

Leucine Zipper Downregulated in Cancer-1 Interacts with Clathrin Adaptors to Control Epidermal Growth Factor Receptor (EGFR) Internalization and Gefitinib Response in EGFR-Mutated Non-Small Cell Lung Cancer


Abstract

1. Introduction
2. Results
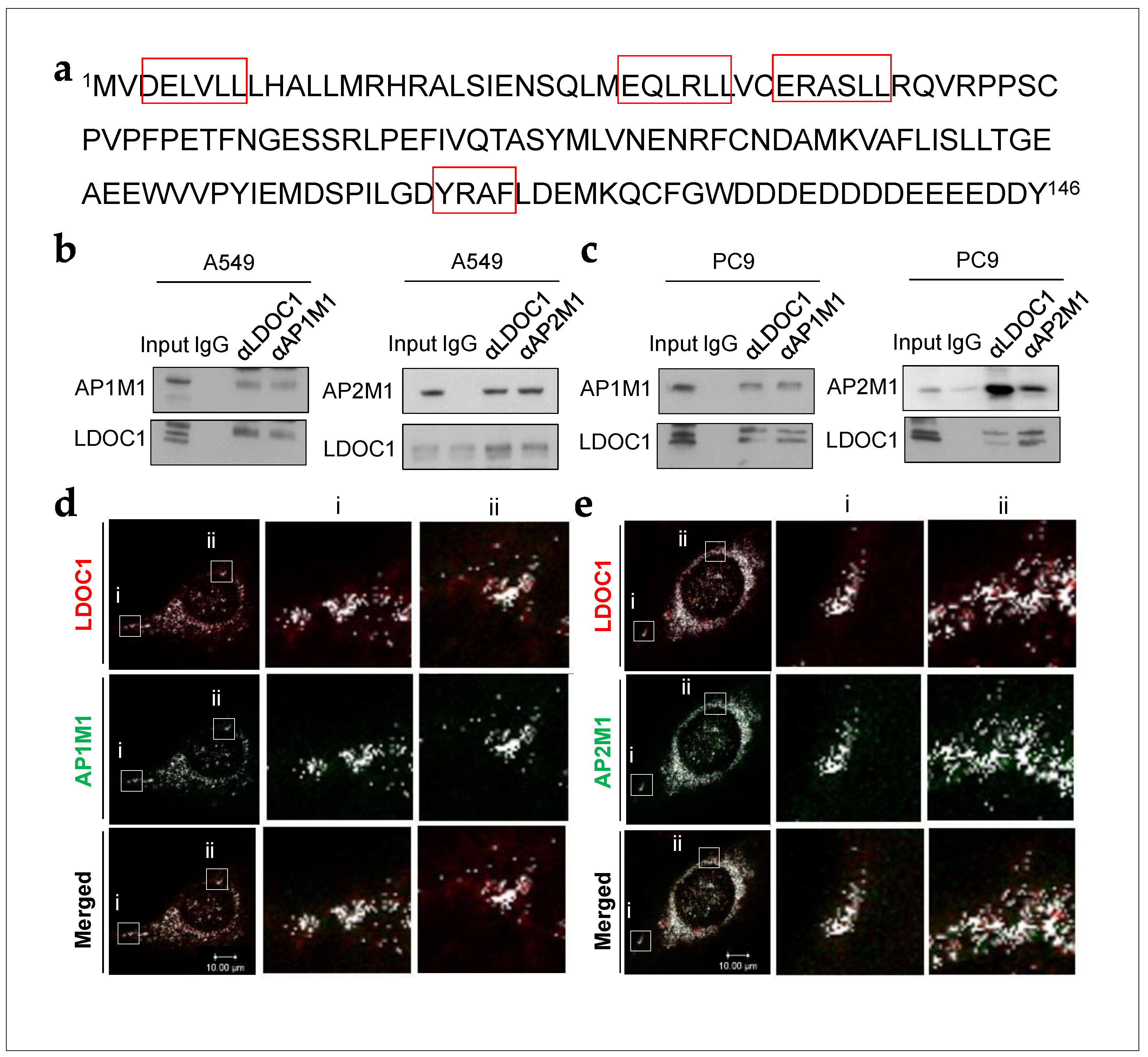
2.1. LDOC1 Bound to Subunits of Clathrin Adaptor Complexes in NSCLC Cells
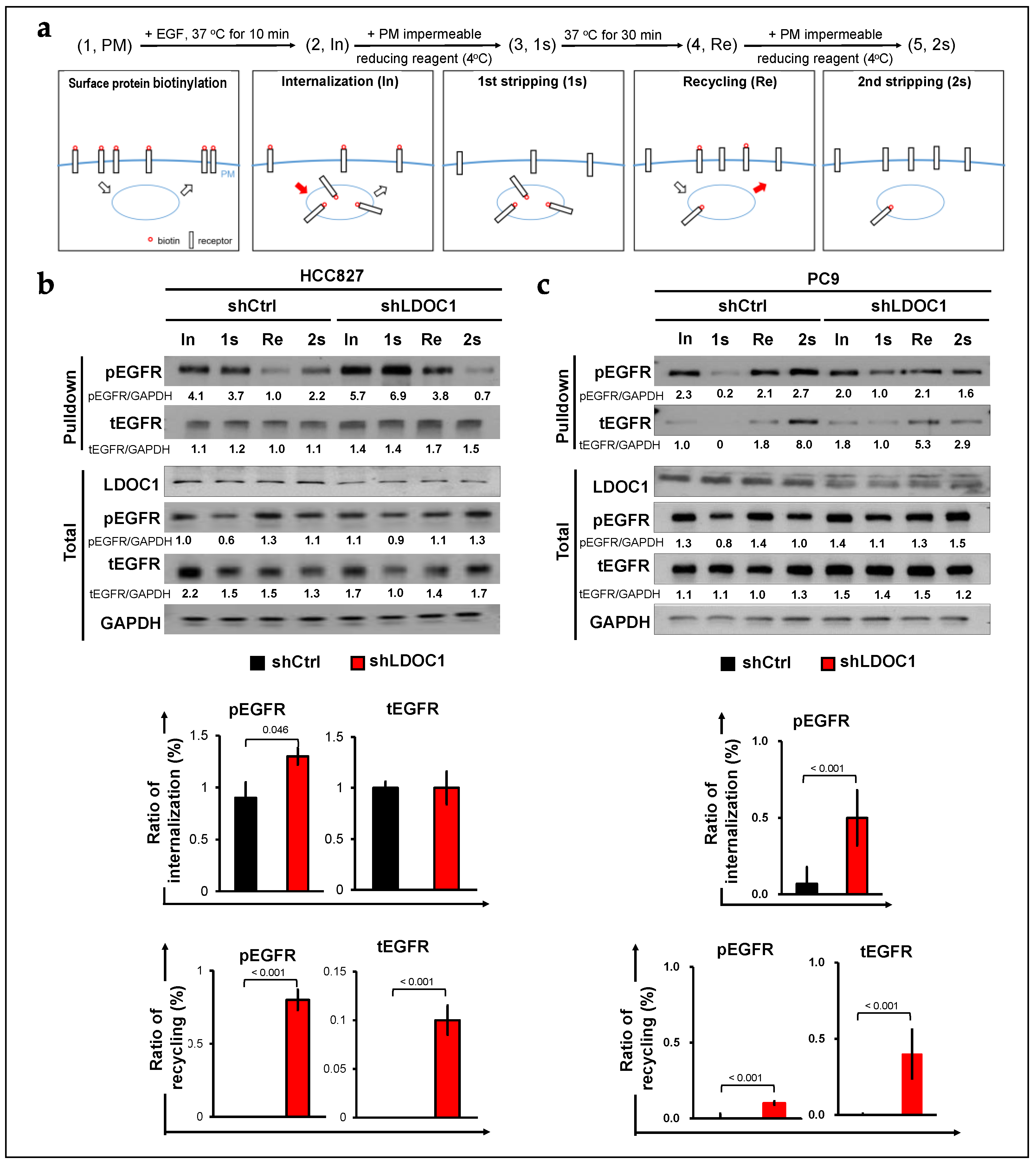
2.2. LDOC1 Depletion Supports Either Internalization or Recycling of EGFR in EGFRM NSCLC Cells
2.3. Depletion of LDOC1 Leads to Upregulation of the Potential EGFR-Interacting RTKs
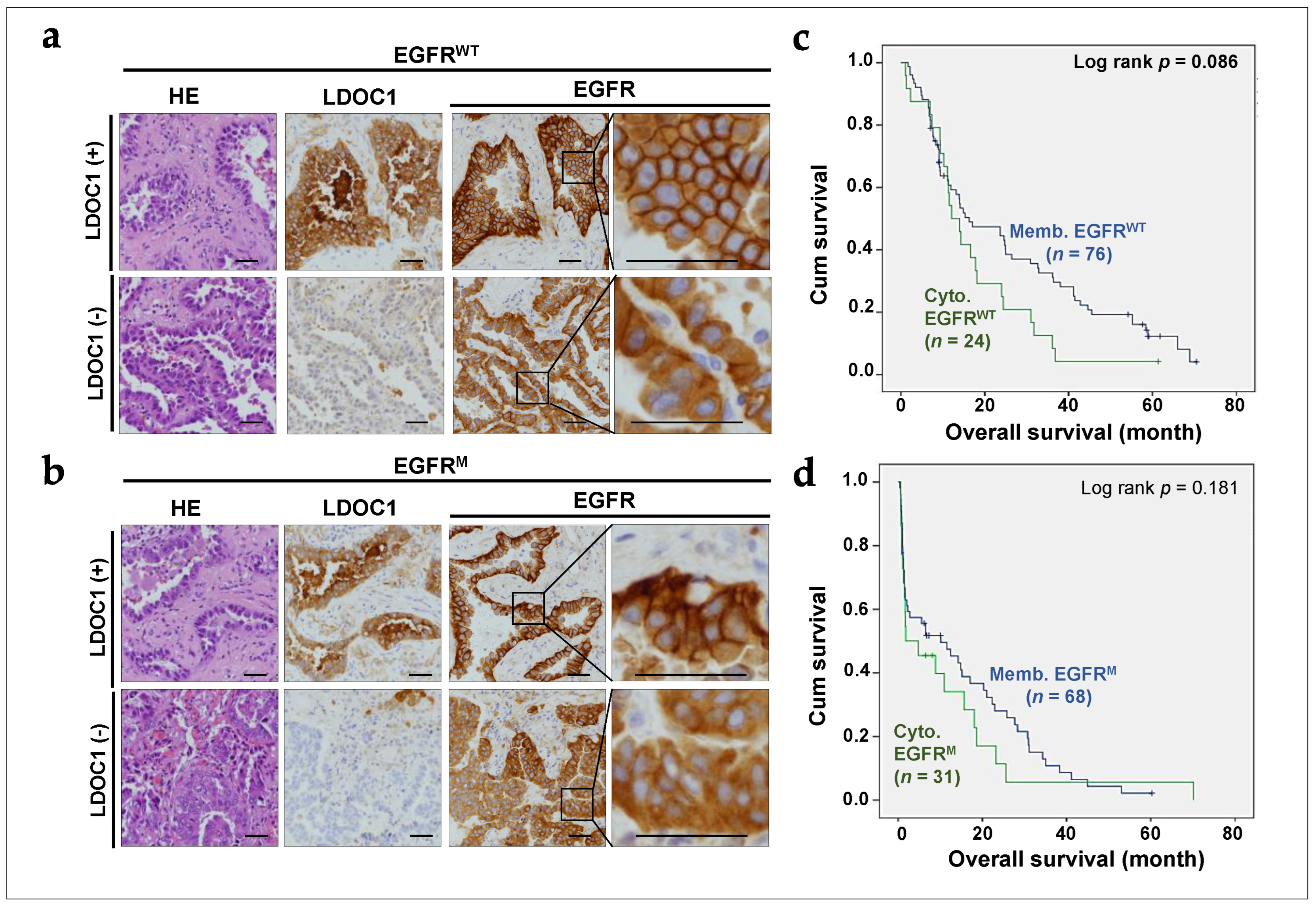
2.4. LDOC1 Downregulation Associated with Cytoplasmic EGFR Expression in NSCLC Tumors
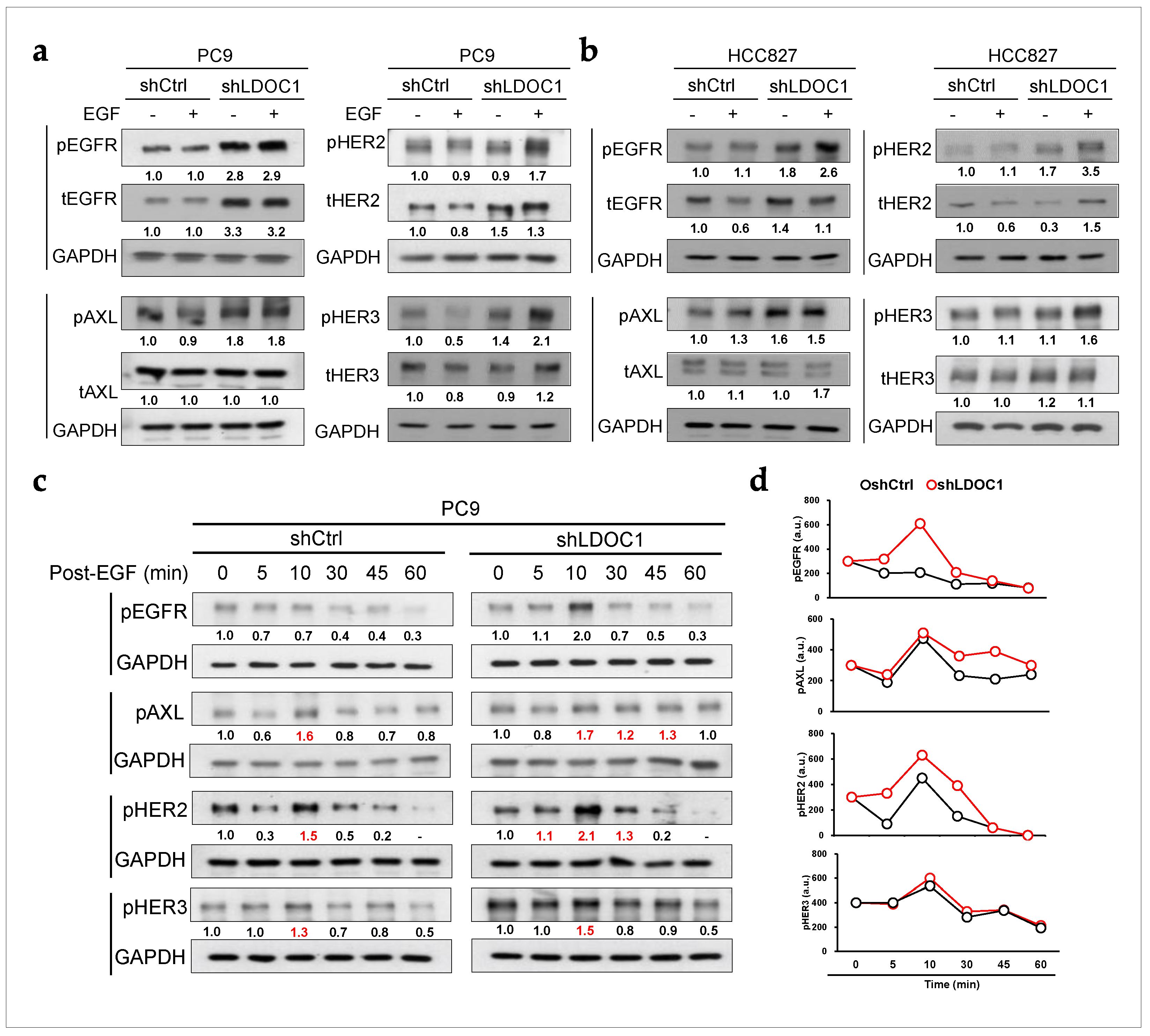
2.5. LDOC1 Depletion Enhances and Sustains Prolonged Activation of EGFR, AXL, and HER2 in EGFRM Cells
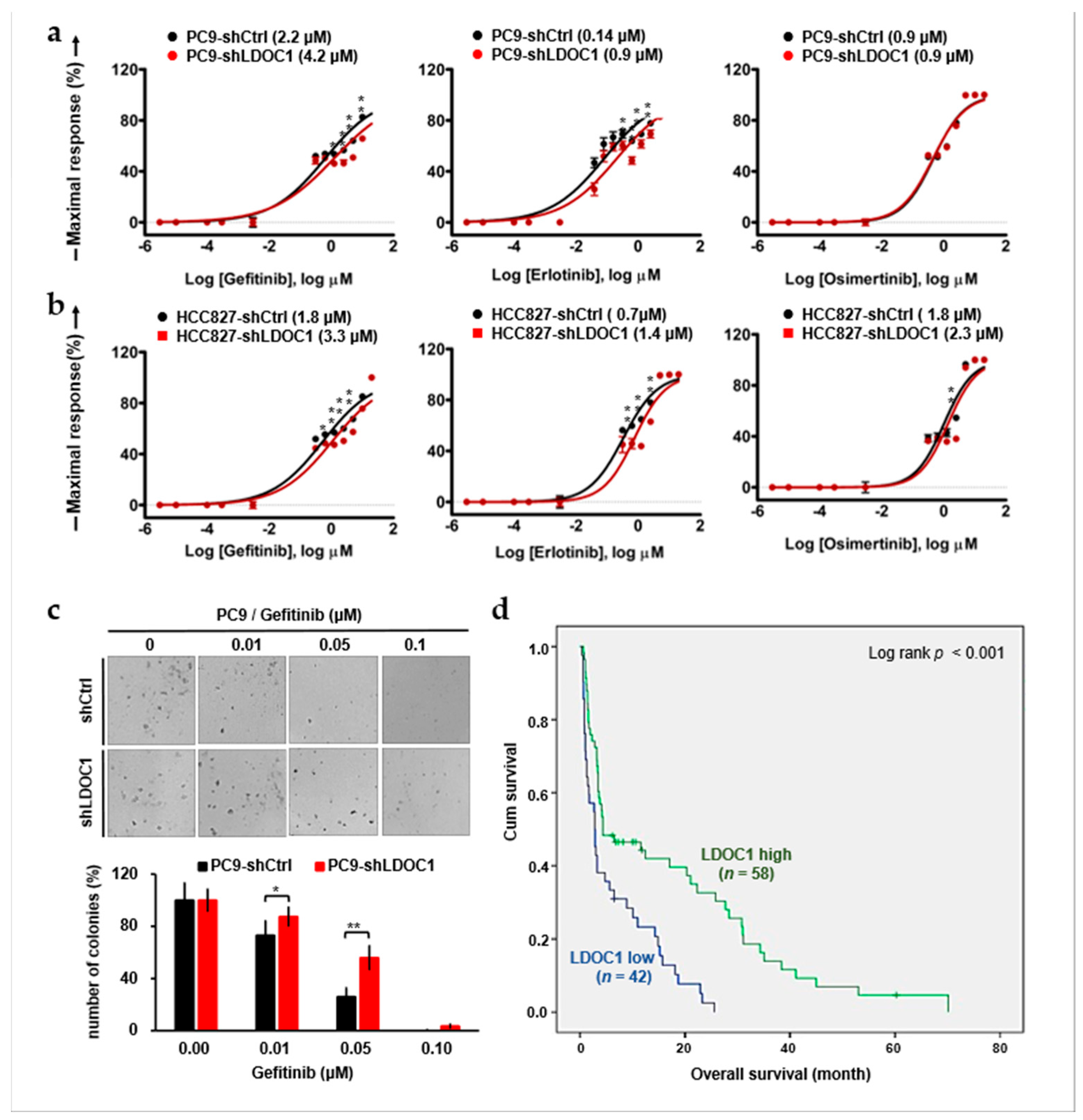
2.6. LDOC1 Downregulation Reduces Sensitivity to First-Generation EGFR-TKIs and Predicts Worse Outcomes in Patients with EGFRM NSCLC Who Are Treated with Gefitinib
3. Discussion
4. Conclusions
5. Materials and Methods
5.1. Patients and Tumor Biopsies
5.2. Cell Culture, EGF Stimulation, and Reagents
5.3. Plasmid Construction, Transfection, and Infection
5.4. Western Blot Analysis and Antibodies
5.5. Coimmunoprecipitation
5.6. Double Immunofluorescence Staining
5.7. Endocytosis Assay for EGFR Internalization and Recycling
5.8. Immunohistochemistry (IHC) Staining and Hematoxylin and Eosin (HE) Staining
5.9. MTT Assay for EGFR-TKI Sensitivity
5.10. Colony Formation Assay for Gefitinib Sensitivity
5.11. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Visbal, A.L.; Williams, B.A.; Nichols, F.C., 3rd; Marks, R.S.; Jett, J.R.; Aubry, M.C.; Edell, E.S.; Wampfler, J.A.; Molina, J.R.; Yang, P. Gender differences in non-small-cell lung cancer survival: An analysis of 4,618 patients diagnosed between 1997 and 2002. Ann. Thorac. Surg. 2004, 78, 209–215; discussion 215. [Google Scholar] [CrossRef] [PubMed]
- Paez, J.G.; Janne, P.A.; Lee, J.C.; Tracy, S.; Greulich, H.; Gabriel, S.; Herman, P.; Kaye, F.J.; Lindeman, N.; Boggon, T.J.; et al. EGFR mutations in lung cancer: Correlation with clinical response to gefitinib therapy. Science 2004, 304, 1497–1500. [Google Scholar] [CrossRef] [PubMed]
- Rosell, R.; Moran, T.; Queralt, C.; Porta, R.; Cardenal, F.; Camps, C.; Majem, M.; Lopez-Vivanco, G.; Isla, D.; Provencio, M.; et al. Screening for epidermal growth factor receptor mutations in lung cancer. N. Engl. J. Med. 2009, 361, 958–967. [Google Scholar] [CrossRef] [PubMed]
- Zhou, C.; Wu, Y.L.; Chen, G.; Feng, J.; Liu, X.Q.; Wang, C.; Zhang, S.; Wang, J.; Zhou, S.; Ren, S.; et al. Erlotinib versus chemotherapy as first-line treatment for patients with advanced EGFR mutation-positive non-small-cell lung cancer (OPTIMAL, CTONG-0802): A multicentre, open-label, randomised, phase 3 study. Lancet Oncol. 2011, 12, 735–742. [Google Scholar] [CrossRef] [PubMed]
- Linardou, H.; Dahabreh, I.J.; Bafaloukos, D.; Kosmidis, P.; Murray, S. Somatic EGFR mutations and efficacy of tyrosine kinase inhibitors in NSCLC. Nat. Rev. Clin. Oncol. 2009, 6, 352–366. [Google Scholar] [CrossRef]
- Lynch, T.J.; Bell, D.W.; Sordella, R.; Gurubhagavatula, S.; Okimoto, R.A.; Brannigan, B.W.; Harris, P.L.; Haserlat, S.M.; Supko, J.G.; Haluska, F.G.; et al. Activating mutations in the epidermal growth factor receptor underlying responsiveness of non-small-cell lung cancer to gefitinib. N. Engl. J. Med. 2004, 350, 2129–2139. [Google Scholar] [CrossRef]
- Sharma, S.V.; Bell, D.W.; Settleman, J.; Haber, D.A. Epidermal growth factor receptor mutations in lung cancer. Nat. Rev. Cancer 2007, 7, 169–181. [Google Scholar] [CrossRef]
- Bartholomew, C.; Eastlake, L.; Dunn, P.; Yiannakis, D. EGFR targeted therapy in lung cancer; an evolving story. Respir. Med. Case Rep. 2017, 20, 137–140. [Google Scholar] [CrossRef]
- Yun, C.H.; Boggon, T.J.; Li, Y.; Woo, M.S.; Greulich, H.; Meyerson, M.; Eck, M.J. Structures of lung cancer-derived EGFR mutants and inhibitor complexes: Mechanism of activation and insights into differential inhibitor sensitivity. Cancer Cell 2007, 11, 217–227. [Google Scholar] [CrossRef]
- Kelly, R.J.; Shepherd, F.A.; Krivoshik, A.; Jie, F.; Horn, L. A phase III, randomized, open-label study of ASP8273 versus erlotinib or gefitinib in patients with advanced stage IIIB/IV non-small-cell lung cancer. Ann. Oncol. 2019, 30, 1127–1133. [Google Scholar] [CrossRef]
- Hong, S.; Gao, F.; Fu, S.; Wang, Y.; Fang, W.; Huang, Y.; Zhang, L. Concomitant Genetic Alterations With Response to Treatment and Epidermal Growth Factor Receptor Tyrosine Kinase Inhibitors in Patients With EGFR-Mutant Advanced Non-Small Cell Lung Cancer. JAMA Oncol. 2018, 4, 739–742. [Google Scholar] [CrossRef]
- Wu, S.G.; Shih, J.Y. Management of acquired resistance to EGFR TKI-targeted therapy in advanced non-small cell lung cancer. Mol. Cancer 2018, 17, 38. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Wang, B.; Chu, H.; Yao, Y. Intrinsic resistance to EGFR tyrosine kinase inhibitors in advanced non-small-cell lung cancer with activating EGFR mutations. Onco Targets Ther. 2016, 9, 3711–3726. [Google Scholar] [CrossRef] [PubMed]
- Jeong, I.; Song, J.; Bae, S.Y.; Lee, S.K. Overcoming the Intrinsic Gefitinib-resistance via Downregulation of AXL in Non-small Cell Lung Cancer. J. Cancer Prev. 2019, 24, 217–223. [Google Scholar] [CrossRef]
- Zhang, Z.; Lee, J.C.; Lin, L.; Olivas, V.; Au, V.; LaFramboise, T.; Abdel-Rahman, M.; Wang, X.; Levine, A.D.; Rho, J.K.; et al. Activation of the AXL kinase causes resistance to EGFR-targeted therapy in lung cancer. Nat. Genet. 2012, 44, 852–860. [Google Scholar] [CrossRef]
- Noronha, A.; Belugali Nataraj, N.; Lee, J.S.; Zhitomirsky, B.; Oren, Y.; Oster, S.; Lindzen, M.; Mukherjee, S.; Will, R.; Ghosh, S.; et al. AXL and Error-Prone DNA Replication Confer Drug Resistance and Offer Strategies to Treat EGFR-Mutant Lung Cancer. Cancer Discov. 2022, 12, 2666–2683. [Google Scholar] [CrossRef] [PubMed]
- Meyer, A.S.; Miller, M.A.; Gertler, F.B.; Lauffenburger, D.A. The receptor AXL diversifies EGFR signaling and limits the response to EGFR-targeted inhibitors in triple-negative breast cancer cells. Sci. Signal 2013, 6, ra66. [Google Scholar] [CrossRef] [PubMed]
- Vouri, M.; Croucher, D.R.; Kennedy, S.P.; An, Q.; Pilkington, G.J.; Hafizi, S. Axl-EGFR receptor tyrosine kinase hetero-interaction provides EGFR with access to pro-invasive signalling in cancer cells. Oncogenesis 2016, 5, e266. [Google Scholar] [CrossRef]
- Takezawa, K.; Pirazzoli, V.; Arcila, M.E.; Nebhan, C.A.; Song, X.; de Stanchina, E.; Ohashi, K.; Janjigian, Y.Y.; Spitzler, P.J.; Melnick, M.A.; et al. HER2 amplification: A potential mechanism of acquired resistance to EGFR inhibition in EGFR-mutant lung cancers that lack the second-site EGFRT790M mutation. Cancer Discov. 2012, 2, 922–933. [Google Scholar] [CrossRef]
- Ortiz-Cuaran, S.; Scheffler, M.; Plenker, D.; Dahmen, L.; Scheel, A.H.; Fernandez-Cuesta, L.; Meder, L.; Lovly, C.M.; Persigehl, T.; Merkelbach-Bruse, S.; et al. Heterogeneous Mechanisms of Primary and Acquired Resistance to Third-Generation EGFR Inhibitors. Clin. Cancer Res. 2016, 22, 4837–4847. [Google Scholar] [CrossRef]
- Yonesaka, K.; Tanizaki, J.; Maenishi, O.; Haratani, K.; Kawakami, H.; Tanaka, K.; Hayashi, H.; Sakai, K.; Chiba, Y.; Tsuya, A.; et al. HER3 Augmentation via Blockade of EGFR/AKT Signaling Enhances Anticancer Activity of HER3-Targeting Patritumab Deruxtecan in EGFR-Mutated Non-Small Cell Lung Cancer. Clin. Cancer Res. 2022, 28, 390–403. [Google Scholar] [CrossRef] [PubMed]
- Scaltriti, M.; Baselga, J. The epidermal growth factor receptor pathway: A model for targeted therapy. Clin. Cancer Res. 2006, 12, 5268–5272. [Google Scholar] [CrossRef] [PubMed]
- Sordella, R.; Bell, D.W.; Haber, D.A.; Settleman, J. Gefitinib-sensitizing EGFR mutations in lung cancer activate anti-apoptotic pathways. Science 2004, 305, 1163–1167. [Google Scholar] [CrossRef]
- Lazzara, M.J.; Lane, K.; Chan, R.; Jasper, P.J.; Yaffe, M.B.; Sorger, P.K.; Jacks, T.; Neel, B.G.; Lauffenburger, D.A. Impaired SHP2-mediated extracellular signal-regulated kinase activation contributes to gefitinib sensitivity of lung cancer cells with epidermal growth factor receptor-activating mutations. Cancer Res. 2010, 70, 3843–3850. [Google Scholar] [CrossRef]
- Burke, P.; Schooler, K.; Wiley, H.S. Regulation of epidermal growth factor receptor signaling by endocytosis and intracellular trafficking. Mol. Biol. Cell 2001, 12, 1897–1910. [Google Scholar] [CrossRef] [PubMed]
- Tomas, A.; Futter, C.E.; Eden, E.R. EGF receptor trafficking: Consequences for signaling and cancer. Trends Cell Biol. 2014, 24, 26–34. [Google Scholar] [CrossRef]
- von Zastrow, M.; Sorkin, A. Signaling on the endocytic pathway. Curr. Opin. Cell Biol. 2007, 19, 436–445. [Google Scholar] [CrossRef]
- Sigismund, S.; Argenzio, E.; Tosoni, D.; Cavallaro, E.; Polo, S.; Di Fiore, P.P. Clathrin-mediated internalization is essential for sustained EGFR signaling but dispensable for degradation. Dev. Cell 2008, 15, 209–219. [Google Scholar] [CrossRef]
- Uemura, T.; Suzuki, T.; Dohmae, N.; Waguri, S. Clathrin adapters AP-1 and GGA2 support expression of epidermal growth factor receptor for cell growth. Oncogenesis 2021, 10, 80. [Google Scholar] [CrossRef]
- Partlow, E.A.; Cannon, K.S.; Hollopeter, G.; Baker, R.W. Structural basis of an endocytic checkpoint that primes the AP2 clathrin adaptor for cargo internalization. Nat. Struct. Mol. Biol. 2022, 29, 339–347. [Google Scholar] [CrossRef]
- Pascolutti, R.; Algisi, V.; Conte, A.; Raimondi, A.; Pasham, M.; Upadhyayula, S.; Gaudin, R.; Maritzen, T.; Barbieri, E.; Caldieri, G.; et al. Molecularly Distinct Clathrin-Coated Pits Differentially Impact EGFR Fate and Signaling. Cell Rep. 2019, 27, 3049–3061.e6. [Google Scholar] [CrossRef] [PubMed]
- Kadlecova, Z.; Spielman, S.J.; Loerke, D.; Mohanakrishnan, A.; Reed, D.K.; Schmid, S.L. Regulation of clathrin-mediated endocytosis by hierarchical allosteric activation of AP2. J. Cell Biol. 2017, 216, 167–179. [Google Scholar] [CrossRef] [PubMed]
- Wu, X.; Zhao, X.; Puertollano, R.; Bonifacino, J.S.; Eisenberg, E.; Greene, L.E. Adaptor and clathrin exchange at the plasma membrane and trans-Golgi network. Mol. Biol. Cell 2003, 14, 516–528. [Google Scholar] [CrossRef] [PubMed]
- Kelly, B.T.; McCoy, A.J.; Spate, K.; Miller, S.E.; Evans, P.R.; Honing, S.; Owen, D.J. A structural explanation for the binding of endocytic dileucine motifs by the AP2 complex. Nature 2008, 456, 976–979. [Google Scholar] [CrossRef]
- Bonifacino, J.S.; Traub, L.M. Signals for sorting of transmembrane proteins to endosomes and lysosomes. Annu. Rev. Biochem. 2003, 72, 395–447. [Google Scholar] [CrossRef]
- Dittmann, K.; Mayer, C.; Kehlbach, R.; Rodemann, H.P. Radiation-induced caveolin-1 associated EGFR internalization is linked with nuclear EGFR transport and activation of DNA-PK. Mol. Cancer 2008, 7, 69. [Google Scholar] [CrossRef]
- Berger, C.; Madshus, I.H.; Stang, E. Cetuximab in combination with anti-human IgG antibodies efficiently down-regulates the EGF receptor by macropinocytosis. Exp. Cell Res. 2012, 318, 2578–2591. [Google Scholar] [CrossRef]
- Nagasaki, K.; Manabe, T.; Hanzawa, H.; Maass, N.; Tsukada, T.; Yamaguchi, K. Identification of a novel gene, LDOC1, down-regulated in cancer cell lines. Cancer Lett. 1999, 140, 227–234. [Google Scholar] [CrossRef]
- Lee, C.H.; Wong, T.S.; Chan, J.Y.; Lu, S.C.; Lin, P.; Cheng, A.J.; Chen, Y.J.; Chang, J.S.; Hsiao, S.H.; Leu, Y.W.; et al. Epigenetic regulation of the X-linked tumour suppressors BEX1 and LDOC1 in oral squamous cell carcinoma. J. Pathol. 2013, 230, 298–309. [Google Scholar] [CrossRef]
- Lee, C.H.; Yang, J.R.; Chen, C.Y.; Tsai, M.H.; Hung, P.F.; Chen, S.J.; Chiang, S.L.; Chang, H.; Lin, P. Novel STAT3 Inhibitor LDOC1 Targets Phospho-JAK2 for Degradation by Interacting with LNX1 and Regulates the Aggressiveness of Lung Cancer. Cancers 2019, 11, 63. [Google Scholar] [CrossRef]
- Notenboom, V.; Hibbert, R.G.; van Rossum-Fikkert, S.E.; Olsen, J.V.; Mann, M.; Sixma, T.K. Functional characterization of Rad18 domains for Rad6, ubiquitin, DNA binding and PCNA modification. Nucleic Acids Res. 2007, 35, 5819–5830. [Google Scholar] [CrossRef] [PubMed]
- Floc’h, N.; Lim, S.; Bickerton, S.; Ahmed, A.; Orme, J.; Urosevic, J.; Martin, M.J.; Cross, D.A.E.; Cho, B.C.; Smith, P.D. Osimertinib, an Irreversible Next-Generation EGFR Tyrosine Kinase Inhibitor, Exerts Antitumor Activity in Various Preclinical NSCLC Models Harboring the Uncommon EGFR Mutations G719X or L861Q or S768I. Mol. Cancer Ther. 2020, 19, 2298–2307. [Google Scholar] [CrossRef] [PubMed]
- Fraser, J.; Simpson, J.; Fontana, R.; Kishi-Itakura, C.; Ktistakis, N.T.; Gammoh, N. Targeting of early endosomes by autophagy facilitates EGFR recycling and signalling. EMBO Rep. 2019, 20, e47734. [Google Scholar] [CrossRef] [PubMed]
- Hsu, L.H.; Liu, K.J.; Tsai, M.F.; Wu, C.R.; Feng, A.C.; Chu, N.M.; Kao, S.H. Estrogen adversely affects the prognosis of patients with lung adenocarcinoma. Cancer Sci. 2015, 106, 51–59. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| No. (%) | ||||
|---|---|---|---|---|
| LDOC1 High Expression | LDOC1 Low Expression | |||
| Characteristics | No. | n = 55 | n = 45 | p Value |
| Age | 0.933 | |||
| ≤75 years | 36 | 20 (36.4) | 16 (35.6) | |
| >75 years | 64 | 35 (63.6) | 29 (64.4) | |
| Gender | 0.841 | |||
| Male | 50 | 27 (49.1) | 23 (51.1) | |
| Female | 50 | 28 (50.9) | 22 (48.9) | |
| Stage | 0.888 | |||
| IIIB + IIIC | 15 | 8 (14.5) | 7 (15.6) | |
| IVA + IVB | 85 | 47 (85.5) | 38 (84.4) | |
| EGFR expression | <0.001 | |||
| Membranous | 76 | 50 (90.9) | 26 (57.8) | |
| Cytoplasmic | 24 | 5 (9.1) | 19 (42.2) | |
| No. (%) | ||||
|---|---|---|---|---|
| LDOC1 High Expression | LDOC1 Low Expression | |||
| Characteristics | No. | n = 58 | n = 42 | p Value |
| Age | 0.276 | |||
| ≤75 years | 54 | 34 (58.6) | 20 (47.6) | |
| >75 years | 46 | 24 (41.4) | 22 (52.4) | |
| Gender | 0.714 | |||
| Male | 45 | 27 (46.6) | 18 (42.9) | |
| Female | 55 | 31 (53.4) | 24 (57.1) | |
| Stage | 0.177 | |||
| IIIB + IIIC | 18 | 13 (22.4) | 5 (11.9) | |
| IVA + IVB | 82 | 45 (77.6) | 37 (88.1) | |
| EGFR expression a | <0.001 | |||
| Membranous | 68 | 51 (87.9) | 17 (40.5) | |
| Cytoplasmic | 31 | 7 (12.1) | 24 (59.5) | |
| Univariate Analysis | Multivariate Analysis | ||||
|---|---|---|---|---|---|
| Covariate | No. | Overall Survival | Overall Survival | ||
| HR a (95% C.I. b) | p Value | HR (95% C.I.) | p Value | ||
| Age | 0.003 | 0.014 | |||
| ≤75 years | 54 | 1 | 1 | ||
| >75 years | 46 | 1.92 (1.24–2.96) | 1.73 (1.12–2.67) | ||
| Gender | 0.697 | ||||
| Male | 45 | 1 | |||
| Female | 55 | 1.09 (0.72–1.65) | |||
| Stage | <0.001 | 0.009 | |||
| IIIB + IIIC | 18 | 1 | 1 | ||
| IVA + IVB | 82 | 3.46 (1.63–7.35) | 2.80 (1.29–6.05) | ||
| LDOC1 expression | <0.001 | 0.002 | |||
| High | 58 | 1 | 1 | ||
| Low | 42 | 2.36 (1.50–3.70) | 2.02 (1.28–3.19) | ||
| EGFR expression c | 0.181 | ||||
| Membranous | 68 | 1 | |||
| Cytoplasmic | 31 | 1.36 (0.87–2.15) | |||
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Huang, H.-N.; Hung, P.-F.; Chen, Y.-P.; Lee, C.-H. Leucine Zipper Downregulated in Cancer-1 Interacts with Clathrin Adaptors to Control Epidermal Growth Factor Receptor (EGFR) Internalization and Gefitinib Response in EGFR-Mutated Non-Small Cell Lung Cancer. Int. J. Mol. Sci. 2024, 25, 1374. https://doi.org/10.3390/ijms25031374
Huang H-N, Hung P-F, Chen Y-P, Lee C-H. Leucine Zipper Downregulated in Cancer-1 Interacts with Clathrin Adaptors to Control Epidermal Growth Factor Receptor (EGFR) Internalization and Gefitinib Response in EGFR-Mutated Non-Small Cell Lung Cancer. International Journal of Molecular Sciences. 2024; 25(3):1374. https://doi.org/10.3390/ijms25031374
Chicago/Turabian StyleHuang, Hsien-Neng, Pin-Feng Hung, Yai-Ping Chen, and Chia-Huei Lee. 2024. "Leucine Zipper Downregulated in Cancer-1 Interacts with Clathrin Adaptors to Control Epidermal Growth Factor Receptor (EGFR) Internalization and Gefitinib Response in EGFR-Mutated Non-Small Cell Lung Cancer" International Journal of Molecular Sciences 25, no. 3: 1374. https://doi.org/10.3390/ijms25031374
APA StyleHuang, H.-N., Hung, P.-F., Chen, Y.-P., & Lee, C.-H. (2024). Leucine Zipper Downregulated in Cancer-1 Interacts with Clathrin Adaptors to Control Epidermal Growth Factor Receptor (EGFR) Internalization and Gefitinib Response in EGFR-Mutated Non-Small Cell Lung Cancer. International Journal of Molecular Sciences, 25(3), 1374. https://doi.org/10.3390/ijms25031374