
Dietary Fermentation with Lactobacillus sp. and Bacillus sp. Modulates Rumen Transcriptomic and Microbiota Profiles in Bos taurus

 and
and 
Abstract

1. Introduction
2. Results
2.1. Animal Growth and Performance by Diet Variations
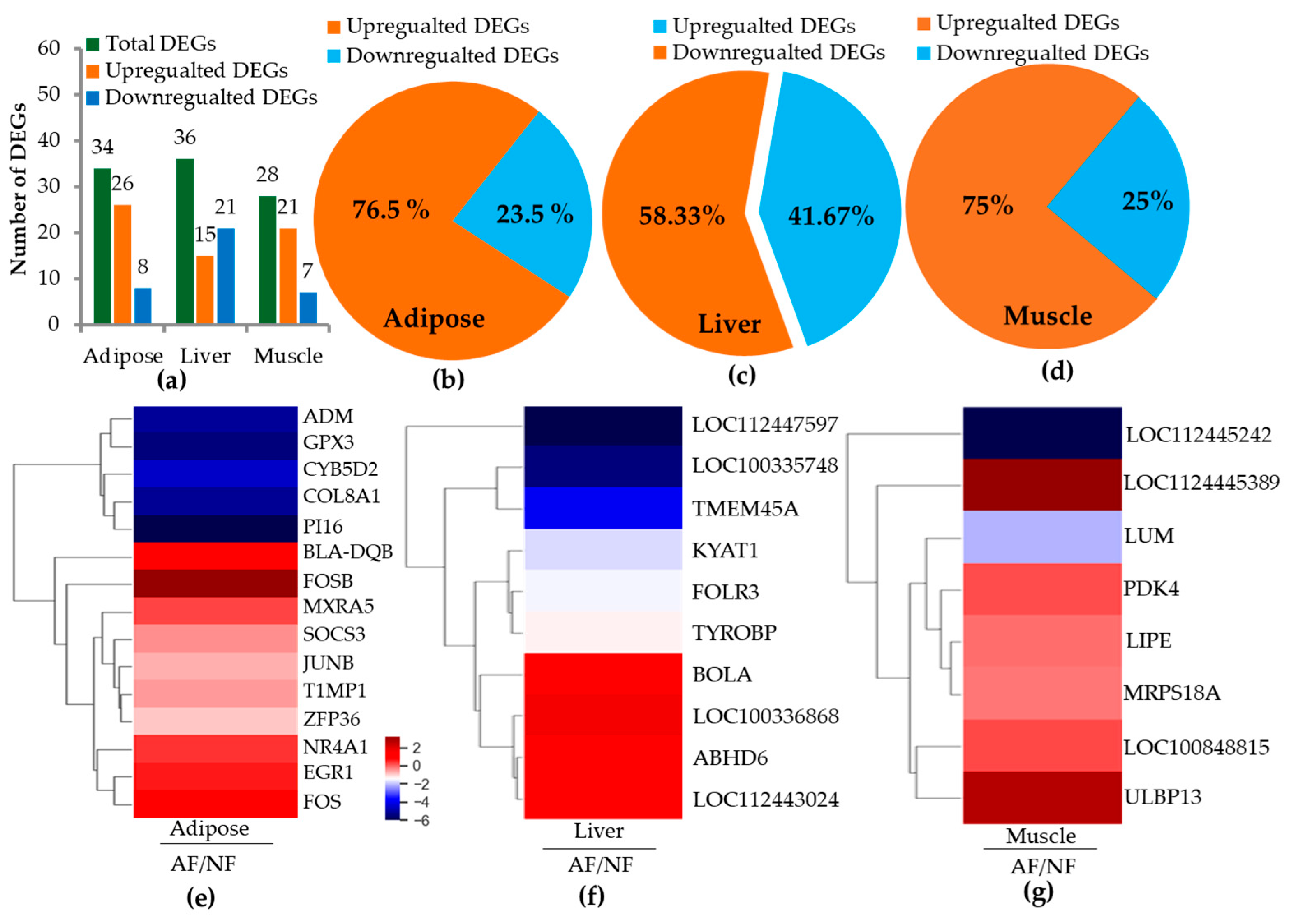
2.2. RNA Sequence Alignment and Differentially Expressed Genes (DEGs)
2.3. Differentially Expressed Genes (DEGs) and Pathways
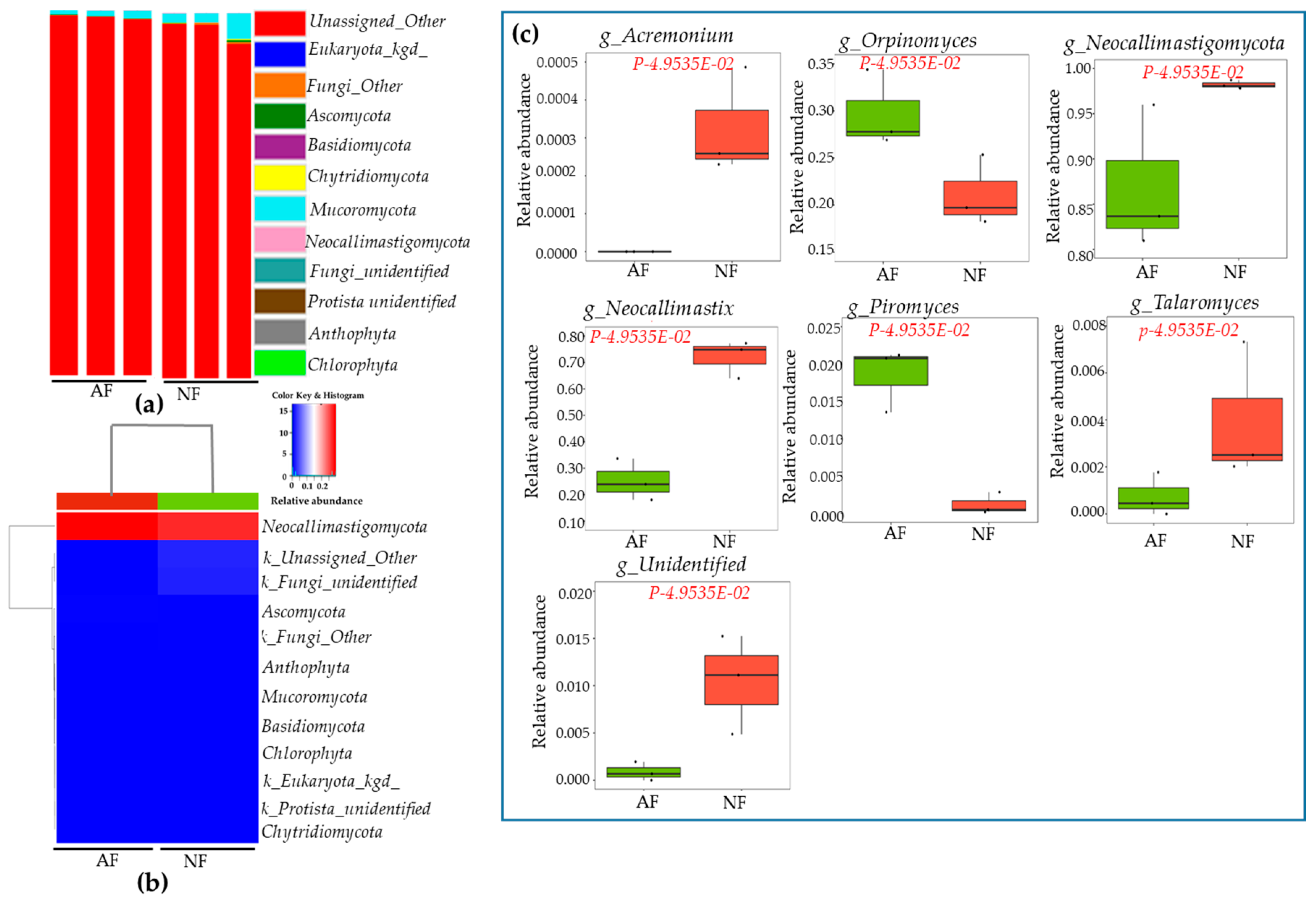
2.4. Sequencing Depth, Coverage, and α-Diversity Index of Bacteria and Fungi
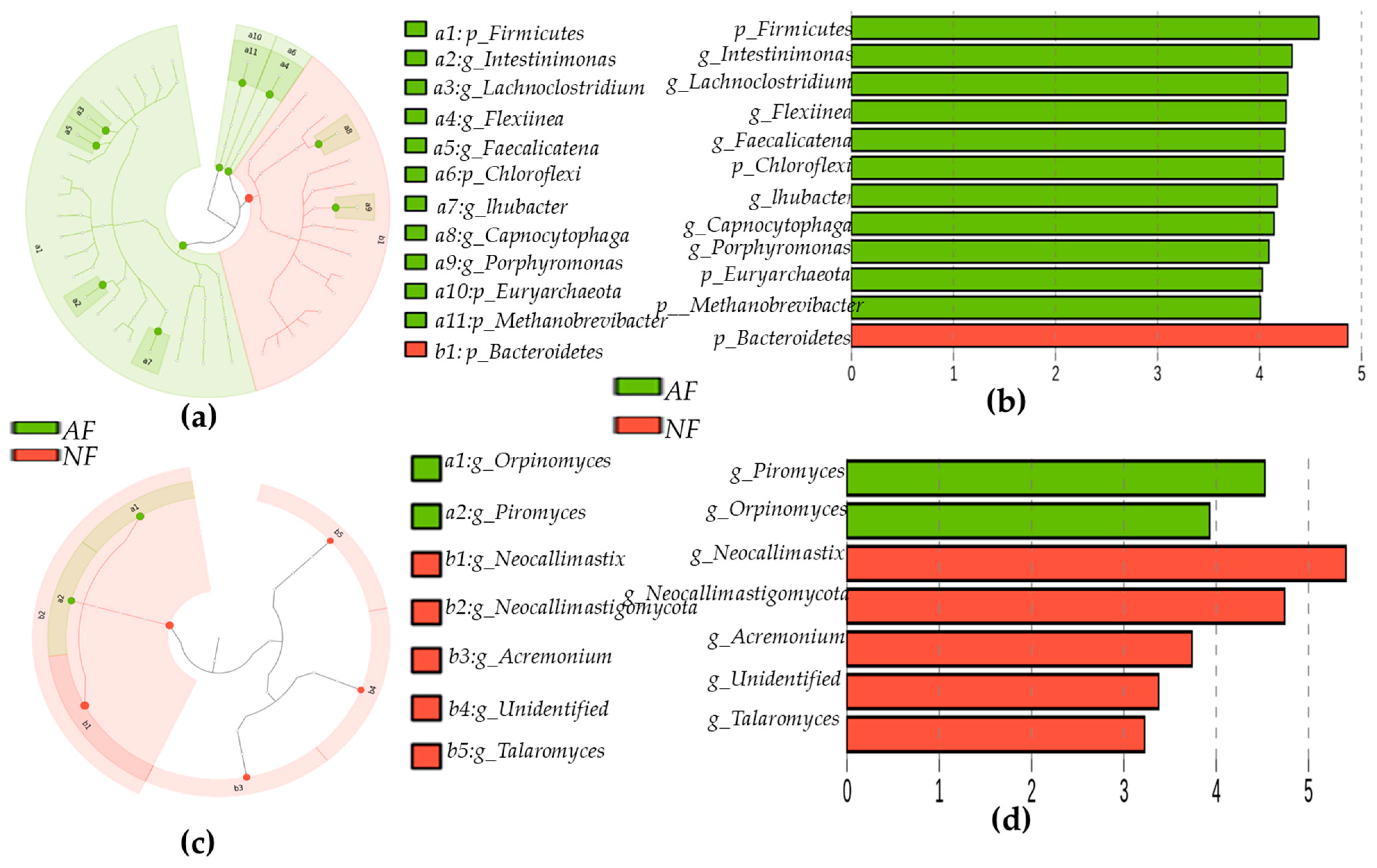
2.5. Rumen Microbiota Community Compositions of NF- and AF-Fed Animals
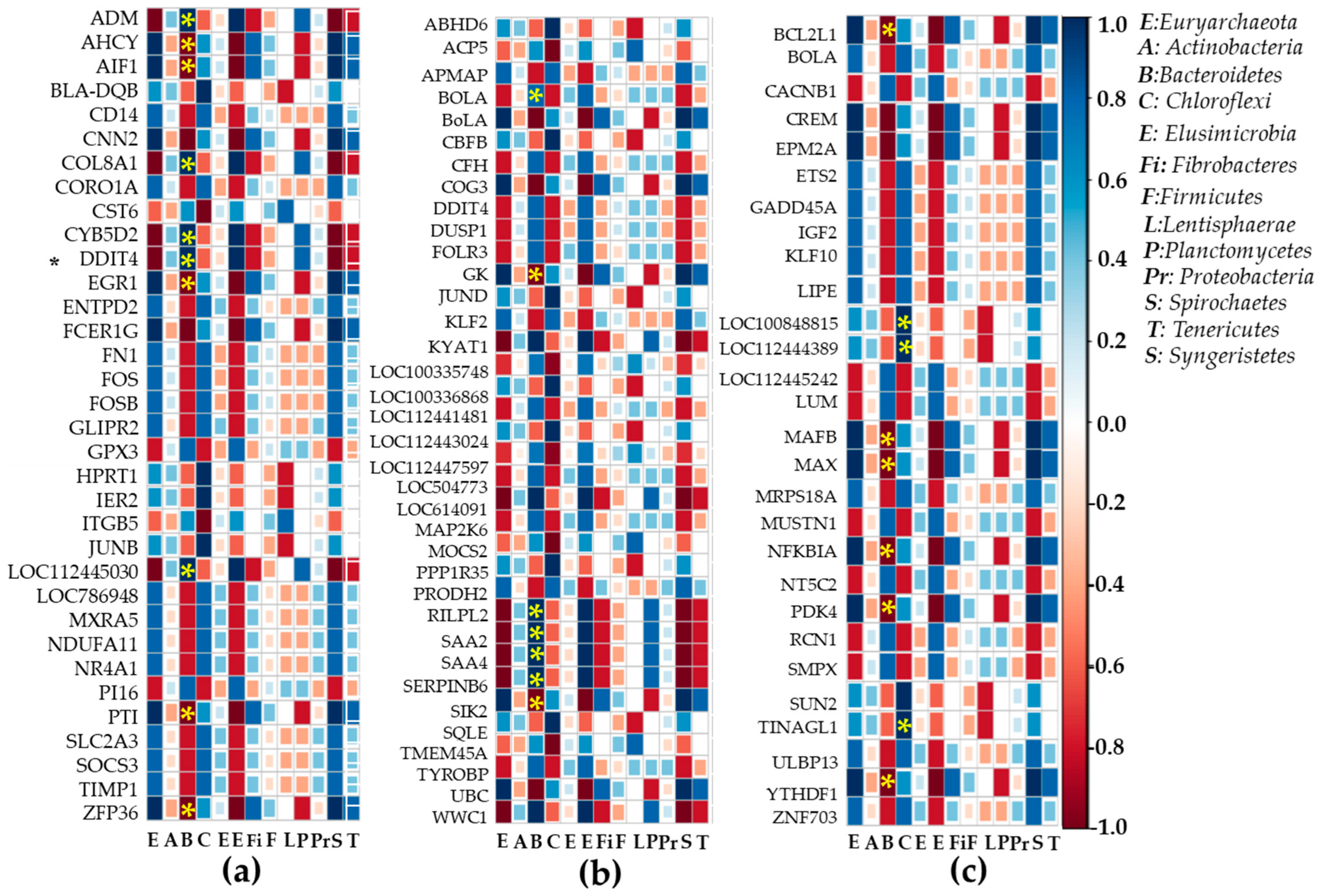
2.6. Heatmap Correlation Studies Between DEGs and Bacteria or Fungi
3. Discussion
4. Materials and Methods
4.1. Crops Collection and Production of Silage with Bacteria
4.2. Fermented Feed Preparation
4.3. Ethics Statements
4.4. Animal and Experimental Design
4.5. RNA Extraction from Different Tissues
4.6. Preparation of Library and Sequencing
4.7. Analysis of Data and Removal of Low-Quality Reads
4.8. Heatmap Correlation and Gene Ontology
4.9. Genomic DNA Extraction
4.10. Library Construction and Sequencing
- V3-F, 5′-TCGTCGGCAGCGTCAGATGTGTATAAGAGACAGCCTACGGGNGGCWGCAG-3;
- V4-R, 5′ TCTCGTGGGCTCGGAGATGTGTATAAGAGACAGGACTACHVGGGTATCTAATCC-3.
- For fungi ITS3 amplicon, PCR forward primer-5
- TCGTCGGCAGCGTCAGATGTGTATAAGAGACAGGCATCGATGAAGAACGCAGC-3 and ITS4 amplicon PCR reverse primer-5 GTCTCGTGGGCTCGGAGATGTGTATAAGAGACAGTCCTCCGCTTATTGATATGC-3 were used.
4.11. Determination of Relative Abundance of Microbial Dynamics
4.12. Integrated Analysis of Interactions Between DEGs in Major Metabolic Organs and Relative Abundance of Bacteria or Fungi in Rumen
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| ABHD6 | Abhydrolase domain containing 6: acylglycerol lipase |
| ACP5 | Acid phosphatase 5, tartrate resistant |
| ADM | Adrenomedullin |
| AF | Alternative diet feed |
| AHCY | Adenosylhomocysteinase |
| AIF1 | Allograft inflammatory factor 1 |
| APMAP | Adipocyte plasma membrane associated protein |
| BLA-DQB | MHC class II antigen |
| BOLA | Major histocompatibility complex, class i,A |
| CACNB1 | Calcium voltage-gated channel auxiliary subunit beta 1 |
| CBFB | Core-binding factor subunit beta |
| CD14 | CD14 molecule |
| CFH | Complement factor H |
| CL2L1 | Bcl2 like 1 |
| CNN2 | Calponin 2 |
| COG3 | Component of oligomeric golgi complex 3 |
| COL8A1 | Collagen type VIII alpha 1 chain |
| CORO1A | Coronin 1A |
| CREM | Camp responsive element modulator |
| CST6 | Cystatin E/M |
| CYB5D2 | Cytochrome b5 domain containing 2 |
| DDIT4 | DNA damage inducible transcript 4 |
| DEGs | Differentially expressed genes |
| DM | Dry matter content |
| DUSP1 | Dual specificity phosphatase 1 |
| EGR1 | Early growth response 1 |
| ENTPD2 | Ectonucleoside triphosphate diphosphohydrolase 2 |
| EPM2A | Epm2a glucan phosphatase, laforin |
| ETS2 | Ets proto-oncogene 2, transcription factor |
| FCER1G | Fc epsilon receptor Ig |
| FCR | Feed conversion ratios |
| FN1 | Fibronectin 1 |
| FOLR3 | Folate receptor 3 |
| FOS | Fos proto-oncogene, AP-1 transcription factor subunit |
| FOSB | Fosb proto-oncogene, AP-1 transcription factor |
| GADD45A | Growth arrest and DNA damage inducible alpha |
| GK | Glycerol kinase |
| GLIPR2 | GLI pathogenesis related 2 |
| GO | Gene Ontology |
| GPX3 | Glutathione peroxidase 3 |
| HPRT1 | Hypoxanthinephosphoribosyltransferase 1 |
| IER2 | Immediate early response 2 |
| IGF2 | Insulin like growth factor 2 |
| ITGB5 | Integrin subunit beta 5 |
| JUNB | Junb proto-oncogene, AP-1 transcription factor |
| JUND | Jund proto-oncogene, AP-1 transcription factor |
| KEGG | Kyoto Encyclopedia of genes and Genomes |
| KLF10 | Klf transcription factor 10 |
| KLF2 | KLF transcription factor 2 |
| KYAT1 | Kynurenine aminotransferase 1 |
| LEfSe | Linear discriminant analysis Effect Size |
| LIPE | Lipase e, hormone sensitive type |
| LOC100335748 | Uncharacterized LOC100335748 |
| LOC100336868 | Complement factor H |
| LOC100848815 | Sla class ii histocompatibility antigen- d alpha chain-like |
| LOC112441481 | Glycine N-phenylacetyltransferase-like |
| LOC112443024 | Uncharacterized |
| LOC112444389 | Small nucleolar RNA SNORA32 |
| LOC112445030 | Four and a half LIM domains protein 1- |
| LOC112445242 | Small nucleolar RNA SNORA70 |
| LOC112447597 | U6 spliceosomal RNA |
| LOC504773 | Regakine 1 |
| LOC614091 | Class I histocompatibility antigen, alpha chain BL3 |
| LOC786948 | Tryptase-2-like |
| LUM | Lumican |
| MAFB | Maf bzip transcription factor B |
| MAP2K6 | Mitogen-activated protein kinase kinase 6 |
| MAX | Myc associated factor x |
| MOCS2 | Molybdenum cofactor synthesis 2 |
| MRPS18A | Mitochondrial ribosomal protein s18a |
| MUSTN1 | Musculoskeletal, embryonic nuclear protein 1 |
| MXRA5 | Matrix remodeling associated 5 |
| NDUFA11 | NADH:ubiquinone oxidoreductase |
| NF | Normal diet feed |
| NFKBIA | Nfkb inhibitor alpha |
| NR4A1 | Nuclear receptor subfamily 4 group A member 1 |
| NT5C2 | Nucleotidase 5′, cytosolic ii |
| OUT | Operational taxonomic unit |
| PDK4 | Pyruvate dehydrogenase kinase 4 |
| PI16 | Peptidase inhibitor 16 |
| PPP1R35 | Protein phosphatase 1 regulatory subunit 35 |
| PRODH2 | Proline dehydrogenase 2 |
| PTI | Pancreatic trypsin inhibitor |
| PUFA | Polyunsaturated fatty acids |
| RCN1 | Reticulocalbin 1 |
| RILPL2 | Rab interacting lysosomal protein like 2 |
| SAA2 | Serum amyloid A2 |
| SAA4 | Serum amyloid A4, constitutive |
| SERPINB6 | Serpin peptidase inhibitor, clade B (ovalbumin), member 6 |
| SIK2 | Salt inducible kinase 2 |
| SLC2A3 | Solute carrier family 2 member 3 |
| SMPX | Small muscle protein x-linked |
| SOCS3 | Suppressor of cytokine signaling 3 |
| SQLE | Squalene epoxidase |
| SUN2 | Sad1 and unc84 domain containing 2 |
| TIMP1 | TIMP metallopeptidase inhibitor 1 |
| TINAGL1 | Tubulointerstitial nephritis antigen like 1 |
| TMEM45A | Transmembrane protein 45A |
| TMR | Total mixed ration |
| TYROBP | Transmembrane immune signaling adaptor TYROBP |
| UBC | Ubiquitin C |
| ULBP13 | Ul16 binding protein 13 |
| VFA | Volatile fatty acids |
| WWC1 | WW and C2 domain containing 1 |
| YTHDF1 | Yth n6-methyladenosine RNA binding protein f1 |
| ZFP36 | ZFP36 ring finger protein |
| ZNF703 | Zinc finger protein 703 |
References
- Yafetto, L.; Odamtten, G.T.; Wiafe-Kwagyan, M. Valorization of agro-industrial wastes into animal feed through microbial fermentation: A review of the global and Ghanaian case. Heliyon 2023, 9, e14814. [Google Scholar] [CrossRef]
- Dentinho, M.T.P.; Paulos, K.; Costa, C.; Costa, J.; Fialho, L.; Cachucho, L.; Portugal, A.P.; Almeida, J.; Rehan, I.; Belo, A.T.; et al. Silages of agro-industrial by-products in lamb diets—Effect on growth performance, carcass, meat quality and in vitro methane emissions. Anim. Feed Sci. Technol. 2023, 298, 115603. [Google Scholar] [CrossRef]
- Uyeno, Y.; Shigemori, S.; Shimosato, T. Effect of Probiotics/Prebiotics on Cattle Health and Productivity. Microbes Environ. 2015, 30, 126–132. [Google Scholar] [CrossRef]
- Zhang, G.; Li, Y.; Fang, X.; Cai, Y.; Zhang, Y. Lactation performance, nitrogen utilization, and profitability in dairy cows fed fermented total mixed ration containing wet corn gluten feed and corn stover in combination replacing a portion of alfalfa hay. Anim. Feed Sci. Technol. 2020, 269, 114687. [Google Scholar] [CrossRef]
- Lee, S.; Ryu, C.H.; Back, Y.C.; Lee, S.D.; Kim, H. Effect of Fermented Concentrate on Ruminal Fermentation, Ruminal and Fecal Microbiome, and Growth Performance of Beef Cattle. Animals 2023, 13, 3622. [Google Scholar] [CrossRef] [PubMed]
- Kim, D.; Jung, J.-S.; Choi, K.-C. A Preliminary Study on Effects of Fermented Feed Supplementation on Growth Performance, Carcass Characteristics, and Meat Quality of Hanwoo Steers during the Early and Late Fattening Period. Appl. Sci. 2021, 11, 5202. [Google Scholar] [CrossRef]
- Skowron, K.; Budzyńska, A.; Grudlewska-Buda, K.; Wiktorczyk-Kapischke, N.; Andrzejewska, M.; Wałecka-Zacharska, E.; Gospodarek-Komkowska, E. Two Faces of Fermented Foods-The Benefits and Threats of Its Consumption. Front. Microbiol. 2022, 13, 845166. [Google Scholar] [CrossRef]
- Cheng, Y.H.; Horng, Y.B.; Chen, W.J.; Hua, K.F.; Dybus, A.; Yu, Y.H. Effect of Fermented Products Produced by Bacillus licheniformis on the Growth Performance and Cecal Microbial Community of Broilers under Coccidial Challenge. Animals 2021, 11, 1245. [Google Scholar] [CrossRef]
- Lian, X.; Shi, M.; Liang, Y.; Lin, Q.; Zhang, L. The Effects of Unconventional Feed Fermentation on Intestinal Oxidative Stress in Animals. Antioxidants 2024, 13, 305. [Google Scholar] [CrossRef]
- Samtiya, M.; Aluko, R.E.; Dhewa, T. Plant food anti-nutritional factors and their reduction strategies: An overview. Food Prod. Process. Nutr. 2020, 2, 6. [Google Scholar] [CrossRef]
- Ortiz-Chura, A.; Corral-Jara, K.F.; Tournayre, J.; Cantalapiedra-Hijar, G.; Popova, M.; Morgavi, D.P. Rumen microbiota associated with feed efficiency in beef cattle are highly influenced by diet composition. Anim. Nutr. 2025, 21, 378–389. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Liu, X.; Xie, K.; Pan, Y.; Liu, F.; Hou, F. Effects of different fiber levels of energy feeds on rumen fermentation and the microbial community structure of grazing sheep. BMC Microbiol. 2025, 25, 180. [Google Scholar] [CrossRef] [PubMed]
- Newbold, C.J.; Ramos-Morales, E. Review: Ruminal microbiome and microbial metabolome: Effects of diet and ruminant host. Animal 2020, 14, s78–s86. [Google Scholar] [CrossRef] [PubMed]
- McCann, J.C.; Wickersham, T.A.; Loor, J.J. High-throughput Methods Redefine the Rumen Microbiome and Its Relationship with Nutrition and Metabolism. Bioinform. Biol. Insights 2014, 8, 109–125. [Google Scholar] [CrossRef]
- Weimer, P.J. Redundancy, resilience, and host specificity of the ruminal microbiota: Implications for engineering improved ruminal fermentations. Front. Microbiol. 2015, 6, 296. [Google Scholar] [CrossRef]
- Wu, Y.; Jiao, C.; Diao, Q.; Tu, Y. Effect of Dietary and Age Changes on Ruminal Microbial Diversity in Holstein Calves. Microorganisms 2023, 12, 12. [Google Scholar] [CrossRef]
- Jiao, J.; Li, X.; Beauchemin, K.A.; Tan, Z.; Tang, S.; Zhou, C. Rumen development process in goats as affected by supplemental feeding v. grazing: Age-related anatomic development, functional achievement and microbial colonisation. Br. J. Nutr. 2015, 113, 888–900. [Google Scholar] [CrossRef]
- Bi, Y.; Zeng, S.; Zhang, R.; Diao, Q.; Tu, Y. Effects of dietary energy levels on rumen bacterial community composition in Holstein heifers under the same forage to concentrate ratio condition. BMC Microbiol. 2018, 18, 69. [Google Scholar] [CrossRef]
- Fernando, S.C.; Purvis, H.T.; Najar, F.Z.; Sukharnikov, L.O.; Krehbiel, C.R.; Nagaraja, T.G.; Roe, B.A.; DeSilva, U. Rumen Microbial Population Dynamics during Adaptation to a High-Grain Diet. Appl. Environ. Microbiol. 2010, 76, 7482–7490. [Google Scholar] [CrossRef]
- Carberry, C.A.; Kenny, D.A.; Han, S.; McCabe, M.S.; Waters, S.M. Effect of phenotypic residual feed intake and dietary forage content on the rumen microbial community of beef cattle. Appl. Environ. Microbiol. 2012, 78, 4949–4958. [Google Scholar] [CrossRef]
- Myer, P.R.; Smith, T.P.L.; Wells, J.E.; Kuehn, L.A.; Freetly, H.C. Rumen Microbiome from Steers Differing in Feed Efficiency. PLoS ONE 2015, 10, e0129174. [Google Scholar] [CrossRef] [PubMed]
- Xu, S.-y.; Feng, X.-r.; Zhao, W.; Bi, Y.-l.; Diao, Q.-y.; Tu, Y. Rumen and hindgut microbiome regulate average daily gain of preweaning Holstein heifer calves in different ways. Microbiome 2024, 12, 131. [Google Scholar] [CrossRef]
- Linde, J.; Schulze, S.; Henkel, S.G.; Guthke, R. Data- and knowledge-based modeling of gene regulatory networks: An update. EXCLI J. 2015, 14, 346–378. [Google Scholar] [CrossRef]
- Cho, D.Y.; Kim, Y.A.; Przytycka, T.M. Chapter 5: Network biology approach to complex diseases. PLoS Comput. Biol. 2012, 8, e1002820. [Google Scholar] [CrossRef]
- Wang, X.; Wang, J.; Raza, S.H.A.; Deng, J.; Ma, J.; Qu, X.; Yu, S.; Zhang, D.; Alshammari, A.M.; Almohaimeed, H.M.; et al. Identification of the hub genes related to adipose tissue metabolism of bovine. Front. Vet. Sci. 2022, 9, 1014286. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.J.; Park, H.S.; Kim, W.; Yoon, D.; Seo, S. Comparison of Metabolic Network between Muscle and Intramuscular Adipose Tissues in Hanwoo Beef Cattle Using a Systems Biology Approach. Int. J. Genom. 2014, 2014, 679437. [Google Scholar] [CrossRef] [PubMed]
- Kenny, D.A.; Fitzsimons, C.; Waters, S.M.; McGee, M. Invited review: Improving feed efficiency of beef cattle—The current state of the art and future challenges. Animal 2018, 12, 1815–1826. [Google Scholar] [CrossRef]
- Greenwood, S.C.; Walker, S.; Baird, H.M.; Parsons, R.; Mehl, S.; Webb, T.L.; Slark, A.T.; Ryan, A.J.; Rothman, R.H. Many Happy Returns: Combining insights from the environmental and behavioural sciences to understand what is required to make reusable packaging mainstream. Sustain. Prod. Consum. 2021, 27, 1688–1702. [Google Scholar] [CrossRef]
- Kim, T.I.; Mayakrishnan, V.; Lim, D.H.; Yeon, J.H.; Baek, K.S. Effect of fermented total mixed rations on the growth performance, carcass and meat quality characteristics of Hanwoo steers. Anim. Sci. J. 2018, 89, 606–615. [Google Scholar] [CrossRef]
- Zhou, X.; Zhang, H.; Li, S.; Jiang, Y.; Kang, L.; Deng, J.; Yang, C.; Zhao, X.; Zhao, J.; Jiang, L.; et al. The effects of fermented feedstuff derived from Citri Sarcodactylis Fructus by-products on growth performance, intestinal digestive enzyme activity, nutrient utilization, meat quality, gut microbiota, and metabolites of broiler chicken. Front. Vet. Sci. 2023, 10, 1231996. [Google Scholar] [CrossRef]
- Diniz, W.J.S.; Crouse, M.S.; Cushman, R.A.; McLean, K.J.; Caton, J.S.; Dahlen, C.R.; Reynolds, L.P.; Ward, A.K. Cerebrum, liver, and muscle regulatory networks uncover maternal nutrition effects in developmental programming of beef cattle during early pregnancy. Sci. Rep. 2021, 11, 2771. [Google Scholar] [CrossRef]
- Wang, D.; Liu, L.; Augustino, S.M.A.; Duan, T.; Hall, T.J.; MacHugh, D.E.; Dou, J.; Zhang, Y.; Wang, Y.; Yu, Y. Identification of novel molecular markers of mastitis caused by Staphylococcus aureus using gene expression profiling in two consecutive generations of Chinese Holstein dairy cattle. J. Anim. Sci. Biotechnol. 2020, 11, 98. [Google Scholar] [CrossRef]
- Abdelbaky, H.H.; Mitsuhashi, S.; Watanabe, K.; Ushio, N.; Miyakawa, M.; Furuoka, H.; Nishikawa, Y. Involvement of chemokine receptor CXCR3 in the defense mechanism against Neospora caninum infection in C57BL/6 mice. Front. Microbiol. 2023, 13, 1045106. [Google Scholar] [CrossRef] [PubMed]
- Alexandre, P.A.; Naval-Sanchez, M.; Porto-Neto, L.R.; Ferraz, J.B.S.; Reverter, A.; Fukumasu, H. Systems Biology Reveals NR2F6 and TGFB1 as Key Regulators of Feed Efficiency in Beef Cattle. Front. Genet. 2019, 10, 230. [Google Scholar] [CrossRef]
- Zarek, C.M.; Lindholm-Perry, A.K.; Kuehn, L.A.; Freetly, H.C. Differential expression of genes related to gain and intake in the liver of beef cattle. BMC Res. Notes 2017, 10, 1. [Google Scholar] [CrossRef]
- Pereira, M.G.; Dyar, K.A.; Nogara, L.; Solagna, F.; Marabita, M.; Baraldo, M.; Chemello, F.; Germinario, E.; Romanello, V.; Nolte, H.; et al. Comparative Analysis of Muscle Hypertrophy Models Reveals Divergent Gene Transcription Profiles and Points to Translational Regulation of Muscle Growth through Increased mTOR Signaling. Front. Physiol. 2017, 8, 968. [Google Scholar] [CrossRef]
- Tizioto, P.C.; Coutinho, L.L.; Oliveira, P.S.N.; Cesar, A.S.M.; Diniz, W.J.S.; Lima, A.O.; Rocha, M.I.; Decker, J.E.; Schnabel, R.D.; Mourão, G.B.; et al. Gene expression differences in Longissimus muscle of Nelore steers genetically divergent for residual feed intake. Sci. Rep. 2016, 6, 39493. [Google Scholar] [CrossRef]
- Keogh, K.; McKenna, C.; Waters, S.M.; Porter, R.K.; Fitzsimons, C.; McGee, M.; Kenny, D.A. Effect of breed and diet on the M. longissimus thoracis et lumborum transcriptome of steers divergent for residual feed intake. Sci. Rep. 2023, 13, 9034. [Google Scholar] [CrossRef]
- Pulakazhi Venu, V.K.; Alston, L.; Iftinca, M.; Tsai, Y.C.; Stephens, M.; Warriyar, K.V.V.; Rehal, S.; Hudson, G.; Szczepanski, H.; von der Weid, P.Y.; et al. Nr4A1 modulates inflammation-associated intestinal fibrosis and dampens fibrogenic signaling in myofibroblasts. Am. J. Physiol. Gastrointest. Liver Physiol. 2021, 321, G280–G297. [Google Scholar] [CrossRef]
- Clark, A.R.; Dean, J.L. The control of inflammation via the phosphorylation and dephosphorylation of tristetraprolin: A tale of two phosphatases. Biochem. Soc. Trans. 2016, 44, 1321–1337. [Google Scholar] [CrossRef]
- Cao, Y.; Huang, W.; Wu, F.; Shang, J.; Ping, F.; Wang, W.; Li, Y.; Zhao, X.; Zhang, X. ZFP36 protects lungs from intestinal I/R-induced injury and fibrosis through the CREBBP/p53/p21/Bax pathway. Cell Death Dis. 2021, 12, 685. [Google Scholar] [CrossRef]
- Porter, J.F.; Shen, S.; Denhardt, D.T. Tissue inhibitor of metalloproteinase-1 stimulates proliferation of human cancer cells by inhibiting a metalloproteinase. Br. J. Cancer 2004, 90, 463–470. [Google Scholar] [CrossRef]
- Cicchetto, A.; Jacobson, E.; Sunshine, H.; Wilde, B.; Krall, A.; Jarrett, K.; Sedgeman, L.; Turner, M.; Plath, K.; Iruela-Arispe, L.; et al. ZFP36-mediated mRNA decay regulates metabolism. Cell Rep. 2023, 42, 112411. [Google Scholar] [CrossRef] [PubMed]
- Derakhshani, H.; Plaizier, J.C.; De Buck, J.; Barkema, H.W.; Khafipour, E. Association of bovine major histocompatibility complex (BoLA) gene polymorphism with colostrum and milk microbiota of dairy cows during the first week of lactation. Microbiome 2018, 6, 203. [Google Scholar] [CrossRef] [PubMed]
- Seo, M.; Kim, K.; Yoon, J.; Jeong, J.Y.; Lee, H.-J.; Cho, S.; Kim, H. RNA-seq analysis for detecting quantitative trait-associated genes. Sci. Rep. 2016, 6, 24375. [Google Scholar] [CrossRef] [PubMed]
- Pralle, R.S.; Li, W.; Murphy, B.N.; Holdorf, H.T.; White, H.M. Novel Facets of the Liver Transcriptome Are Associated with the Susceptibility and Resistance to Lipid-Related Metabolic Disorders in Periparturient Holstein Cows. Animals 2021, 11, 2558. [Google Scholar] [CrossRef]
- Ku, H.C.; Cheng, C.F. Master Regulator Activating Transcription Factor 3 (ATF3) in Metabolic Homeostasis and Cancer. Front. Endocrinol. 2020, 11, 556. [Google Scholar] [CrossRef]
- Micallef, P.; Vujičić, M.; Wu, Y.; Peris, E.; Wang, Y.; Chanclón, B.; Ståhlberg, A.; Cardell, S.L.; Wernstedt Asterholm, I. C1QTNF3 is Upregulated During Subcutaneous Adipose Tissue Remodeling and Stimulates Macrophage Chemotaxis and M1-Like Polarization. Front. Immunol. 2022, 13, 914956. [Google Scholar] [CrossRef]
- Marima, J.K.; Nel, C.L.; Marufu, M.C.; Jonsson, N.N.; Dube, B.; Dzama, K. A genetic and immunological comparison of tick-resistance in beef cattle following artificial infestation with Rhipicephalus ticks. Exp. Appl. Acarol. 2020, 80, 569–590. [Google Scholar] [CrossRef]
- Mukiibi, R.; Vinsky, M.; Keogh, K.A.; Fitzsimmons, C.; Stothard, P.; Waters, S.M.; Li, C. Transcriptome analyses reveal reduced hepatic lipid synthesis and accumulation in more feed efficient beef cattle. Sci. Rep. 2018, 8, 7303. [Google Scholar] [CrossRef]
- Graja, A.; Garcia-Carrizo, F.; Jank, A.M.; Gohlke, S.; Ambrosi, T.H.; Jonas, W.; Ussar, S.; Kern, M.; Schürmann, A.; Aleksandrova, K.; et al. Loss of periostin occurs in aging adipose tissue of mice and its genetic ablation impairs adipose tissue lipid metabolism. Aging Cell 2018, 17, e12810. [Google Scholar] [CrossRef] [PubMed]
- Goad, D.W.; Goad, C.L.; Nagaraja, T.G. Ruminal microbial and fermentative changes associated with experimentally induced subacute acidosis in steers. J. Anim. Sci. 1998, 76, 234–241. [Google Scholar] [CrossRef] [PubMed]
- Perez, H.G.; Stevenson, C.K.; Lourenco, J.M.; Callaway, T.R. Understanding Rumen Microbiology: An Overview. Encyclopedia 2024, 4, 148–157. [Google Scholar] [CrossRef]
- Kumar, M.; Yadav, A.N.; Saxena, R.; Paul, D.; Tomar, R.S. Biodiversity of pesticides degrading microbial communities and their environmental impact. Biocatal. Agric. Biotechnol. 2021, 31, 101883. [Google Scholar] [CrossRef]
- Boone, D.R.; Castenholz, R.W.; Garrity, G.M. Bergey’s Manual of Systematic Bacteriology; Springer: Berlin/Heidelberg, Germany, 2001. [Google Scholar]
- Zhang, J.; Shi, H.; Wang, Y.; Cao, Z.; Yang, H.; Li, S. Effect of Limit-Fed Diets With Different Forage to Concentrate Ratios on Fecal Bacterial and Archaeal Community Composition in Holstein Heifers. Front. Microbiol. 2018, 9, 976. [Google Scholar] [CrossRef]
- Bui, T.P.; Shetty, S.A.; Lagkouvardos, I.; Ritari, J.; Chamlagain, B.; Douillard, F.P.; Paulin, L.; Piironen, V.; Clavel, T.; Plugge, C.M.; et al. Comparative genomics and physiology of the butyrate-producing bacterium Intestinimonas butyriciproducens. Environ. Microbiol. Rep. 2016, 8, 1024–1037. [Google Scholar] [CrossRef]
- Durand, G.; Afouda, P.; Raoult, D.; Dubourg, G. “Intestinimonas massiliensis” sp. nov, a new bacterium isolated from human gut. New Microbes New Infect. 2017, 15, 1–2. [Google Scholar] [CrossRef]
- Purushe, J.; Fouts, D.E.; Morrison, M.; White, B.A.; Mackie, R.I.; Coutinho, P.M.; Henrissat, B.; Nelson, K.E. Comparative genome analysis of Prevotella ruminicola and Prevotella bryantii: Insights into their environmental niche. Microb. Ecol. 2010, 60, 721–729. [Google Scholar] [CrossRef]
- Matsui, H.; Ogata, K.; Tajima, K.; Nakamura, M.; Nagamine, T.; Aminov, R.I.; Benno, Y. Phenotypic Characterization of Polysaccharidases Produced by Four Prevotella Type Strains. Curr. Microbiol. 2000, 41, 45–49. [Google Scholar] [CrossRef]
- Han, X.; Li, B.; Wang, X.; Chen, Y.; Yang, Y. Effect of dietary concentrate to forage ratios on ruminal bacterial and anaerobic fungal populations of cashmere goats. Anaerobe 2019, 59, 118–125. [Google Scholar] [CrossRef]
- Cheng, Y.; Shi, Q.; Sun, R.; Liang, D.; Li, Y.; Li, Y.; Jin, W.; Zhu, W. The biotechnological potential of anaerobic fungi on fiber degradation and methane production. World J. Microbiol. Biotechnol. 2018, 34, 155. [Google Scholar] [CrossRef]
- Haitjema, C.H.; Solomon, K.V.; Henske, J.K.; Theodorou, M.K.; O’Malley, M.A. Anaerobic gut fungi: Advances in isolation, culture, and cellulolytic enzyme discovery for biofuel production. Biotechnol. Bioeng. 2014, 111, 1471–1482. [Google Scholar] [CrossRef]
- Lee, S.M.; Guan, L.L.; Eun, J.S.; Kim, C.H.; Lee, S.J.; Kim, E.T.; Lee, S.S. The effect of anaerobic fungal inoculation on the fermentation characteristics of rice straw silages. J. Appl. Microbiol. 2015, 118, 565–573. [Google Scholar] [CrossRef]
- Denman, S.E.; Nicholson, M.J.; Brookman, J.L.; Theodorou, M.K.; McSweeney, C.S. Detection and monitoring of anaerobic rumen fungi using an ARISA method. Lett. Appl. Microbiol. 2008, 47, 492–499. [Google Scholar] [CrossRef] [PubMed]
- Hooker, C.A.; Hillman, E.T.; Overton, J.C.; Ortiz-Velez, A.; Schacht, M.; Hunnicutt, A.; Mosier, N.S.; Solomon, K.V. Hydrolysis of untreated lignocellulosic feedstock is independent of S-lignin composition in newly classified anaerobic fungal isolate, Piromyces sp. UH3-1. Biotechnol. Biofuels 2018, 11, 293. [Google Scholar] [CrossRef] [PubMed]
- Arasu, M.V.; Jung, M.-W.; Kim, D.H.; Ilavenil, S.; Jane, M.; Park, H.S.; Al-Dhabi, N.A.; Jeon, B.T.; Choi, K.C. Enhancing Nutritional Quality of Silage by Fermentation with Lactobacillus plantarum. Indian. J. Microbiol. 2014, 54, 396–402. [Google Scholar] [CrossRef] [PubMed]
- Simon, A. FastQC: A Quality Control Tool for High Throughput Sequence Data; Babraham Bioinformatics: Cambridge, UK, 2010. [Google Scholar]
- Bushnell, B. BBMap: A Fast, Accurate, Splice-Aware Aligner; Joint Genome Institute: Walnut Creek, CA, USA, 2014. [Google Scholar]
- Trapnell, C.; Pachter, L.; Salzberg, S.L. TopHat: Discovering splice junctions with RNA-Seq. Bioinformatics 2009, 25, 1105–1111. [Google Scholar] [CrossRef]
- R Development Core Team. A Language and Environment for Statistical Computing; Foundation for Statistical Computing: Vienna, Austria, 2020. [Google Scholar]
- Roberts, A.; Trapnell, C.; Donaghey, J.; Rinn, J.L.; Pachter, L. Improving RNA-Seq expression estimates by correcting for fragment bias. Genome Biol. 2011, 12, R22. [Google Scholar] [CrossRef]
- Best, D.J.; Roberts, D.E. The Upper Tail Probabilities of Spearman’s RHO. J. R. Stat. Soc. Ser. C Appl. Stat. 1975, 24, 377–379. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| S.Nos | Gene_Symbol | Gene Name | Fold | p Values |
|---|---|---|---|---|
| 1. | ADM | Adrenomedullin | 0.396 | 0.017 |
| 2. | AHCY | Adenosylhomocysteinase | 1.780 | 0.010 |
| 3. | AIF1 | Allograft inflammatory factor 1 | 1.806 | 0.040 |
| 4. | BLA-DQB | MHC class II antigen | 4.589 | 0.014 |
| 5. | CD14 | CD14 molecule | 1.576 | 0.046 |
| 6. | CNN2 | Calponin 2 | 1.548 | 0.034 |
| 7. | COL8A1 | Collagen type VIII alpha 1 chain | 0.381 | 0.007 |
| 8. | CORO1A | Coronin 1A | 1.648 | 0.047 |
| 9. | CST6 | Cystatin E/M | 0.604 | 0.024 |
| 10. | CYB5D2 | Cytochrome b5 domain containing 2 | 0.494 | 0.040 |
| 11. | DDIT4 | DNA damage inducible transcript 4 | 0.616 | 0.036 |
| 12. | EGR1 | Early growth response 1 | 3.915 | 0.039 |
| 13. | ENTPD2 | Ectonucleoside triphosphate diphosphohydrolase 2 | 1.560 | 0.036 |
| 14. | FCER1G | Fc epsilon receptor Ig | 1.734 | 0.045 |
| 15. | FN1 | Fibronectin 1 | 1.617 | 0.007 |
| 16. | FOS | Fos proto-oncogene, AP-1 transcription factor subunit | 4.819 | 0.013 |
| 17. | FOSB | Fosb proto-oncogene, AP-1 transcription factor | 11.001 | 0.007 |
| 18. | GLIPR2 | GLI pathogenesis related 2 | 1.702 | 0.049 |
| 19. | GPX3 | Glutathione peroxidase 3 | 0.339 | 0.028 |
| 20. | HPRT1 | Hypoxanthinephosphoribosyltransferase 1 | 1.611 | 0.012 |
| 21. | IER2 | Immediate early response 2 | 1.881 | 0.009 |
| 22. | ITGB5 | Integrin subunit beta 5 | 0.641 | 0.000 |
| 23. | JUNB | Junb proto-oncogene, AP-1 transcription factor | 2.319 | 0.017 |
| 24. | LOC112445030 | Four and a half LIM domains protein 1- | 0.610 | 0.037 |
| 25. | LOC786948 | Tryptase-2-like | 1.811 | 0.008 |
| 26. | MXRA5 | Matrix remodeling associated 5 | 3.365 | 0.014 |
| 27. | NDUFA11 | NADH:ubiquinone oxidoreductase | 1.609 | 0.020 |
| 28. | NR4A1 | Nuclear receptor subfamily 4 group A member 1 | 3.575 | 0.010 |
| 29. | PI16 | Peptidase inhibitor 16 | 0.268 | 0.047 |
| 30. | PTI | Pancreatic trypsin inhibitor | 1.974 | 0.007 |
| 31. | SLC2A3 | Solute carrier family 2 member 3 | 1.696 | 0.040 |
| 32. | SOCS3 | Suppressor of cytokine signaling 3 | 2.595 | 0.037 |
| 33. | TIMP1 | TIMP metallopeptidase inhibitor 1 | 2.499 | 0.014 |
| 34. | ZFP36 | ZFP36 ring finger protein | 2.106 | 0.001 |
| S. Nos | Gene_Symbol | Gene Name | Fold | p Values |
|---|---|---|---|---|
| 1. | CL2L1 | Bcl2 like 1 | 1.580 | 0.033 |
| 2. | BOLA | Major histocompatibility complex, class i, A | 1.996 | 0.012 |
| 3. | CACNB1 | Calcium voltage-gated channel auxiliary subunit beta 1 | 0.603 | 0.025 |
| 4. | CREM | Camp responsive element modulator | 1.697 | 0.043 |
| 5. | EPM2A | Epm2a glucan phosphatase, laforin | 1.535 | 0.024 |
| 6. | ETS2 | Ets proto-oncogene 2, transcription factor | 1.950 | 0.030 |
| 7. | GADD45A | Growth arrest and DNA damage inducible alpha | 1.804 | 0.028 |
| 8. | IGF2 | Insulin like growth factor 2 | 1.653 | 0.035 |
| 9. | KLF10 | Klf transcription factor 10 | 1.536 | 0.019 |
| 10. | LIPE | Lipase e, hormone sensitive type | 2.198 | 0.007 |
| 11. | LOC100848815 | Sla class ii histocompatibility antigen- d alpha chain-like | 2.872 | 0.042 |
| 12. | LOC112444389 | Small nucleolar RNA SNORA32 | 27.484 | 0.013 |
| 13. | LOC112445242 | Small nucleolar RNA SNORA70 | 0.024 | 0.034 |
| 14. | LUM | Lumican | 0.480 | 0.031 |
| 15. | MAFB | Maf bzip transcription factor B | 1.711 | 0.036 |
| 16. | MAX | Myc associated factor x | 1.923 | 0.039 |
| 17. | MRPS18A | Mitochondrial ribosomal protein s18a | 2.114 | 0.030 |
| 18. | MUSTN1 | Musculoskeletal, embryonic nuclear protein 1 | 0.580 | 0.048 |
| 19. | NFKBIA | Nfkb inhibitor alpha | 1.505 | 0.032 |
| 20. | NT5C2 | 5′-nucleotidase, cytosolic ii | 0.638 | 0.042 |
| 21. | PDK4 | Pyruvate dehydrogenase kinase 4 | 2.733 | 0.005 |
| 22. | RCN1 | Reticulocalbin 1 | 0.519 | 0.009 |
| 23. | SMPX | Small muscle protein x-linked | 0.663 | 0.043 |
| 24. | SUN2 | Sad1 and unc84 domain containing 2 | 1.576 | 0.040 |
| 25. | TINAGL1 | Tubulointerstitial nephritis antigen like 1 | 1.607 | 0.001 |
| 26. | ULBP13 | Ul16 binding protein 13 | 19.040 | 0.001 |
| 27. | YTHDF1 | Yth n6-methyladenosine RNA binding protein f1 | 1.597 | 0.035 |
| 28. | ZNF703 | Zinc finger protein 703 | 1.649 | 0.008 |
| S.Nos | Gene_Symbol | Gene Name | Fold | p Values |
|---|---|---|---|---|
| 1. | ABHD6 | Abhydrolase domain containing 6, acylglycerol lipase | 2.322 | 0.035 |
| 2. | ACP5 | Acid phosphatase 5, tartrate resistant | 0.623 | 0.022 |
| 3. | APMAP | Adipocyte plasma membrane associated protein | 1.776 | 0.012 |
| 4. | BoLA | Major histocompatibility complex, class I, antigen | 2.392 | 0.003 |
| 5. | BoLA | Major histocompatibility complex, class I, A | 0.585 | 0.025 |
| 6. | CBFB | Core-binding factor subunit beta | 1.860 | 0.036 |
| 7. | CFH | Complement factor H | 0.642 | 0.036 |
| 8. | COG3 | Component of oligomeric golgi complex 3 | 1.828 | 0.010 |
| 9. | DDIT4 | DNA damage inducible transcript 4 | 0.526 | 0.009 |
| 10. | DUSP1 | Dual specificity phosphatase 1 | 0.662 | 0.016 |
| 11. | FOLR3 | Folate receptor 3 | 0.365 | 0.027 |
| 12. | GK | Glycerol kinase | 1.636 | 0.006 |
| 13. | JUND | Jund proto-oncogene, AP-1 transcription factor | 1.929 | 0.042 |
| 14. | KLF2 | KLF transcription factor 2 | 1.898 | 0.029 |
| 15. | KYAT1 | Kynurenine aminotransferase 1 | 0.303 | 0.045 |
| 16. | LOC100335748 | Uncharacterized LOC100335748 | 0.024 | 0.036 |
| 17. | LOC100336868 | Complement factor H | 3.347 | 0.035 |
| 18. | LOC112441481 | Glycine N-phenylacetyltransferase-like | 0.505 | 0.037 |
| 19. | LOC112443024 | Uncharacterized | 2.021 | 0.033 |
| 20. | LOC112447597 | U6 spliceosomal RNA | 0.016 | 0.000 |
| 21. | LOC504773 | Regakine 1 | 0.578 | 0.038 |
| 22. | LOC614091 | Class I histocompatibility antigen, alpha chain BL3 | 0.619 | 0.016 |
| 23. | MAP2K6 | Mitogen-activated protein kinase kinase 6 | 0.621 | 0.009 |
| 24. | MOCS2 | Molybdenum cofactor synthesis 2 | 0.636 | 0.007 |
| 25. | PPP1R35 | Protein phosphatase 1 regulatory subunit 35 | 1.596 | 0.003 |
| 26. | PRODH2 | Proline dehydrogenase 2 | 1.924 | 0.017 |
| 27. | RILPL2 | Rab interacting lysosomal protein like 2 | 0.629 | 0.023 |
| 28. | SAA2 | Serum amyloid A2 | 0.597 | 0.038 |
| 29. | SAA4 | Serum amyloid A4, constitutive | 0.550 | 0.003 |
| 30. | SERPINB6 | Serpin peptidase inhibitor, clade B (ovalbumin), member 6 | 0.620 | 0.047 |
| 31. | SIK2 | Salt inducible kinase 2 | 1.747 | 0.027 |
| 32. | SQLE | Squalene epoxidase | 1.598 | 0.034 |
| 33. | TMEM45A | Transmembrane protein 45A | 0.002 | |
| 34. | TYROBP | Transmembrane immune signaling adaptor TYROBP | 0.418 | 0.011 |
| 35. | UBC | Ubiquitin C | 1.678 | 0.015 |
| 36. | WWC1 | WW and C2 domain containing 1 | 0.652 | 0.016 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Jung, J.S.; Kim, D.; Singaravelu, A.; Soundharrajan, I.; Choi, K.C. Dietary Fermentation with Lactobacillus sp. and Bacillus sp. Modulates Rumen Transcriptomic and Microbiota Profiles in Bos taurus. Int. J. Mol. Sci. 2025, 26, 6816. https://doi.org/10.3390/ijms26146816
Jung JS, Kim D, Singaravelu A, Soundharrajan I, Choi KC. Dietary Fermentation with Lactobacillus sp. and Bacillus sp. Modulates Rumen Transcriptomic and Microbiota Profiles in Bos taurus. International Journal of Molecular Sciences. 2025; 26(14):6816. https://doi.org/10.3390/ijms26146816
Chicago/Turabian StyleJung, Jeong Sung, Dahye Kim, Anand Singaravelu, Ilavenil Soundharrajan, and Ki Choon Choi. 2025. "Dietary Fermentation with Lactobacillus sp. and Bacillus sp. Modulates Rumen Transcriptomic and Microbiota Profiles in Bos taurus" International Journal of Molecular Sciences 26, no. 14: 6816. https://doi.org/10.3390/ijms26146816
APA StyleJung, J. S., Kim, D., Singaravelu, A., Soundharrajan, I., & Choi, K. C. (2025). Dietary Fermentation with Lactobacillus sp. and Bacillus sp. Modulates Rumen Transcriptomic and Microbiota Profiles in Bos taurus. International Journal of Molecular Sciences, 26(14), 6816. https://doi.org/10.3390/ijms26146816