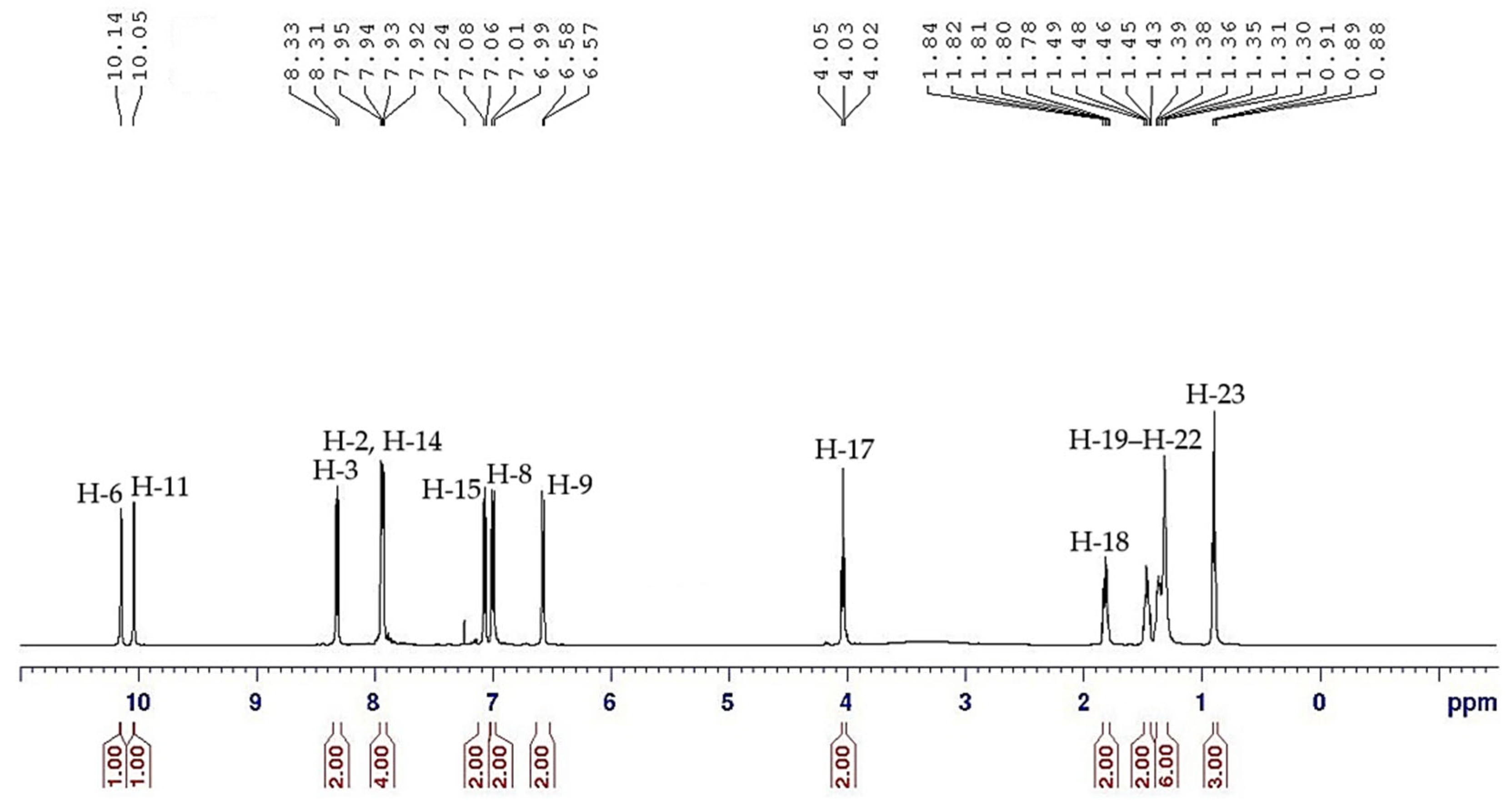
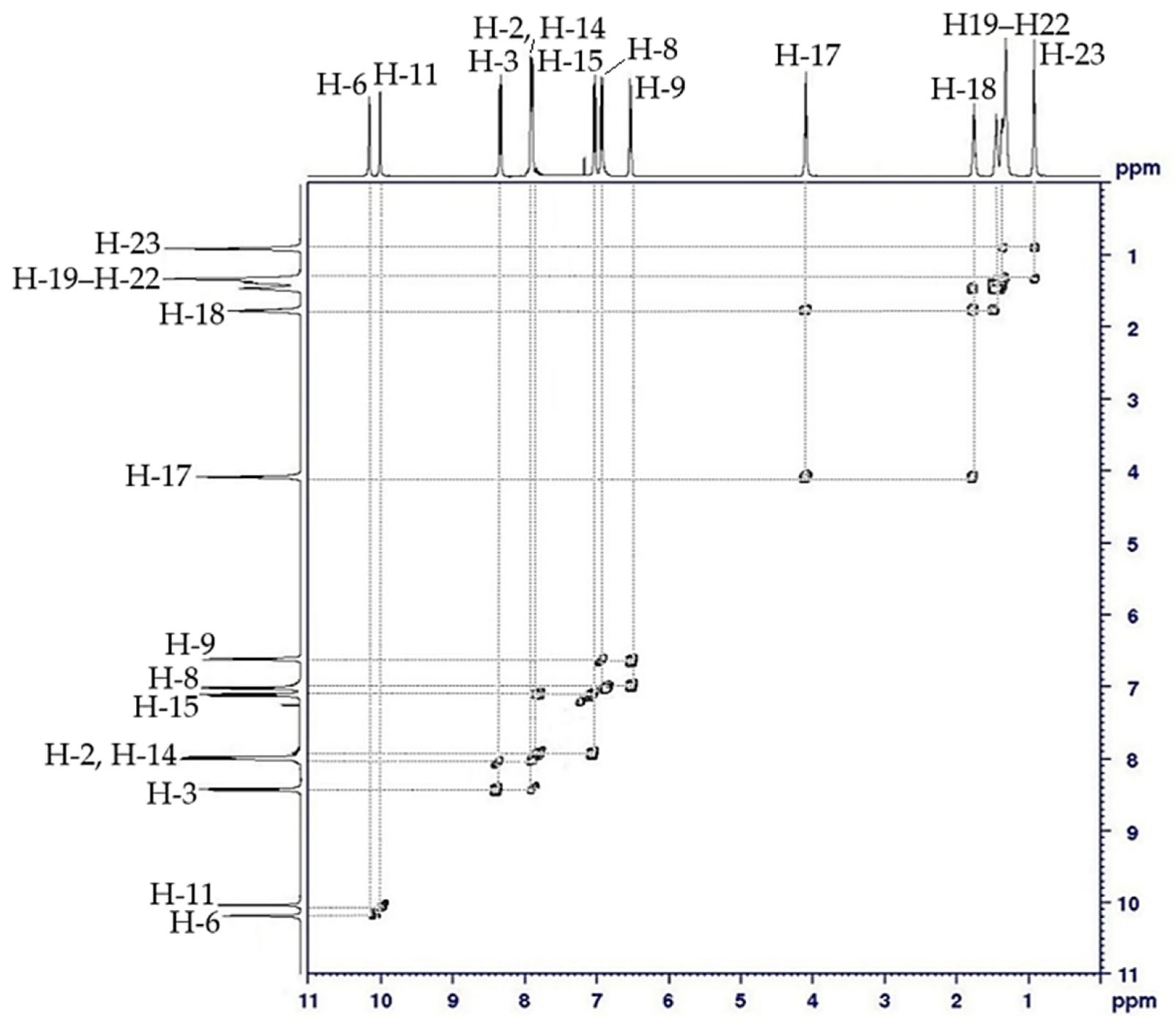
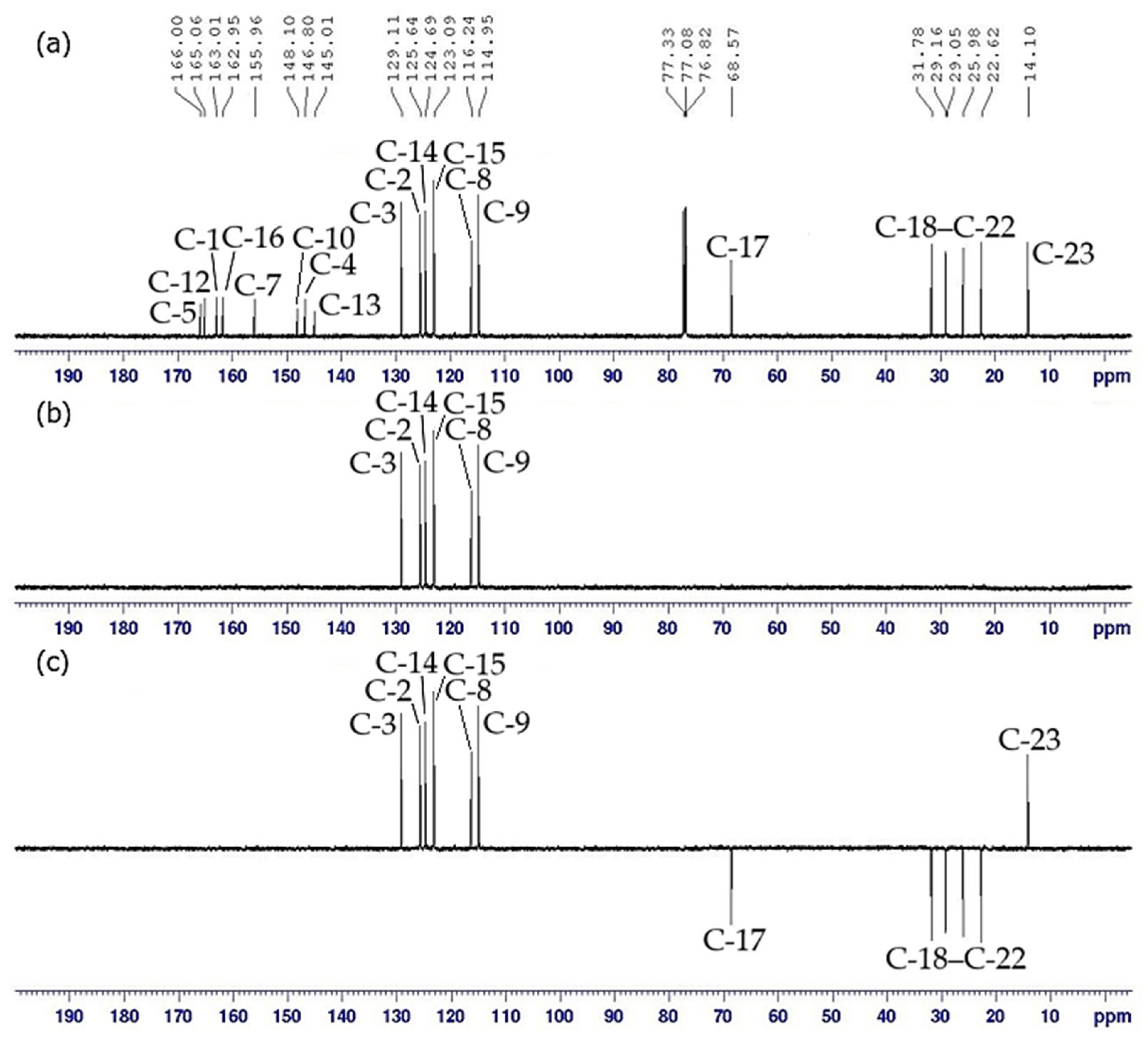
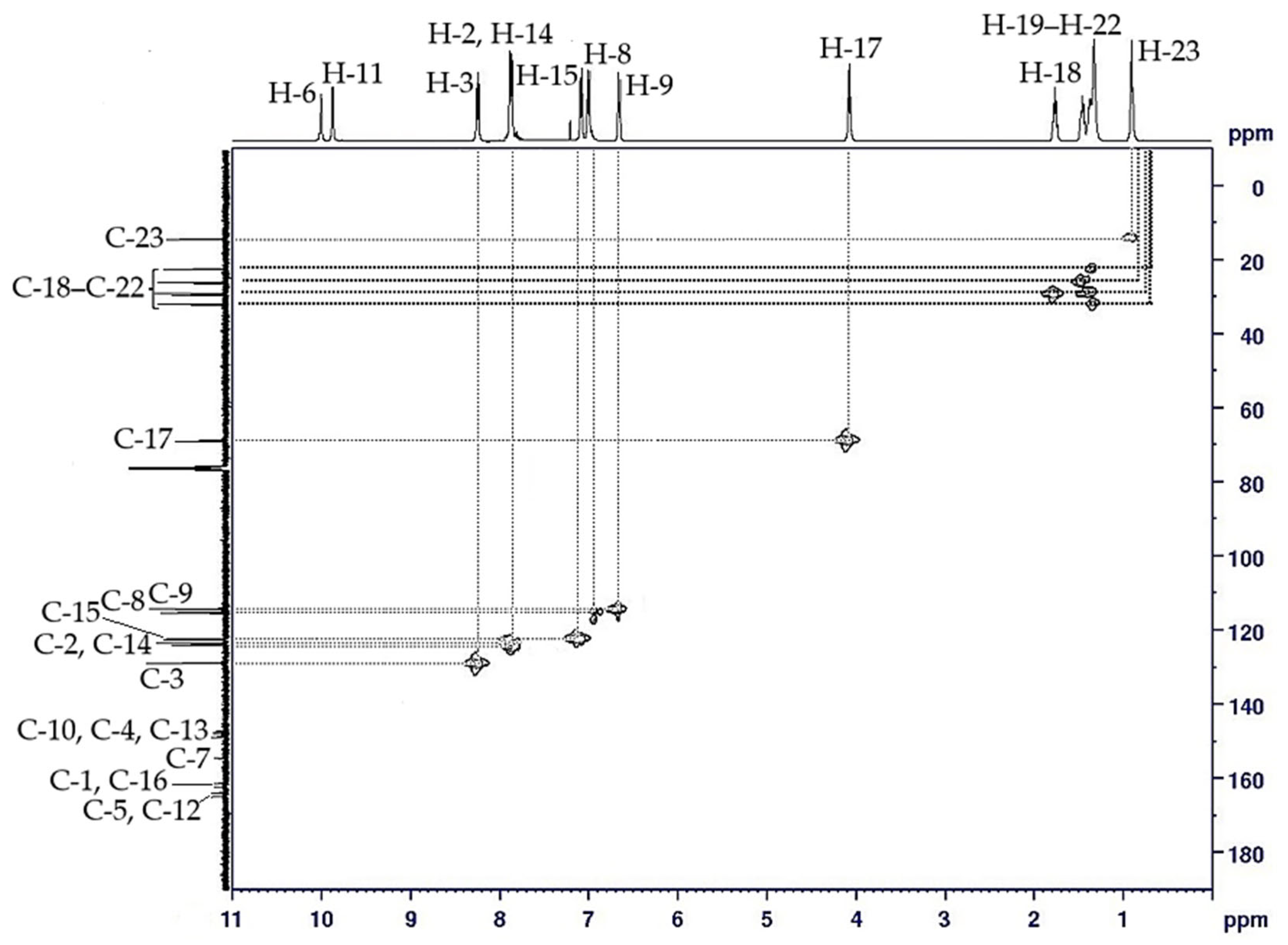
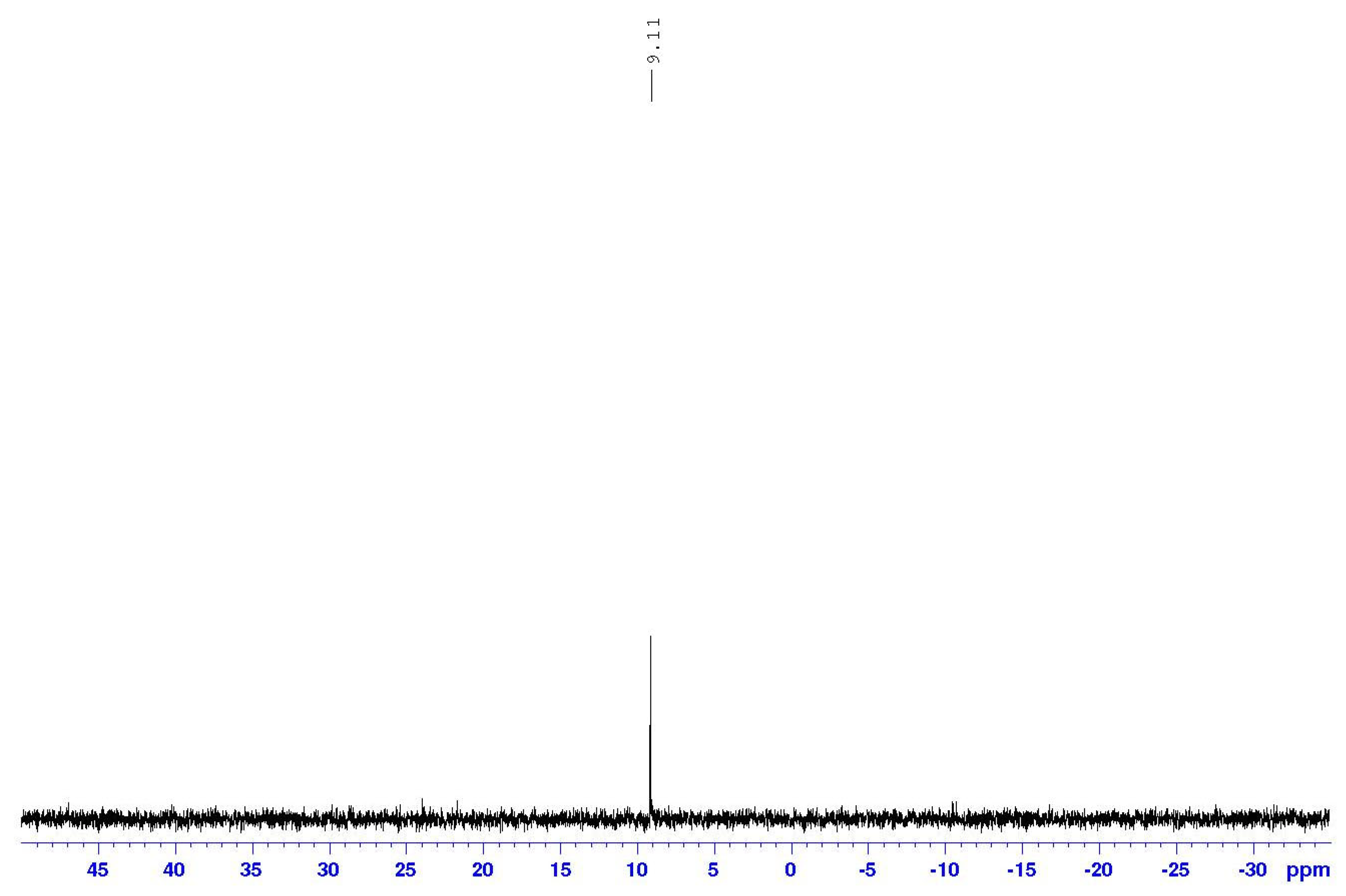
2.3. NMR Spectral Discussion
3. Materials and Methods
3.1. Chemicals
3.2. Instrumentation
3.3. Synthesis Method
3.3.1. Synthesis of 4-(Alkoxy)benzoic Acid (1a-c)
4-(Heptyloxy)benzoic Acid (1a)
4-(Decyloxy)benzoic Acid (1b)
4-(Tetradecyloxy)benzoic Acid (1c)
3.3.2. Synthesis of 4-(Substituted)-N-(4-nitrophenyl)benzamide (2a-c)
4-(Heptyloxy)-N-(4-nitrophenyl)benzamide (2a)
4-(Decyloxy)-N-(4-nitrophenyl)benzamide (2b)
4-(Tetradecyloxy)-N-(4-nitrophenyl)benzamide (2c)
3.3.3. Synthesis of N-(4-aminophenyl)-4-(substituted)benzamide, 3a-c
N-(4-aminophenyl)-4-(heptyloxy)benzamide (3a)
N-(4-aminophenyl)-4-(decyloxy)benzamide (3b)
N-(4-aminophenyl)-4-(tetradecyloxy)benzamide (3c)
3.3.4. Synthesis of Hexa(oxy-4-benzoate)cyclotriphosphazene, 4
3.3.5. Synthesis of Hexa(oxy-4-carboxy)cyclotriphosphazene, 5
3.3.6. Synthesis of hexakis((4-substituted-phenyl)benzamide)cyclotriphosphazene, 6a–c
Hexakis-((4-heptyl-phenyl)benzamide)cyclotriphosphazene (6a)
Hexakis-((4-decyl-phenyl)benzamide)cyclotriphosphazene (6b)
Hexakis-((4-tetradecyl)phenyl)benzamide)cyclotriphosphazene (6c)
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Ahmad, M.; Nawaz, T.; Hussain, I.; Chen, X.; Imran, M.; Hussain, R.; Assiri, M.A.; Ali, S.; Wu, Z. Phosphazene cyclomatrix network-based polymer: Chemistry, synthesis, and applications. ACS Omega 2022, 7, 28694–28707. [Google Scholar] [CrossRef]
- Qu, T.; Yang, N.; Hou, J.; Li, G.; Yao, Y.; Zhang, Q.; He, L.; Wu, D.; Qu, X. Flame retarding epoxy composites with poly(phosphazene-: Co -bisphenol A)-coated boron nitride to improve thermal conductivity and thermal stability. RSC Adv. 2017, 7, 6140–6151. [Google Scholar] [CrossRef]
- Beytur, A.; Tekin, Ç.; Çalışkan, E.; Tekin, S.; Koran, K.; Orhan Görgülü, A.; Sandal, S. Hexa-substituted cyclotriphosphazene derivatives containing hetero-ring chalcones: Synthesis, in vitro cytotoxic activity and their DNA damage determination. Bioorg. Chem. 2022, 127, 105997. [Google Scholar] [CrossRef] [PubMed]
- Palabıyık, D.; Mutlu Balcı, C.; Tümay, S.O.; Sengul, I.F.; Beşli, S. New design of cyclotriphosphazene derivatives bearing carbazole units: The syntheses, characterization, and photophysical properties. Inorg. Chim. Acta 2022, 539, 121022. [Google Scholar] [CrossRef]
- Waldin, N.A.; Jamain, Z. Synthesis and mechanical property of hexasubstituted cyclotriphosphazene derivatives attached to hydrazine-bridge linkage with high fire retardancy. J. Mol. Struct. 2023, 1284, 135330. [Google Scholar] [CrossRef]
- Wang, D.; Xu, X.; Qiu, Y.; Wang, J.; Meng, L. Cyclotriphosphazene based materials: Structure, functionalization and applications. Prog. Mater. Sci. 2024, 142, 101232. [Google Scholar] [CrossRef]
- Saini, A.; Bhedi, D.; Dhanwant, K.; Thirumoorthi, R. Multi-azobenzene moieties on rigid cyclotriphosphazene core: Synthesis, structural characterization, electrochemistry, and photoisomerization study. J. Photochem. Photobiol. A 2024, 452, 115603. [Google Scholar] [CrossRef]
- Shariatinia, Z.; Javeri, N.; Shekarriz, S. Flame retardant cotton fibers produced using novel synthesized halogen-free phosphoramide nanoparticles. Carbohydr. Polym. 2015, 118, 183–198. [Google Scholar] [CrossRef]
- Davarci, D.; Doganci, S. Liquid crystal phosphazenes. J. Mol. Struct. 2022, 1269, 1–18. [Google Scholar] [CrossRef]
- Cheng, J.; Wang, J.; Yang, S.; Zhang, Q.; Huo, S.; Zhang, Q.; Hu, Y.; Ding, G. Benzimidazolyl-substituted cyclotriphosphazene derivative as latent flame-retardant curing agent for one-component epoxy resin system with excellent comprehensive performance. Compos. Part B Eng. 2019, 177, 107440. [Google Scholar] [CrossRef]
- Dagdag, O.; El Bachiri, A.; Hamed, O.; Haldhar, R.; Verma, C.; Ebenso, E.; El Gouri, M. Dendrimeric Epoxy Resins Based on Hexachlorocyclotriphosphazene as a Reactive Flame Retardant Polymeric Materials: A Review. J. Inorg. Organomet. Polym. Mater. 2021, 31, 3240–3261. [Google Scholar] [CrossRef]
- Cheng, J.; Wang, J.; Yang, S.; Zhang, Q.; Hu, Y.; Ding, G.; Huo, S. Aminobenzothiazole-substituted cyclotriphosphazene derivative as reactive flame retardant for epoxy resin. React. Funct. Polym. 2020, 146, 104412. [Google Scholar] [CrossRef]
- Dagdag, O.; Kim, H. Recent advances for poly(cyclotriphosphazene) functionalized graphene oxide composites: Synthesis, properties and applications. J. Ind. Eng. Chem. 2024, 136, 89–122. [Google Scholar] [CrossRef]
- Shaban, A.; Al-Shukri, S.M.; Kahlaf, H.I.; Al Hanbali, O.A. Synthesis, characterization, thermal and fire retardant properties of new homo- And block copolymers of polyacrylate and epoxy resin with cyclotriphosphazene core. Polimery/Polymers 2019, 64, 578–591. [Google Scholar] [CrossRef]
- Shin, Y.J.; Shin, M.J.; Shin, J.S. Flame Retardant Properties of Cyclotriphosphazene Derivatives for ABS. Polym. Polym. Compos. 2018, 26, 309–314. [Google Scholar] [CrossRef]
- Mohd Taip, N.A.; Jamain, Z.; Palle, I. Fire-retardant property of hexasubstituted cyclotriphosphazene derivatives with Schiff base linking unit applied as an additives in polyurethane coating for wood fabrication. Polymers 2022, 14, 3768. [Google Scholar] [CrossRef]
- Jamain, Z.; Khairuddean, M.; Kamaruddin, K.; Rui, Y. Synthesis, structural elucidation and mesophase behavior of hexasubstituted cyclotriphosphazene molecules with amide linking unit. Malays. J. Chem. 2021, 23, 213–225. [Google Scholar]
- Andrienko, D. Introduction to liquid crystals. J. Mol. Liq. 2018, 267, 520–541. [Google Scholar] [CrossRef]
- Jamain, Z.; Khairuddean, M. Synthesis and mesophase behaviour of benzylidene-based molecules containing two azomethine units. J. Phys. Conf. Ser. 2021, 1882, 012120. [Google Scholar] [CrossRef]
- Joshi, V.K.; Katariya, K.D.; Nakum, K.J. Synthesis, mesomorphic behaviour, and DFT studies of biphenyl bis-ester Schiff base liquid crystals. J. Mol. Struct. 2024, 1311, 138338. [Google Scholar] [CrossRef]
- Wu, W.N.; Jiang, Y.M.; Fei, Q.; Du, H.T.; Yang, M.F. Synthesis and antifungal activity of novel 1,2,4-triazole derivatives containing an amide moiety. J. Heterocycl. Chem. 2020, 57, 1379–1386. [Google Scholar] [CrossRef]
- Jamain, Z.; Khairuddean, M.; Guan-Seng, T. Synthesis of novel liquid crystalline and fire retardant molecules based on six-armed cyclotriphosphazene core containing Schiff base and amide linking units. RSC Adv. 2020, 10, 28918–28934. [Google Scholar] [CrossRef]
- Singh, S.; Dunmur, D.A. Liquid Crystals: Fundamentals; World Scientific Publishing Co. Pte. Ltd.: London, UK, 2002. [Google Scholar]
- Kozmík, V.; Horčic, M.; Svoboda, J.; Novotná, V.; Pociecha, D. 3-Aminophenol based bent-shaped liquid crystals with an amide linking group. Liq. Cryst. 2012, 39, 943–955. [Google Scholar] [CrossRef]
- Guardià, J.; Reina, J.A.; Giamberini, M.; Montané, X. An Up-to-Date Overview of Liquid Crystals and Liquid Crystal Polymers for Different Applications: A Review. Polymers 2024, 16, 2293. [Google Scholar] [CrossRef] [PubMed]
- Joaquín, B.; Josefina, J.; Antonio, L.; Luis, O.; Sonia, P.; José, L.S. Cyclotriphosphazene as a dendritic core for the preparation of columnar supermolecular liquid crystals. Chem. Mater. 2006, 18, 5437–5445. [Google Scholar] [CrossRef]
- Sharma, M.K.; Parashar, S.; Sharma, D.; Jakhar, K.; Lal, K.; Pandya, N.U.; Om, H. Synthesis, characterization, docking and antimicrobial studies of binol based amide linked symmetrical bistriazoles. J. Indian Chem. Soc. 2023, 100, 100973. [Google Scholar] [CrossRef]
- Jamain, Z.; Habil, S.; Makmud, M.Z.H.; Khairuddean, M. Synthesis, Structural and Dielectric Characteristics of Liquid Crystalline Azo-Based Compounds with Different Terminal Length. In Proceedings of the 2021 IEEE International Conference on the Properties and Applications of Dielectric Materials (ICPADM), Johor Bahru, Malaysia, 12–14 July 2021; pp. 49–52. [Google Scholar]
- Kılıç, S.Y.; Taşci, N.; Demirkan, M.F.; Yuksel, F.; Çiftçi, G.Y. The new polycyclotriphosphazenes: Synthesis, characterization, thermal and photophysical properties. J. Mol. Struct. 2024, 1309, 138161. [Google Scholar] [CrossRef]
- Valadbeigi, Y. Comparison of Effects of Charge Delocalization and Π-Electron Delocalization on The Stability of Monocyclic Compounds. J. Mol. Graph. Model. 2018, 80, 104–112. [Google Scholar] [CrossRef]
- Martin, N.H.; Allen, N.W.; Brown, J.D.; Kmiec, D.M.; Vo, L. An NMR shielding model for protons above the plane of a carbonyl group. J. Mol. Graph. Model. 2003, 22, 127–131. [Google Scholar] [CrossRef]
- Kupka, T.; Dziuk, B.; Ejsmont, K.; Makieieva, N.; Fizer, L.; Monka, N.; Konechna, R.; Stadnytska, N.; Vasyliuk, S.; Lubenets, V. Impact of crystal and molecular structure of three novel thiosulfonate crystals on their vibrational and NMR parameters. J. Mol. Struct. 2024, 1313, 138642. [Google Scholar] [CrossRef]
- Kara, Y.S.; Yıldız, B. Synthesis and substituent effect study on 13C NMR chemical shifts of 4-(substitue-phenyl)-6-methyl-3-phenyl-4H-1,2,4-oxadiazin-5(6H)-one. J. Mol. Struct. 2022, 1250, 131787. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Proton | 1H [δ (ppm), Multiplicity, Coupling Constant (Hz)] | COSY (1H–1H) Correlation | HSQC (1H–13C) Correlation (δ, ppm) |
|---|---|---|---|
| H-6 | 10.05 (s) | - | - |
| H-11 | 10.14 (s) | - | - |
| H-2 | 7.95 (d, J = 5.0 Hz, 2H) | H-3 | C-2 (125.64) |
| H-3 | 8.32 (d, J = 10.0 Hz, 2H) | H-2 | C-3 (129.11) |
| H-8 | 7.00 (d, J = 10.0 Hz, 2H) | H-9 | C-7 (116.24) |
| H-9 | 6.58 (d, J = 5.0 Hz, 2H) | H-8 | C-8 (114.95) |
| H-14 | 7.93 (d, J = 5.0 Hz, 2H) | H-15 | C-14 (124.69) |
| H-15 | 7.07 (d, J = 10.0 Hz, 2H) | H-14 | C-15 (123.09) |
| H-17 | 4.03 (t, J = 7.5 Hz, 2H) | H-18 | C-17 (68.57) |
| H-18 | 1.78–1.84 (m, 2H) | H-17, H-1–H-9 | C-18 (31.78) |
| H-19–H-22 | 1.30–1.49 (m, 2H) | H-18–H-23 | C-19 (29.16) |
| C-20 (29.05) | |||
| C-21 (25.98) | |||
| C-22 (22.62) | |||
| H-23 | 0.89 (t, J = 7.5 Hz, 3H) | H-22 | C-23 (14.10) |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Abdul Rahim, K.; Jamain, Z. Synthesis and Characterization of Amide-Based Cyclotriphosphazene Derivatives with Alkoxy Terminal Groups. Molbank 2025, 2025, M2039. https://doi.org/10.3390/M2039
Abdul Rahim K, Jamain Z. Synthesis and Characterization of Amide-Based Cyclotriphosphazene Derivatives with Alkoxy Terminal Groups. Molbank. 2025; 2025(3):M2039. https://doi.org/10.3390/M2039
Chicago/Turabian StyleAbdul Rahim, Khairunnisa, and Zuhair Jamain. 2025. "Synthesis and Characterization of Amide-Based Cyclotriphosphazene Derivatives with Alkoxy Terminal Groups" Molbank 2025, no. 3: M2039. https://doi.org/10.3390/M2039
APA StyleAbdul Rahim, K., & Jamain, Z. (2025). Synthesis and Characterization of Amide-Based Cyclotriphosphazene Derivatives with Alkoxy Terminal Groups. Molbank, 2025(3), M2039. https://doi.org/10.3390/M2039