
Can the Supplementation of Oocytes with Extra Copies of mtDNA Impact Development Without Being Transmitted? A Molecular Account


Abstract
1. Introduction
2. Results
2.1. Assessment of the mtDNA Integrity of Autologous Offspring
2.2. Assessment of the mtDNA Integrity of the Heterologous Cohort
2.3. Assessment of Mitochondrial Preparations for Genetic Integrity
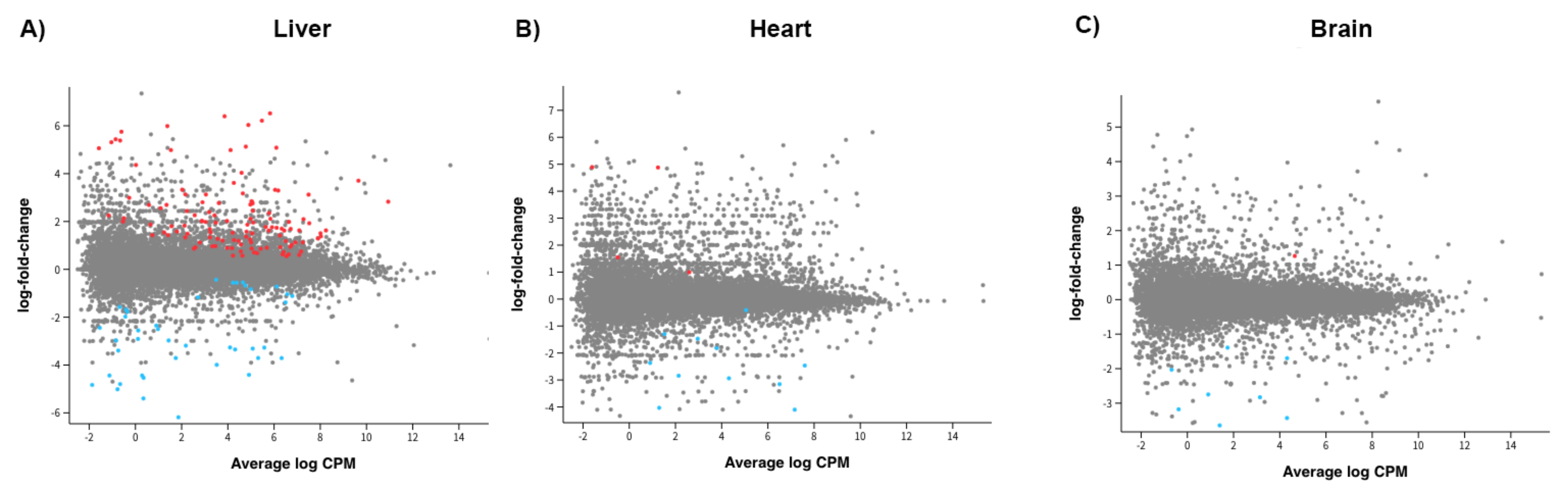
2.4. Gene Expression for All Supplemented Offspring
2.5. Gene Expression Based on Source of mtDNA Supplementation
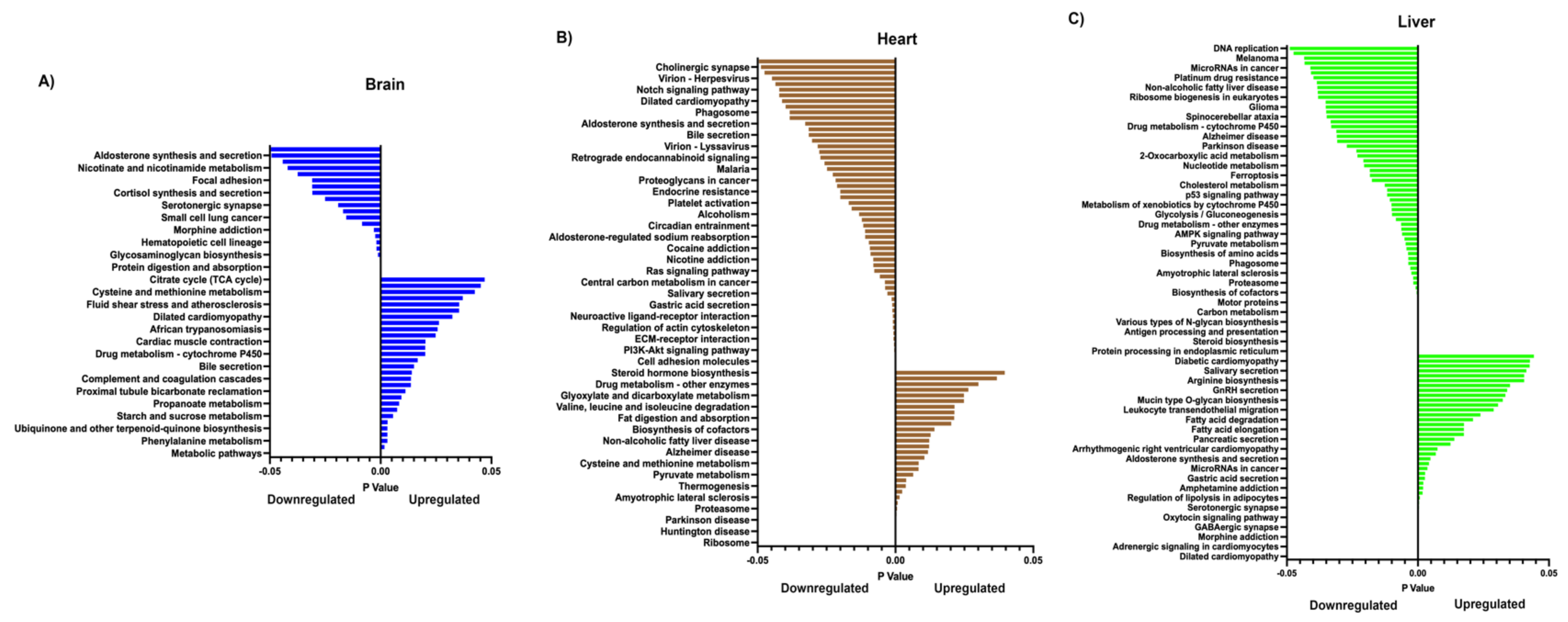
2.6. Reactome Pathway Analysis
2.7. DNA Methylation Analysis
2.8. Overlap of Differentially DNA Methylated Regions and Differentially Expressed Genes
2.9. Origin of the Differential DNA Methylation and Gene Expression
3. Discussion
4. Materials and Methods
4.1. Animal Ethics
4.2. Collection of Sus Scrofa Cumulus–Oocyte Complexes and In Vitro Maturation
4.3. Isolation of Mitochondria from Oocytes
4.4. Generation of Zygotes and Blastocysts
4.5. Synchronisation of Recipient Pigs
4.6. Surgery and Embryo Transfer
4.7. Post-Operative Care and Determination of Pregnancy
4.8. Farrowing and Assessment at Birth
4.9. RNA Extraction from Heart, RNAseq Library Construction, and Next-Generation Sequencing
4.10. DNA Extraction and Reduced Representation Bisulfite Sequencing
4.11. Gene Ontology, Functional Pathway Enrichment, and Gene Network Analysis
4.12. mtDNA Amplification
4.13. mtDNA Next Generation Sequencing
4.14. mtDNA Mapping
4.15. Analysis of Heteroplasmic Load
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- St John, J.C. Epigenetic Regulation of the Nuclear and Mitochondrial Genomes: Involvement in Metabolism, Development, and Disease. Annu. Rev. Anim. Biosci. 2021, 9, 203–224. [Google Scholar] [CrossRef] [PubMed]
- Tachibana, M.; Sparman, M.; Sritanaudomchai, H.; Ma, H.; Clepper, L.; Woodward, J.; Li, Y.; Ramsey, C.; Kolotushkina, O.; Mitalipov, S. Mitochondrial gene replacement in primate offspring and embryonic stem cells. Nature 2009, 461, 367–372. [Google Scholar] [CrossRef] [PubMed]
- Hyslop, L.A.; Blakeley, P.; Craven, L.; Richardson, J.; Fogarty, N.M.; Fragouli, E.; Lamb, M.; Wamaitha, S.E.; Prathalingam, N.; Zhang, Q.; et al. Towards clinical application of pronuclear transfer to prevent mitochondrial DNA disease. Nature 2016, 534, 383–386. [Google Scholar] [CrossRef] [PubMed]
- Steinborn, R.; Zakhartchenko, V.; Wolf, E.; Muller, M.; Brem, G. Non-balanced mix of mitochondrial DNA in cloned cattle produced by cytoplast-blastomere fusion. FEBS Lett. 1998, 426, 357–361. [Google Scholar] [CrossRef]
- Cagnone, G.L.; Tsai, T.S.; Makanji, Y.; Matthews, P.; Gould, J.; Bonkowski, M.S.; Elgass, K.D.; Wong, A.S.; Wu, L.E.; McKenzie, M.; et al. Restoration of normal embryogenesis by mitochondrial supplementation in pig oocytes exhibiting mitochondrial DNA deficiency. Sci. Rep. 2016, 6, 23229. [Google Scholar] [CrossRef]
- McIlfatrick, S.; O’Leary, S.; Okada, T.; Penn, A.; Nguyen, V.H.T.; McKenny, L.; Huang, S.Y.; Andreas, E.; Finnie, J.; Kirkwood, R.; et al. Does supplementation of oocytes with additional mtDNA influence developmental outcome? iScience 2023, 26, 105956. [Google Scholar] [CrossRef]
- Morimoto, Y.; Gamage, U.S.K.; Yamochi, T.; Saeki, N.; Morimoto, N.; Yamanaka, M.; Koike, A.; Miyamoto, Y.; Tanaka, K.; Fukuda, A.; et al. Mitochondrial Transfer into Human Oocytes Improved Embryo Quality and Clinical Outcomes in Recurrent Pregnancy Failure Cases. Int. J. Mol Sci. 2023, 24, 2738. [Google Scholar] [CrossRef]
- Anderson, S.; Bankier, A.T.; Barrell, B.G.; de Bruijn, M.H.; Coulson, A.R.; Drouin, J.; Eperon, I.C.; Nierlich, D.P.; Roe, B.A.; Sanger, F.; et al. Sequence and organization of the human mitochondrial genome. Nature 1981, 290, 457–465. [Google Scholar] [CrossRef]
- Moyes, C.D.; Battersby, B.J.; Leary, S.C. Regulation of muscle mitochondrial design. J. Exp. Biol. 1998, 201, 299–307. [Google Scholar] [CrossRef]
- McFarland, R.; Taylor, R.W.; Turnbull, D.M. Mitochondrial disease—Its impact, etiology, and pathology. Curr. Top. Dev. Biol. 2007, 77, 113–155. [Google Scholar] [CrossRef]
- Kucej, M.; Butow, R.A. Evolutionary tinkering with mitochondrial nucleoids. Trends Cell Biol. 2007, 17, 586–592. [Google Scholar] [CrossRef]
- Giles, R.E.; Blanc, H.; Cann, H.M.; Wallace, D.C. Maternal inheritance of human mitochondrial DNA. Proc. Natl. Acad. Sci. USA 1980, 77, 6715–6719. [Google Scholar] [CrossRef] [PubMed]
- Spikings, E.C.; Alderson, J.; St John, J.C. Regulated mitochondrial DNA replication during oocyte maturation is essential for successful porcine embryonic development. Biol. Reprod. 2007, 76, 327–335. [Google Scholar] [CrossRef] [PubMed]
- Evans, M.J.; Kaufman, M.H. Establishment in culture of pluripotential cells from mouse embryos. Nature 1981, 292, 154–156. [Google Scholar] [CrossRef] [PubMed]
- Piliszek, A.; Grabarek, J.B.; Frankenberg, S.R.; Plusa, B. Cell fate in animal and human blastocysts and the determination of viability. Mol. Hum. Reprod. 2016, 22, 681–690. [Google Scholar] [CrossRef]
- Hance, N.; Ekstrand, M.I.; Trifunovic, A. Mitochondrial DNA polymerase gamma is essential for mammalian embryogenesis. Hum. Mol. Genet. 2005, 14, 1775–1783. [Google Scholar] [CrossRef]
- Larsson, N.G.; Wang, J.; Wilhelmsson, H.; Oldfors, A.; Rustin, P.; Lewandoski, M.; Barsh, G.S.; Clayton, D.A. Mitochondrial transcription factor A is necessary for mtDNA maintenance and embryogenesis in mice. Nat. Genet. 1998, 18, 231–236. [Google Scholar] [CrossRef]
- Bowles, E.J.; Lee, J.H.; Alberio, R.; Lloyd, R.E.; Stekel, D.; Campbell, K.H.; St John, J.C. Contrasting effects of in vitro fertilization and nuclear transfer on the expression of mtDNA replication factors. Genetics 2007, 176, 1511–1526. [Google Scholar] [CrossRef]
- Takeda, K.; Tasai, M.; Iwamoto, M.; Akita, T.; Tagami, T.; Nirasawa, K.; Hanada, H.; Onishi, A. Transmission of mitochondrial DNA in pigs and progeny derived from nuclear transfer of Meishan pig fibroblast cells. Mol. Reprod. Dev. 2006, 73, 306–312. [Google Scholar] [CrossRef]
- Costa-Borges, N.; Nikitos, E.; Spath, K.; Miguel-Escalada, I.; Ma, H.; Rink, K.; Coudereau, C.; Darby, H.; Koski, A.; Van Dyken, C.; et al. First pilot study of maternal spindle transfer for the treatment of repeated in vitro fertilization failures in couples with idiopathic infertility. Fertil. Steril. 2023, 119, 964–973. [Google Scholar] [CrossRef]
- Larsen, M.O.; Rolin, B. Use of the Gottingen minipig as a model of diabetes, with special focus on type 1 diabetes research. ILAR J. 2004, 45, 303–313. [Google Scholar] [CrossRef] [PubMed]
- Bode, G.; Clausing, P.; Gervais, F.; Loegsted, J.; Luft, J.; Nogues, V.; Sims, J. The utility of the minipig as an animal model in regulatory toxicology. J. Pharmacol. Toxicol. Methods 2010, 62, 196–220. [Google Scholar] [CrossRef]
- Olivennes, F.; Fanchin, R.; Ledee, N.; Righini, C.; Kadoch, I.J.; Frydman, R. Perinatal outcome and developmental studies on children born after IVF. Hum. Reprod. Update 2002, 8, 117–128. [Google Scholar] [CrossRef] [PubMed]
- Pandey, S.; Shetty, A.; Hamilton, M.; Bhattacharya, S.; Maheshwari, A. Obstetric and perinatal outcomes in singleton pregnancies resulting from IVF/ICSI: A systematic review and meta-analysis. Hum. Reprod. Update 2012, 18, 485–503. [Google Scholar] [CrossRef]
- Leunens, L.; Celestin-Westreich, S.; Bonduelle, M.; Liebaers, I.; Ponjaert-Kristoffersen, I. Cognitive and motor development of 8-year-old children born after ICSI compared to spontaneously conceived children. Hum. Reprod. 2006, 21, 2922–2929. [Google Scholar] [CrossRef]
- Liapi, S.; Polychronopoulou, S. Cognitive and socio-emotional development and manifestation of learning disabilities of 8- to 10-year-old children born after intracytoplasmatic sperm injection compared to naturally conceived children. Clin. Exp. Obstet. Gynecol. 2017, 44, 104–109. [Google Scholar] [CrossRef]
- Hong, G.; Zhang, W.; Li, H.; Shen, X.; Guo, Z. Separate enrichment analysis of pathways for up- and downregulated genes. J. Royal Soc. Interface 2014, 11, 950. [Google Scholar] [CrossRef] [PubMed]
- Penn, A.; Andreas, E.; Okada, T.; St John, J.C. Nuclear transfer leads to aberrant cell cycle regulation and translation, as determined by gene expression, and selection of mitochondrial DNA in porcine blastocysts. NAR Mol. Med. 2024, 1, ugae018. [Google Scholar] [CrossRef]
- Ivanova, E.; Canovas, S.; Garcia-Martinez, S.; Romar, R.; Lopes, J.S.; Rizos, D.; Sanchez-Calabuig, M.J.; Krueger, F.; Andrews, S.; Perez-Sanz, F.; et al. DNA methylation changes during preimplantation development reveal inter-species differences and reprogramming events at imprinted genes. Clin. Epigenet. 2020, 12, 64. [Google Scholar] [CrossRef]
- Okada, T.; McIlfatrick, S.; Hin, N.; Aryamanesh, N.; Breen, J.; St John, J.C. Mitochondrial supplementation of Sus scrofa metaphase II oocytes alters DNA methylation and gene expression profiles of blastocysts. Epigenet. Chromatin 2022, 15, 12. [Google Scholar] [CrossRef]
- Tsai, T.S.; Tyagi, S.; St John, J.C. The molecular characterisation of mitochondrial DNA deficient oocytes using a pig model. Hum. Reprod. 2018, 33, 942–953. [Google Scholar] [CrossRef]
- Woods, D.C.; Khrapko, K.; Tilly, J.L. Influence of Maternal Aging on Mitochondrial Heterogeneity, Inheritance, and Function in Oocytes and Preimplantation Embryos. Genes 2018, 9, 265. [Google Scholar] [CrossRef]
- Chappel, S. The role of mitochondria from mature oocyte to viable blastocyst. Obstet. Gynecol. Int. 2013, 2013, 183024. [Google Scholar] [CrossRef]
- Piko, L.; Taylor, K.D. Amounts of mitochondrial DNA and abundance of some mitochondrial gene transcripts in early mouse embryos. Dev. Biol. 1987, 123, 364–374. [Google Scholar] [CrossRef]
- Stigliani, S.; Persico, L.; Lagazio, C.; Anserini, P.; Venturini, P.L.; Scaruffi, P. Mitochondrial DNA in Day 3 embryo culture medium is a novel, non-invasive biomarker of blastocyst potential and implantation outcome. Mol. Hum. Reprod. 2014, 20, 1238–1246. [Google Scholar] [CrossRef]
- Houghton, F.D. Energy metabolism of the inner cell mass and trophectoderm of the mouse blastocyst. Differentiation 2006, 74, 11–18. [Google Scholar] [CrossRef]
- Chen, X.; Prosser, R.; Simonetti, S.; Sadlock, J.; Jagiello, G.; Schon, E.A. Rearranged mitochondrial genomes are present in human oocytes. Am. J. Hum. Genet. 1995, 57, 239–247. [Google Scholar]
- Hsieh, R.H.; Tsai, N.M.; Au, H.K.; Chang, S.J.; Wei, Y.H.; Tzeng, C.R. Multiple rearrangements of mitochondrial DNA in unfertilized human oocytes. Fertil. Steril. 2002, 77, 1012–1017. [Google Scholar] [CrossRef]
- St John, J.C.; Makanji, Y.; Johnson, J.L.; Tsai, T.S.; Lagondar, S.; Rodda, F.; Sun, X.; Pangestu, M.; Chen, P.; Temple-Smith, P. The transgenerational effects of oocyte mitochondrial supplementation. Sci. Rep. 2019, 9, 6694. [Google Scholar] [CrossRef]
- Riley, J.S.; Tait, S.W. Mitochondrial DNA in inflammation and immunity. EMBO Rep. 2020, 21, e49799. [Google Scholar] [CrossRef]
- De Gaetano, A.; Solodka, K.; Zanini, G.; Selleri, V.; Mattioli, A.V.; Nasi, M.; Pinti, M. Molecular Mechanisms of mtDNA-Mediated Inflammation. Cells 2021, 10, 2898. [Google Scholar] [CrossRef]
- Lepelley, A.; Della Mina, E.; Van Nieuwenhove, E.; Waumans, L.; Fraitag, S.; Rice, G.I.; Dhir, A.; Fremond, M.L.; Rodero, M.P.; Seabra, L.; et al. Enhanced cGAS-STING-dependent interferon signaling associated with mutations in ATAD3A. J. Exp. Med. 2021, 218, 20201560. [Google Scholar] [CrossRef]
- Liu, M.; Long, X.; Fu, S.; Zhang, Y.; Liu, Z.; Xu, X.; Wu, M. Mitochondrial DNA copy number and the risk of autoimmune diseases: A Mendelian randomization study with meta-analysis. J. Transl. Autoimmun. 2024, 9, 100251. [Google Scholar] [CrossRef]
- Gammill, H.S.; Nelson, J.L. Naturally acquired microchimerism. Int. J. Dev. Biol. 2010, 54, 531–543. [Google Scholar] [CrossRef]
- Song, W.H.; Yi, Y.J.; Sutovsky, M.; Meyers, S.; Sutovsky, P. Autophagy and ubiquitin-proteasome system contribute to sperm mitophagy after mammalian fertilization. Proc. Natl. Acad. Sci. USA 2016, 113, E5261–E5270. [Google Scholar] [CrossRef]
- St John, J.C.; Schatten, G. Paternal mitochondrial DNA transmission during nonhuman primate nuclear transfer. Genetics 2004, 167, 897–905. [Google Scholar] [CrossRef]
- Sutovsky, P.; Moreno, R.D.; Ramalho-Santos, J.; Dominko, T.; Simerly, C.; Schatten, G. Ubiquitin tag for sperm mitochondria. Nature 1999, 402, 371–372. [Google Scholar] [CrossRef]
- Rossant, J. Genetic Control of Early Cell Lineages in the Mammalian Embryo. Annu. Rev. Genet. 2018, 52, 185–201. [Google Scholar] [CrossRef]
- El-Gohary, Y.; Gittes, G.K. Embryologic development of the liver, biliary tract, and pancreas. In Embryologic Development of the Liver, Biliary Tract, and Pancreas, 12th ed.; Jarnagin, W.R., Blumgart, L.H., Eds.; Elsevier: Amsterdam, The Netherlands, 2012; pp. 18–33. [Google Scholar]
- Lee, W.T.; Sun, X.; Tsai, T.S.; Johnson, J.L.; Gould, J.A.; Garama, D.J.; Gough, D.J.; McKenzie, M.; Trounce, I.A.; St John, J.C. Mitochondrial DNA haplotypes induce differential patterns of DNA methylation that result in differential chromosomal gene expression patterns. Cell Death Discov. 2017, 3, 17062. [Google Scholar] [CrossRef]
- Montgomery, T.; Uh, K.; Lee, K. TET enzyme driven epigenetic reprogramming in early embryos and its implication on long-term health. Front. Cell Dev. Biol. 2024, 12, 1358649. [Google Scholar] [CrossRef]
- Doherty, A.S.; Mann, M.R.; Tremblay, K.D.; Bartolomei, M.S.; Schultz, R.M. Differential effects of culture on imprinted H19 expression in the preimplantation mouse embryo. Biol. Reprod. 2000, 62, 1526–1535. [Google Scholar] [CrossRef]
- Andreas, E.; Penn, A.; Okada, T.; St John, J.C. Supplementation of Oocytes by Microinjection with Extra Copies of mtDNA Alters Metabolite Profiles and Interactions with Expressed Genes in a Tissue-Specific Manner. Biomolecules 2024, 14, 1477. [Google Scholar] [CrossRef]
- Koussounadis, A.; Langdon, S.P.; Um, I.H.; Harrison, D.J.; Smith, V.A. Relationship between differentially expressed mRNA and mRNA-protein correlations in a xenograft model system. Sci. Rep. 2015, 5, 10775. [Google Scholar] [CrossRef]
- Kosti, I.; Jain, N.; Aran, D.; Butte, A.J.; Sirota, M. Cross-tissue Analysis of Gene and Protein Expression in Normal and Cancer Tissues. Sci. Rep. 2016, 6, 24799. [Google Scholar] [CrossRef]
- Inoue, A.; Jiang, L.; Lu, F.; Suzuki, T.; Zhang, Y. Maternal H3K27me3 controls DNA methylation-independent imprinting. Nature 2017, 547, 419–424. [Google Scholar] [CrossRef]
- Okada, T.; Penn, A.; St John, J.C. Mitochondrial DNA Supplementation of Oocytes Has Downstream Effects on the Transcriptional Profiles of Sus scrofa Adult Tissues with High mtDNA Copy Number. Int. J. Mol Sci. 2023, 24, 7545. [Google Scholar] [CrossRef]
- Deshmukh, R.S.; Ostrup, O.; Ostrup, E.; Vejlsted, M.; Niemann, H.; Lucas-Hahn, A.; Petersen, B.; Li, J.; Callesen, H.; Hyttel, P. DNA methylation in porcine preimplantation embryos developed in vivo and produced by in vitro fertilization, parthenogenetic activation and somatic cell nuclear transfer. Epigenetics 2011, 6, 177–187. [Google Scholar] [CrossRef]
- Srirattana, K.; St John, J.C. Additional mitochondrial DNA influences the interactions between the nuclear and mitochondrial genomes in a bovine embryo model of nuclear transfer. Sci. Rep. 2018, 8, 7246. [Google Scholar] [CrossRef]
- Tzeng, C.R.; Hsieh, R.H.; Au, H.K.; Yen, Y.H.; Chang, S.J.; Cheng, Y.F. Mitochondria transfer (MIT) into oocyte from autologous cumulus granulosa cells (cGCs). Fertil. Steril. 2004, 82, S53. [Google Scholar] [CrossRef]
- Cohen, J.; Scott, R.; Schimmel, T.; Levron, J.; Willadsen, S. Birth of infant after transfer of anucleate donor oocyte cytoplasm into recipient eggs. Lancet 1997, 350, 186–187. [Google Scholar] [CrossRef]
- Zhang, J.; Liu, H.; Luo, S.; Lu, Z.; Chavez-Badiola, A.; Liu, Z.; Yang, M.; Merhi, Z.; Silber, S.J.; Munne, S.; et al. Live birth derived from oocyte spindle transfer to prevent mitochondrial disease. Reprod. Biomed. Online 2017, 34, 361–368. [Google Scholar] [CrossRef]
- Tachibana, M.; Amato, P.; Sparman, M.; Woodward, J.; Sanchis, D.M.; Ma, H.; Gutierrez, N.M.; Tippner-Hedges, R.; Kang, E.; Lee, H.S.; et al. Towards germline gene therapy of inherited mitochondrial diseases. Nature 2013, 493, 627–631. [Google Scholar] [CrossRef] [PubMed]
- Feng, Z.; Shi, J.; Ren, J.; Luo, L.; Liu, D.; Guo, Y.; Sun, B.; Liu, G.; Deng, M.; Li, Y. Mitochondria-Targeted Antioxidant MitoQ Improves In Vitro Maturation and Subsequent Embryonic Development from Culled Cows. Animals 2024, 14, 2929. [Google Scholar] [CrossRef]
- Ardehjani, N.A.; Agha-Hosseini, M.; Nashtaei, M.S.; Khodarahmian, M.; Shabani, M.; Jabarpour, M.; Fereidouni, F.; Rastegar, T.; Amidi, F. Resveratrol ameliorates mitochondrial biogenesis and reproductive outcomes in women with polycystic ovary syndrome undergoing assisted reproduction: A randomized, triple-blind, placebo-controlled clinical trial. J. Ovarian Res. 2024, 17, 143. [Google Scholar] [CrossRef] [PubMed]
- Walker, S.C.; Shin, T.; Zaunbrecher, G.M.; Romano, J.E.; Johnson, G.A.; Bazer, F.W.; Piedrahita, J.A. A highly efficient method for porcine cloning by nuclear transfer using in vitro-matured oocytes. Cloning Stem Cells 2002, 4, 105–112. [Google Scholar] [CrossRef] [PubMed]
- Dobin, A.; Davis, C.A.; Schlesinger, F.; Drenkow, J.; Zaleski, C.; Jha, S.; Batut, P.; Chaisson, M.; Gingeras, T.R. STAR: Ultrafast universal RNA-seq aligner. Bioinformatics 2013, 29, 15–21. [Google Scholar] [CrossRef]
- Krueger, F.; Andrews, S.R. Bismark: A flexible aligner and methylation caller for Bisulfite-Seq applications. Bioinformatics 2011, 27, 1571–1572. [Google Scholar] [CrossRef]
- Kanehisa, M.; Goto, S. KEGG: Kyoto Encyclopedia of Genes and Genomes. Nucleic Acids Res. 2000, 28, 27–30. [Google Scholar] [CrossRef]
- Milacic, M.; Beavers, D.; Conley, P.; Gong, C.; Gillespie, M.; Griss, J.; Haw, R.; Jassal, B.; Matthews, L.; May, B.; et al. The Reactome Pathway Knowledgebase 2024. Nucleic Acids Res. 2024, 52, D672–D678. [Google Scholar] [CrossRef]
- Ursing, B.M.; Arnason, U. The Complete Mitochondrial DNA Sequence of the Pig (Sus scrofa). J. Mol. Evol. 1998, 47, 302–306. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Pig ID | Mitochondrial Preparation | Autologous or Heterologous | Assigned mtDNA Haplotype |
|---|---|---|---|
| Pig 3.1 | MP3 | Heterologous | KT279758 |
| Pig 3.2 | MP3 | Heterologous | KT279759 * |
| Pig 3.3 | MP3 | Heterologous | KT261430 |
| Pig 3.4 | MP3 | Autologous | KT261429 |
| Pig 3.5 | MP3 | Heterologous | KT279759 * |
| 12.1 | MP12 | Autologous | KT279760 |
| 12.2 | MP9 | Autologous | KT261430 |
| 12.3 | MP13 | Autologous | KT279758 |
| 12.4 | MP12 | Autologous | KT279760 |
| 17.1 | MP14 | Autologous | Unassigned |
| 17.2 | MP14 | Autologous | Unassigned |
| 17.3 | MP14 | Autologous | Unassigned |
| Brain | Heart | Liver | Function |
|---|---|---|---|
| LOC110260659 | LOC110260659 | LOC110260659 | Translationally controlled tumour protein pseudogene |
| LOC100518848 | LOC100518848 | LOC100518848 | 40S ribosomal protein S21 |
| LOC110258138 | LOC110258138 | non-coding RNA | |
| SGCA | SGCA | Sarcoglycan Alpha (stability of muscle fibre membranes) | |
| LOC106505031 | LOC106505031 | non-coding RNA | |
| PDZD9 | PDZD9 | PDZ Domain Containing 9 (Mitochondrial Complex V Deficiency, Nuclear Type 3 and Meckel Syndrome, Type 4) |
| Brain | Heart | Liver | |
|---|---|---|---|
| ACSM5 | ACSM5 | Acyl-Coenzyme A Synthetase ACSM5, Mitochondrial | |
| LOC102164558 | LOC102164558 | LOC102164558 | ncRNA |
| LOC100525311 | LOC100525311 | LOC100525311 | Golgi phosphoprotein 3 (GOLPH3) |
| IGF2BP3 | IGF2BP3 | Insulin Like Growth Factor 2 MRNA Binding Protein 3 | |
| LOC100152036 | LOC100152036 | LOC100152036 | SLAM family member 9-like (SLAMF9) |
| KIRREL | KIRREL | Kirre Like Nephrin Family Adhesion Molecule 1 | |
| LOC106508088 | LOC106508088 | ncRNA | |
| TLE4 | TLE4 | TLE Family Member 4, Transcriptional Corepressor | |
| NTRK2 | NTRK2 | Neurotrophic Receptor Tyrosine Kinase 2 | |
| TRIM7 | TRIM7 | Tripartite Motif Containing 7 | |
| NEXN | NEXN | Nexilin F-Actin Binding Protein | |
| LOC106509396 | LOC106509396 | Uncharacterised, Protein coding | |
| COG7 | COG7 | Component Of Oligomeric Golgi Complex 7 | |
| LOC110261018 | LOC110261018 | Uncharacterised, Protein coding | |
| EMILIN3 | EMILIN3 | Elastin Microfibril Interface-Located Protein 3 | |
| LOC106506003 | LOC106506003 | ncRNA | |
| RRBP1 | RRBP1 | Ribosome Binding Protein 1 | |
| LOC102163231 | LOC102163231 | ncRNA | |
| FLRT3 | FLRT3 | Fibronectin Leucine Rich Transmembrane Protein 3 | |
| LOC110258845 | LOC110258845 | ncRNA | |
| LOC106506130 | LOC106506130 | ncRNA | |
| SPAG16 | SPAG16 | Sperm Associated Antigen 16 |
| Downregulated | ||
|---|---|---|
| Brain | Heart | Liver |
| Inhibition of TSC complex formation by PKB | Resolution of AP sites via the single-nucleotide replacement pathway | Regulation of cytoskeletal remodelling and cell spreading by IPP complex components |
| APC/C:Cdh1-mediated degradation of Cdc20 and other APC/C:Cdh1 targeted proteins in late mitosis/early G1 | Synthesis of PS | The AIM2 inflammasome |
| Defective C1GALT1C1 causes TNPS | Hydrolysis of LPC | The IPAF inflammasome |
| VxPx cargo-targeting to cilium | Glycerophospholipid biosynthesis | MET promotes cell motility |
| TNFR2 non-canonical NF-kB pathway | Abasic sugar-phosphate removal via the single-nucleotide replacement pathway | Collagen biosynthesis and modifying enzymes |
| TNFs bind their physiological receptors | SUMOylation of DNA replication proteins | Phospholipase C-mediated cascade; FGFR3 |
| tRNA modification in the nucleus and cytosol | NPAS4 regulates expression of target genes | PLC-gamma1 signalling |
| Ethanol oxidation | Host Interactions of HIV factors | MET activates PTK2 signalling |
| Defective F8 binding to von Willebrand factor | Regulation of signalling by NODAL | Alternative complement activation |
| Upregulated | ||
| Brain | Heart | Liver |
| Elevation of cytosolic Ca2+ levels | Presynaptic depolarization and calcium channel opening | Vif-mediated degradation of APOBEC3G |
| Autodegradation of Cdh1 by Cdh1:APC/C | Organic cation transport | Regulation of apoptosis |
| APC/C:Cdc20-mediated degradation of Securin | Dual incision in GG-NER | Regulation of activated PAK-2p34 by proteasome-mediated degradation |
| Transport and synthesis of PAPS | Organic cation/anion/zwitterion transport | Branched-chain amino acid catabolism |
| APC/C:Cdc20-mediated degradation of mitotic proteins | Choline catabolism | Abnormal conversion of 2-oxoglutarate to 2-hydroxyglutarate |
| Activation of APC/C and APC/C:Cdc20-mediated degradation of mitotic proteins | Ciprofloxacin ADME | Ubiquitin-mediated degradation of phosphorylated Cdc25A |
| APC:Cdc20-mediated degradation of cell cycle proteins prior to satisfaction of the cell cycle checkpoint | Transcription-coupled nucleotide excision repair (TC-NER) | p53-independent DNA damage response |
| Downregulation of TGF-beta receptor signalling | Defective MMAA causes MMA, cblA type | p53-independent G1/S DNA damage checkpoint |
| Metabolism of ingested H2SeO4 and H2SeO3 into H2Se | Defective MUT causes MMAM | Cross-presentation of soluble exogenous antigens (endosomes) |
| DSCAM interactions | Diseases of mitochondrial beta oxidation | Hh mutants abrogate ligand secretion |
| Downregulated | ||
|---|---|---|
| Brain | Heart | Liver |
| Methionine salvage pathway | Phase II—conjugation of compounds | Opioid signalling |
| Mitochondrial iron–sulfur cluster biogenesis | Cytosolic sulfonation of small molecules | Calmodulin-induced events |
| Amino acid conjugation | Transfer of LPS from LBP carrier to CD14 | Cam-PDE 1 activation |
| L13a-mediated translational silencing of ceruloplasmin expression | Nucleotide-binding domain, leucine-rich repeat-containing receptor (NLR) signalling pathways | Ca-dependent events |
| Conjugation of carboxylic acids | DDX58/IFIH1-mediated induction of interferon-alpha/beta | CaM pathway |
| Conjugation of salicylate with glycine | Regulation of pyruvate–dehydrogenase (PDH) complex | G-protein mediated events |
| Signalling by FGFR2 amplification mutants | Biological oxidations | PLC beta mediated events |
| Activated point mutants of FGFR2 | Defective GGT1 causes GLUTH | Neurotransmitter receptors and postsynaptic signal transmission |
| MicroRNA (miRNA) biogenesis | RHOG GTPase cycle | Cross-presentation of particulate exogenous antigens (phagosomes) |
| Regulation of gene expression in endocrine-committed (NEUROG3+) progenitor cells | Defective GGT1 in aflatoxin detoxification causes GLUTH | SHC1 events in ERBB2 signalling |
| Upregulated | ||
| Brain | Heart | Liver |
| Cam-PDE 1 activation | Collagen degradation | ER-Phagosome pathway |
| G-protein-mediated events | Degradation of the extracellular matrix | Antigen processing cross presentation |
| PLC beta-mediated events | Collagen formation | Signalling by NOTCH |
| Presynaptic depolarization and calcium channel opening | Elastic fibre formation | Disease |
| Nuclear signalling by ERBB4 | Activation of matrix metalloproteinases | Complement cascade |
| Tandem of pore domain in a weak inwardly rectifying K+ channels (TWIK) | Sulphide oxidation to sulphate | Activation of C3 and C5 |
| Sperm motility and taxes | Glycosaminoglycan metabolism | Pre-NOTCH processing in Golgi |
| Regulation of signalling by NODAL | Collagen biosynthesis and modifying enzymes | Pre-NOTCH expression and processing |
| ARMS-mediated activation | Complement cascade | Cholesterol biosynthesis |
| FGFR1c and Klotho ligand binding and activation | NGF processing | Downstream TCR signalling |
| All Offspring | Heterologous v Autologous | ||||
|---|---|---|---|---|---|
| Brain | Heart | Liver | Brain | Heart | Liver |
| ADRM1 | CHRNA4 | PTK6 | ACSM5 | ACSM5 | KCND3 |
| RPS21 | MYT1 | KIF1A | SLAMF9 | TOP1MT | PARVB |
| ABCA13 | DGKI | DLGAP2 | |||
| NOS2 | PRKAR1B | SGF29 | |||
| DSCAM | MYT1L | SLC26A2 | |||
| NAV3 | CDS1 | NAV2 | |||
| SDK1 | MLYCD | TTBK1 | |||
| HS3ST2 | CLDN10 | ||||
| MICU2 | SYT13 | ||||
| CRELD2 | |||||
| PC | |||||
| ATP8A2 | |||||
| CACNB2 | |||||
| LRRC8D | |||||
| SMURF1 | |||||
| PPFIBP2 | |||||
| RGS17 | |||||
| MBOAT2 | |||||
| CFAP46 | |||||
| Three Tissues | Two Tissues | ||
|---|---|---|---|
| All Offspring | Heterologous vs. Autologous | All Offspring | Heterologous vs. Autologous |
| TAFA5 | RALGDS | SHANK2 | ARID1B |
| DST | JAKMIP2 | FSTL4 | CDK5RAP2 |
| PITRM1 | ARHGEF37 | GSE1 | PPP6R3 |
| COG3 | PPARGC1B | SACS | DPP3 |
| BNIP3 | PDE6A | ABCA13 | MACROH2A1 |
| COL4A4 | HMGXB3 | TCERG1L | HTR4 |
| STMN3 | CSF1R | NDUFA10 | ABLIM3 |
| PTPRN2 | CDX1 | CDH4 | SUN1 |
| TCOF1 | TPD52L2 | SDK1 | |
| SYNPO | SHROOM2 | AUTS2 | |
| FOXK1 | DCDC2C | IQCK | |
| RADIL | NAV3 | TXNDC11 | |
| TRRAP | SORCS2 | CIITA | |
| GTF3C1 | ADARB2 | TBC1D8 | |
| SNX29 | ST18 | ||
| CLEC16A | TBC1D22A | ||
| DNMT3A | TTLL1 | ||
| OSR1 | USP10 | ||
| ZNHIT6 | PLCG2 | ||
| CBFA2T3 | CDYL2 | ||
| PEPD | CAMTA1 | ||
| SORCS2 | LUZP1 | ||
| LEF1 | RGS12 | ||
| PITRM1 | TBC1D1 | ||
| PFKP | PPFIBP2 | ||
| ADARB2 | NMNAT2 | ||
| DIP2C | LNX2 | ||
| SACS | SLC7A1 | ||
| MIPEP | KCNJ2 | ||
| ATP8A2 | MSI2 | ||
| DNAH17 | SLC5A10 | ||
| SEPTIN9 | MAP2K3 | ||
| MGAT5B | HSPA12A | ||
| SPECC1 | DLGAP2 | ||
| MTUS2 | INPP5D | ||
| USP36 | SLC12A7 | ||
| MGMT | PCBP3 | ||
| AGAP1 | PDE1C | ||
| NDUFA10 | COG3 | ||
| CDH4 | KSR1 | ||
| PTPRN2 | C10orf90 | ||
| DPP6 | HDAC4 | ||
| DGKI | NDUFS6 | ||
| PHACTR3 | |||
| SND1 | |||
| AFAP1L1 | |||
| SMURF1 | |||
| EPS8L3 | |||
| LRRC8D | |||
| TAFA5 | |||
| CRISPLD2 | |||
| WWOX | |||
| STX18 | |||
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
St. John, J.C.; Andreas, E.; Penn, A. Can the Supplementation of Oocytes with Extra Copies of mtDNA Impact Development Without Being Transmitted? A Molecular Account. Int. J. Mol. Sci. 2025, 26, 2746. https://doi.org/10.3390/ijms26062746
St. John JC, Andreas E, Penn A. Can the Supplementation of Oocytes with Extra Copies of mtDNA Impact Development Without Being Transmitted? A Molecular Account. International Journal of Molecular Sciences. 2025; 26(6):2746. https://doi.org/10.3390/ijms26062746
Chicago/Turabian StyleSt. John, Justin C., Eryk Andreas, and Alexander Penn. 2025. "Can the Supplementation of Oocytes with Extra Copies of mtDNA Impact Development Without Being Transmitted? A Molecular Account" International Journal of Molecular Sciences 26, no. 6: 2746. https://doi.org/10.3390/ijms26062746
APA StyleSt. John, J. C., Andreas, E., & Penn, A. (2025). Can the Supplementation of Oocytes with Extra Copies of mtDNA Impact Development Without Being Transmitted? A Molecular Account. International Journal of Molecular Sciences, 26(6), 2746. https://doi.org/10.3390/ijms26062746