
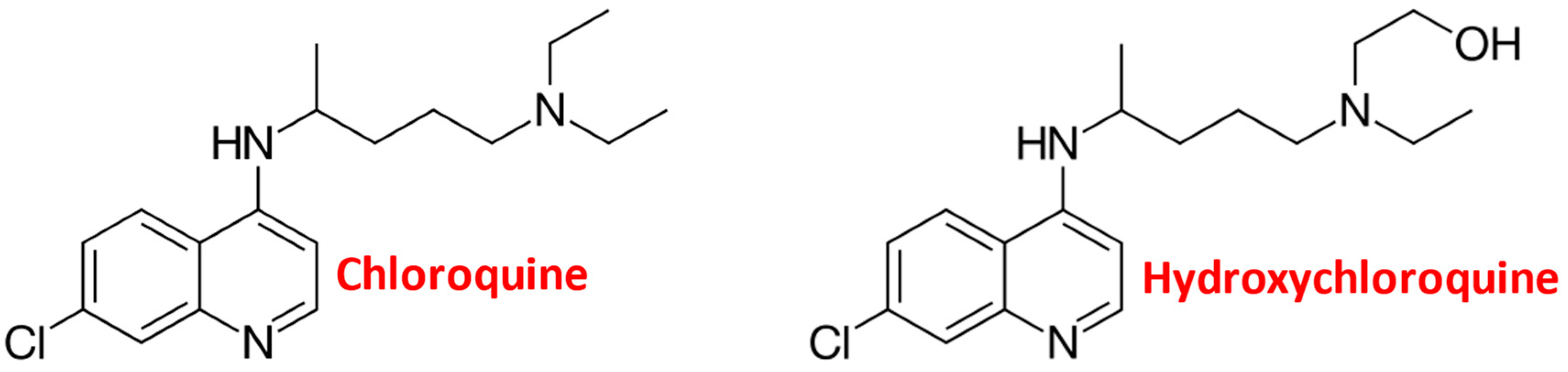
Chloroquine and Chemotherapeutic Compounds in Experimental Cancer Treatment


Abstract
1. Introduction
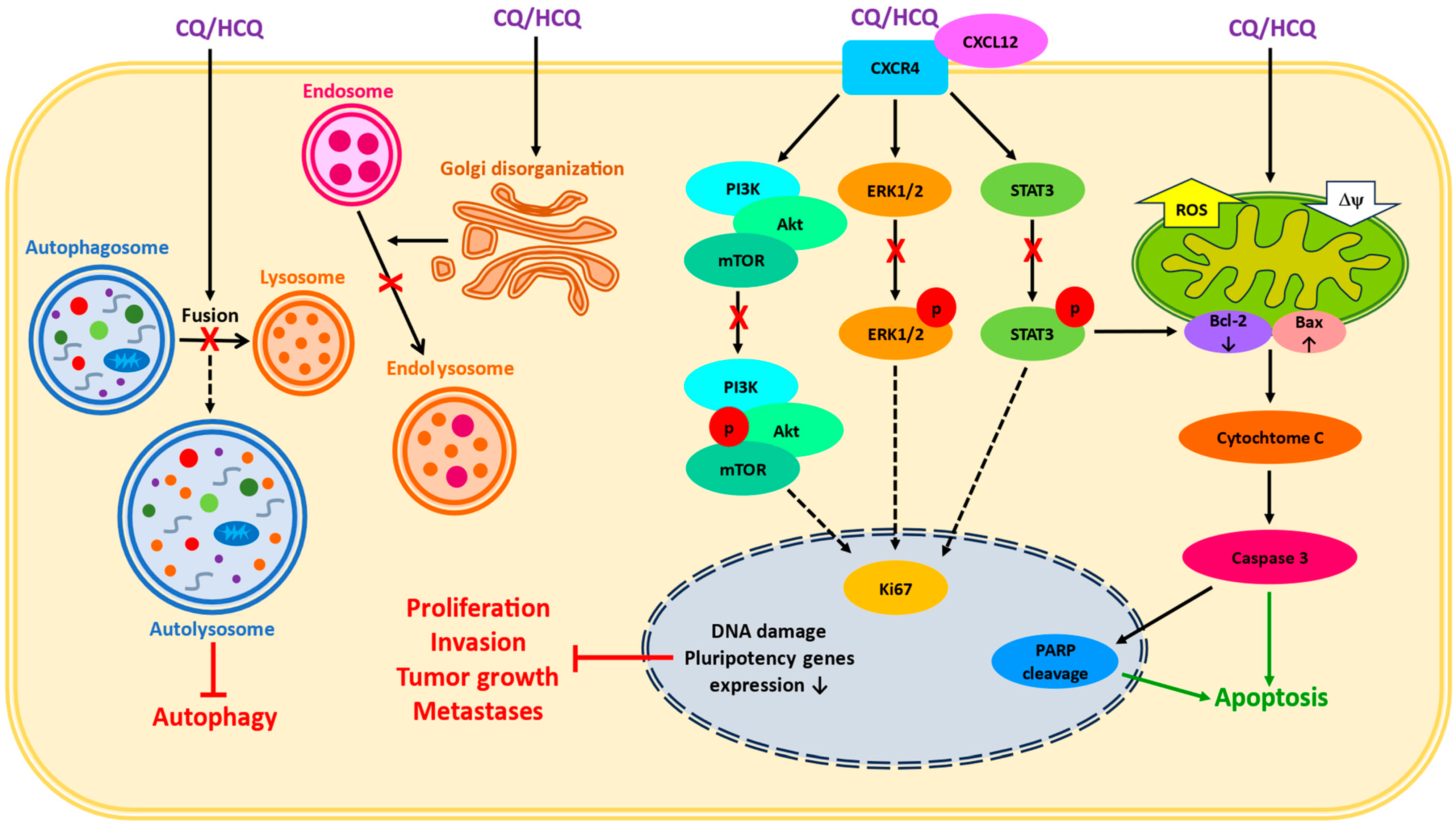
2. Cellular Chloroquine Effects
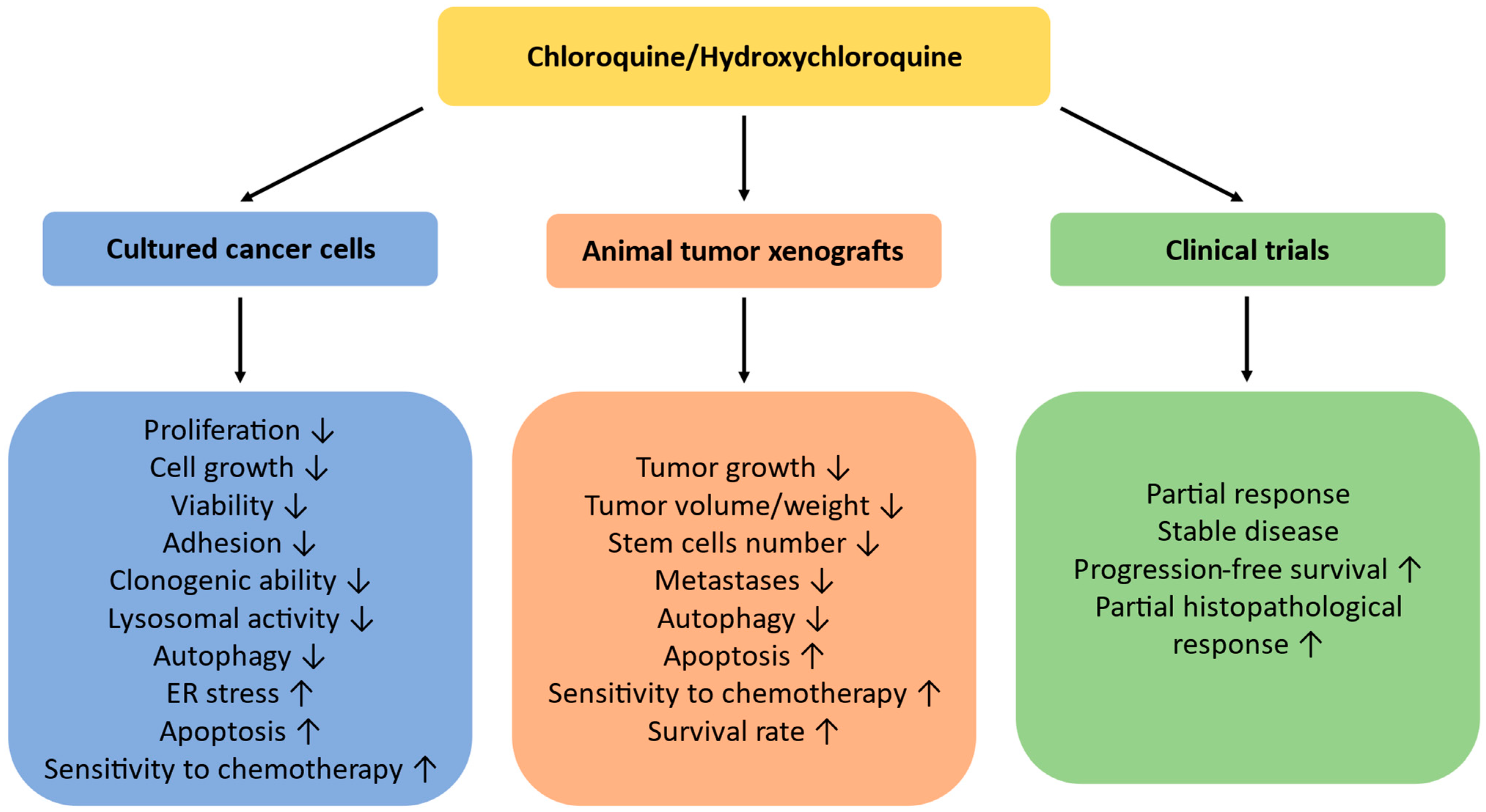
3. Chloroquine as a Single Treatment

{kind=link}
{kind=link}
{kind=link}
| Agent | Experimental System | Treatment Regime | Effects | Molecular Markers | Reference |
|---|---|---|---|---|---|
| CQ | Glioma U87MG, U251, G120, G130, and G44 cells | 10–40 µg/mL for 24–72 h | ↓Cell growth ↓Viability | ↑Caspase 3 ↑p53 ↑Bax | [25] |
| CQ | Melanoma SK-MEL23 and VMM39 cells | 25–50 µM for 5–28 h | ↓Viability, ↓Lysosomal activity ↓Autophagy ↑Apoptosis | ↑Caspase 3 ↑PUMA ↑p62 ↑LC3 | [26] |
| CQ | Primary pancreatic cancer cells | 10 µM for 7 days | ↓CSCs number ↓Sphere-forming ability ↓CSCs pool in spheres ↓Invasiveness | ↓CXCL12/CXCR4 signaling ↓Hedgehog signaling ↓p-ERK and p-STAT3 ↓Expression of pluripotency- related genes OCT4, SOX2, NANOG, and cyclins D1 and E1 | [27] |
| CQ | HepG2 and Huh7 human liver cancer cells | 10–30 µM for 24–72 h | ↓Proliferation, ↑Apoptosis G0/G1 cell cycle arrest | DNA damage ↑Caspase-3, cleaved PARP, and Bim ↓Mitochondrial membrane potential | [28] |
| CQ | Pancreatic neuroendocrine neoplasm | ↑ER stress ↑Apoptosis | ↑PERK, eIF2α, ATF4, and CHOP | [29] | |
| CQ | Patient-derived glioblastoma stem cell lines no. 993, G112SP and no. 1095 | CQ 30 µM for 24–72 h | ↓Proliferation ↓Viability | ↓Ki67 ↑SubG1 fraction ↑p53, p21, and caspase-3 ↓HIPK2 and ATM ↓p-Akt ↑LC3-II and p62 | [30] |
| CQ | Human cervical cancer HeLa cells | 100 µM for 2–5 h | ↓Autophagy | ↑Autophagosomes Disorganization of Golgi and endo-lysosomal systems | [31] |
| CQ | Osteosarcoma U2OS cells | 100 µM for 2–5 h | ↓Autophagy | Disorganization of Golgi and endo-lysosomal systems ↑LC3-II, p62/SQSTM1, and LAMP | [31] |
| CQ | Triple-negative breast cancer Hs578t, MDAMB231, and SUM159PT cells | 1 µM for 48 h | ↓Mammosphere-forming efficiency ↓CD44+/CD24−/low stem cells population ↓Autophagy ↓DNA methylation | ↑Autophagosomes ↑LC3, p62, and caspase-3 ↓STAT3 and Jak2 phosphorylation ↓DNMT1 | [32] |
| CQ | Triple-negative breast cancer Hs578t, MDAMB231, and SUM159 cells | 10–20 μM for 48 h | ↓Autophagy ↓CD44+/CD24−/low CSCs number Mitochondrial damage Cristae vacuolization DNA damage | Mitochondrial membrane depolarization Cytochrome C release ↑LC3 and p62 ↑Superoxide ↓Cytochrome C oxidase and NQO1 ↑γ-H2AX | [33] |
| CQ HCQ | Adult T-cell leukemia/lymphoma (ATLL) cell lines | CQ 50 µM or HCQ 25 µM for 6–24 h | ↓Viability and growth ↓Autophagy ↑Apoptosis | ↑Caspase-3, LC3 ↑Autophagosomes ↑p47 and IκBα ↓NEMO, CADM1 | [34] |
| CQ | Endometrial cancer AN3CA, KLE, and Ishikawa cells | 0.5–20 µM for 24–72 h | ↓Proliferation ↓Colony formation ↓Autophagy ↑Apoptosis Cell cycle arrest | ↑Cleaved caspase-3 ↑LC3-I, LC3-II, and p62 ↑Autophagosomes and endosomes | [35] |
| CQ, HCQ | Bladder cancer RT4, 5637, and T24 cells | CQ 25 µM or HCQ 20 µM for 24–72 h | ↓Viability ↓Clonogenic ability ↓Autophagy ↑Apoptosis | ↑Caspase3/7 activity; ↑Cleaved PARP ↑LC3-II and p62 ↓Lysosome fusion DNA fragmentation | [36] |
| CQ | Vemurafenib-resistant brain tumor 794R and AM38R cells | CQ 5 or 10 μM for 6 or 96 h | ↑LC3-II | [37] | |
| CQ | Epithelial ovarian CSCs | 10–50 µM for 72 h or 2–10 µM for week | ↓Viability ↓Adhesion ↓Spheroid cell viability and diameter | [38] | |
| CQ | Breast cancer MCF-7 cells | 16–256 µM for 48 h | ↓Viability and growth | [39] | |
| CQ | Breast cancer MCF-7 cells | 32.5 µM for 48 h | ↓Viability and growth ↑Apoptosis ↓Autophagy | DNA damage Cytochrome C release ↑Autophagosomes ↑Bax, p53 ↑Caspases 3 and 9 mRNA | [40] |
| CQ | Thyroid cancer TPC1, ATC1, and KTC1 cells | 50 µM for 48 h | ↓Viability ↓Autophagy ↑Apoptosis | ↑LC3 and p62 DNA damage | [41] |
4. Chloroquine and Chemotherapy Drugs
4.1. Chloroquine and Doxorubicin (DOX)
| Agent | Experimental System | Treatment Regime | Effects | Molecular Markers | Reference |
|---|---|---|---|---|---|
| CQ + DOX | Breast cancer MCF-7 cells | DOX 0.05–0.2 µM + CQ 16–64 µM for 48 h | ↑Sensitivity to DOX ↓Viability and growth | [39] | |
| CQ + DOX | Breast cancer MCF-7 cells | DOX 3.38 µM + CQ 32.5 µM for 48 h | ↑Sensitivity to DOX ↓Viability and growth ↑Apoptosis ↓Autophagy | DNA damage Cytochrome C release ↑Autophagosomes ↑Bax, p53, and caspases 3 and 9 ↑Beclin-1, ATG7, LC3-II, and p62 ↓PI3K, Akt, mTOR, and Bcl-2 | [40] |
| CQ + DOX | Hepatocellular cancer HepG2, Huh7, SNU387, and SNU449 cells | DOX 0.25–1 μg/mL + CQ 20 μM for 48 h | ↑DOX cytotoxicity ↓Viability ↓Autophagy | ↑LC3 and p62 | [47] |
| CQ + DOX | Melanoma SK-MEL-5, SK-MEL-28, and A-375 cells | DOX 1–2.5 μM + CQ 20 μM for 24 h | ↑Pyroptosis ↓Autophagy ↓Viability | ↑Cleaved caspase-3 ↑N-DFNA5 | [48] |
| CQ + DOX | Breast cancer MCF-7 cells | DOX 0.17 µM + CQ 16–256 µM for 48 h | ↓Viability and proliferation | ↓Viability ↓PPT1 expression | [49] |
| CQ + DOX | Cervical cancer HeLa cells | DOX 40 nM + CQ 40 µM | ↑Sensitivity to DOX ↑Apoptosis ↓Autophagy | ↑p62, LC3-II, caspase-3, and PARP ↓LAMP-2, Syntaxin 17, Rab 5, and Rab 7 | [51] |
| CQ + DOX | Human umbilical vein endothelial cells (HUVECs) | DOX 01–1 µM + CQ 0.25–32 µM for 48 h | ↑Anti-angiogenic effect of DOX | [54] | |
| CQ + SpHL-DOX | Cervical cancer HeLa cells | SpHDL-DOX 3.22 µM + CQ 20 μM for 4 h | ↓Viability ↑Apoptosis | [60] | |
| CQ + DOX@ FP-MoS2 | Cervical cancer HeLa-R cells | DOX 5 μg/mL + CQ 5 μg/mL + FP-MoS2 40 μg/mL for 48 h | ↓Viability ↑Transfer and accumulation in tumor cells | [61] | |
| CQ + DOX HCl in DC-DIV/C | DOX-resistant MCF-7/ADR and K562/ADR cells | DOX 5 μg/mL + CQ 10 μg/mL for 24–48 h | ↑Sensitivity to DOX ↑Apoptosis ↓Autophagy | ↑Autophagosomes ↑LC3-II and p62 | [62] |
| CQ + PTX | TNBC Hs578t, MDAMB231, and SUM159PT cells | PTX 5 nM + CQ 1 µM for 48 h | ↑Sensitivity to PTX ↓Autophagy ↓CD44+/CD24−/low stem cells population ↓Sphere-forming capacity ↓DNA methylation | ↑Autophagosomes ↑Cleaved caspase-3 ↑LC-3II and p62 ↓p-STAT3 and p-Jak2 ↑SOCS1 and SOCS3 ↓DNMT1 | [32] |
| CQ + PTX | Breast cancer MCF-7 cells | PTX 1.5–3 nM + CQ 32–64 µM for 48 h | ↓Viability and growth | [39] | |
| CQ + CIS | CIS-resistant endometrial cancer Ishikawa cells | CIS 0.01–100 µM + CQ 1 µM for 72 h | ↑Sensitivity to CIS | [35] | |
| CQ + CIS | Thyroid TPC1, ACT1, and KTC1 cells | CIS 2 µM + CQ 50 µM for 48 h | ↑Apoptosis ↓Autophagy | ↑LC3 and p62 | [41] |
| CQ + CIS | Human neuroblastoma SH-SY5Y | CIS 2 µM + CQ 15 µM for 48 h | ↑Apoptosis ↑CIS sensitivity | ↑LC3-II/LC3-I and p62 | [63] |
| CQ + CIS | Epithelial ovarian cancer SKOV3 and hey cells | CIS 2.5–10 µM + CQ 5–10 µM for 24–48 h | ↓Viability, migration and invasion ↑Apoptosis | ↑Autophagosomes ↑Bax and LC3-II/LC3-I ↑Cleaved caspase-3 and PARP ↓Bcl-2 and Bcl-XL | [64] |
| HCQ + CIS | Human neuroblastoma SH-SY5Y | CIS 0.5–2 µM + HCQ 1 µg/mL for 24–48 h | ↑Apoptosis ↓Autophagy | ↑LC3-II ↑ROS | [65] |
| CQ + CPT | TNBC SUM159 SCSs | CPT 10 µM + 10 µM CQ for 48 h | Additive CQ effect ↓CD44+/CD24−/low DNA damage | ↓Rad50 and Rad51 ↑Cleaved PARP and Bcl-2 | [33] |
| CQ + OXP | Hepatocellular carcinoma HepG2 transfected with ATG7 shRNA | OXP 18 µM + CQ 80 µM for 12–48 h | ↑Apoptosis | ↑AVOs ↑LC3 ↑caspase-3 | [66] |
| CQ + OXP | Colon cancer HT29 cells | OXP 0.95–1.6 µM + CQ 1–5 µM for 24 h | ↑Sensitivity to OXP ↓Autophagy | ↓LC3 staining | [67] |
| TH-NP with HCQ + OXP | Hepatocellular carcinoma HepG2, Huh-7, and HCCLM3 cells | OXP 20 µM + HCQ 10 µM for 24 h | ↓Autophagy ↓Proliferation ↓Colony formation ↓Invasion and migration | ↑LC3-I, LC3-II, and p62 ↑E-cadherin, Paxillin, and PARP ↑Autophagosomes | [68] |
| CQ + GEM | Gallbladder cancer cells GBC-SD, SGC-996, and NOZ | GEM 20 µM + CQ 10 µM for 48 h | ↑Antitumor GEM effect ↑Apoptosis ↓Viability ↓Colony formation Cell cycle arrest | ↑Bax, LC3-II/LC3-I, and p62 ↓Bcl-2 and PARP ↓p-Akt and p-mTOR | [69] |
| CQ + GEM | Pancreatic cancer PANC-1 cells | GEM 20 µM + CQ 10 µM for 72 h | ↓Viability | [70] | |
| PDGL-GEM@CAP/CQ | PDAC Pan 02 cells | GEM 0.5 µg/mL + CQ 2.5 µg/mL for 48 h | ↓Viability ↓Migration or invasion ↓Proliferation | ↑LC3-II/LC3-I and p62; ↑Autophagosomes ↓Degradation of paxillin and MMP-2 | [25] |
| CQ + IMA | CML K562 cells | IMA 0.25–0.5 µM + 25 µM CQ for 48 h | ↑IMA-induced cell death ↓Autophagy | ↑LC3-II | [71] |
| CQ + IMA | IMA-resistant BaF3/E255K and BaF3/T315I lymphoid cells | IMA 5–10 µM + 25 µM CQ for 48 h | ↑IMA-induced cell death ↓Autophagy | ↑LC3-II | [71] |
| CQ + IMA | CML K562 cells | IMA 5 µM + CQ 25 µM for 24 h and up to 5 days | ↑Sensitivity to IMA ↓Viability ↓Autophagy ↑Necrosis Cell shrinkage | ↓Beclin-1 ↑LC3 Nuclei fragmentation | [72] |
| CQ + IMA | GIST-T1 cells | IMA 1 µM + CQ 50 µM for 72 h or IMA 0.1 µM + CQ 5 µM for 14 d | ↓Cell growth ↓Colony formation ↑Apoptosis | ↑Caspases 3/7 ↑CC-3 staining | [73] |
| CQ + IMA | GIST GIST882 cells | IMA 0.5–5 µM for 48 h | ↓Cell growth ↑Apoptosis ↓Viability | ↓p-ERK/ERK and p-Kit/Kit ↓LC3-II/LC3-I ↑Caspases 3/7 | [74] |
| CQ + Lenvatinib | Papillary thyroid cancer K1 and BCPAP cells | Lenvatinib 10–25 µM + CQ 50 µM for 24 h | ↑Inhibitory effect of Lenvatinib ↑Apoptosis ↓Viability and proliferation ↓Angiogenesis | ↑LC3-I and LC3-II ↓VEGFA level | [75] |
| CQ + Apatinib | Anaplastic thyroid cancer KHM-5M and C643 cells | Apatinib 20 µM + CQ 10 µM for 24 h | ↓Autophagy ↑Apoptosis | ↑LC3-II/LC3-I and p62 ↑Cleaved PARP ↓p-mTOR and p-Akt ↓Autophagosomes | [76] |
| CQ + Apatinib | Esophageal squamous cell carcinoma ECA-109 and KYSE-150 lines | Apatinib 25 µM + CQ 10 µM for 24 h | ↑Apoptosis ↓Autophagy ↓Viability and proliferation ↓Formation of ESCC clones | ↑LC3-II/LC3-I and p62 ↑ Bax, ↓Bcl-2, p-Akt, p-mTOR ↓Autophagosomes | [77] |
| CQ + RAPA | Osteosarcoma MG63 cells | RAPA 20 μM + CQ 20 μM for 24 h | ↑Effects of RAPA ↑Apoptosis ↓Proliferation ↓Autophagy | ↑LC3-I/II and p62 ↑Cleaved caspases 3 and 9 ↑PARP ↑Autophagosomes | [78] |
| CQ + RAPA | Human well differentiated liposarcoma 93T449 cells | RAPA 6 µM + CQ 80 µM for 24 h | ↓Viability | DNA damage ↑Autophagosomes ↑LC3-II ↑TUNEL-positive cells | [79] |
| CQ + Salid- roside | Hepatocellular cancer HepG2 and 97H cells | Salidroside 80 µM + CQ 5–20 µM for 48 h | ↑Apoptosis ↓Viability ↓Autophagy Changes in cell morphology Chromatin condensation | ↑ROS ↓Mitochondrial membrane potential ↑Bax, and cleaved caspase-3 ↓Bcl-2 and Beclin-1 ↑p62, p-mTOR/mTOR, p-PI3K/PI3K, and p-Akt/Akt | [80] |
| Lys05 + Dacto- lisib | Lung cancer A549 cells | Dactolisib 0.05 µM + Lys05 3.19 µM | ↓Autophagy ↑Apoptosis ↓Proliferation | ↓ATG4B, LC3A, LC3B, and KI67 genes ↑CASP3 ↑LC3B/LC3A and p62 | [81] |
| CQ + Evero- limus | Renal adenocarcinoma A498, RXF393, 769P, and SN12C cells | Everolimus 1.3–19.3 µM + CQ 2.4–19.3 µM for 72 h | Synergic growth inhibition ↑Apoptosis ↓Autophagy | ↓Bcl-2 ↓Beclin-1/Bcl-2 complex formation ↓p-4EBP1 and ERK1/2 ↑Caspases 3 and 9 | [82] |
| CQ + Pd(II) complex | Prostate cancer PC-3 and LNCaP cells | Pd (II) complex 12.5 µM + CQ 5 µM for 12–48 h | ↓Viability ↑Apoptosis ↓Autophagy ↑ROS | ↑Caspases 3/7 ↓Atg5, Beclin-1, LC3, and p62 ↓p-Akt/p-mTOR, p-STAT5, and p-CREB | [83] |
| CQ + Tamoxifen | Antiestrogen-resistant breast carcinoma MCF7-RR, LCC9 cells | 1 μM CQ, 10–1000 nM Tamoxifen for 6 days | ↓Cell growth ↓ Autophagy ↑Cell death | ↑Autophagosomes ↑LC3-II and p62 | [84] |
| CQ + Faslodex | Antiestrogen-resistant breast carcinoma MCF7-RR, LCC9 cells | 1 μM CQ, 10–1000 nM Faslodex for 6 days | ↓Cell growth ↓ Autophagy ↑Cell death | ↑Autophagosomes ↑LC3-II and p62 | [84] |
| CQ + Ipata-sertib | MDAMB231, MDAM468, MCF7, and SKBR3 breast cancer cell lines | Ipatasertib 1–10 μM + CQ 1–10 μM | ↑Apoptosis ↓Autophagy ↓Proliferation ↓Clonogenic capacity ↓Spheroid-forming capacity | ↑Cleaved PARP ↑LC3-II and p62 ↑Autophagosomes | [85] |
| CQ + Taselisib | MDAMB231, MDAM468, MCF7, and SKBR3 breast cancer cell lines | Taselisib 1–10 μM + CQ 1–10 μM | ↑Apoptosis ↓Autophagy ↓Proliferation ↓Clonogenic capacity ↓Spheroid-forming capacity | ↑Cleaved PARP ↑LC3-II and p62 ↑Autophagosomes | [85] |
| CQ + IR | Glioblastoma no. 993, no. 1095 and G112SP cells | CQ 30 μM + IR 2.5 Gy for 72 h | ↓Proliferation ↑Cell death Cell cycle arrest | ↑LC3B-II and p62 ↓Akt and Ki67 ↑SubG1 population | [30] |
| CQ + Vemu- rafenib | Glioblastoma 794 and AM38 cells | Vemurafenib 1 μM + CQ 5 μM | ↓Clonogenic growth | [37] | |
| CQ + Trame tinib | Glioblastoma 794 and AM38 cells | Trametinib 7.5–30 nM + CQ 5 μM | ↓Growth ↓Clonogenic growth | [37] | |
| CQ + Vemu- rafenib | Patient-derived glioblastoma cells | Vemurafenib 1–2 μM + CQ 10–20 μM for 72 h | ↓Autophagy ↓Tumor growth | ↑LC3B-II, p-ERK/ERK ↑Caspases 3/7 ↓p-Akt and pS6 | [37] |
| CQ + Sorafenib | Thyroid cancer TPC1, ACT1, and KTC1 cell lines | Sorafenib 100 nM + 50 μM CQ for 48 h | ↑Apoptosis ↓Autophagy | ↑LC3B-II and p62 | [41] |
| HCQ + Temozo-lomide | Glioblastoma U-87 Mg cells | TMZ 100 µg/mL + HCQ 1 µg/mL for 24 h | ↑Apoptosis ↓Autophagy | ↑LC3-II ↑ROS | [65] |
| CQ + PTX + Apatinib | Esophageal carcinoma ECA-109 and KYSE-150 cells | PTX 5 μM + CQ 10 μM + Apatinib 25 μM for 24–72 h | ↑Sensitivity to PTX ↑Apoptosis ↓Proliferation ↓Colony formation | ↑Bax and cleaved caspase-3 ↓Bcl-2, p-Akt, and p-mTOR | [77] |
4.2. Chloroquine and Paclitaxel (PTX)
4.3. Chloroquine- and Platinum-Based Anticancer Drugs
4.4. Chloroquine and Gemcitabine (GEM)
| Agent | Experimental System | Treatment Regime | Effect | Molecular Markers | Reference |
|---|---|---|---|---|---|
| CQ | Glioblastoma U87MG xenografts of NMRI nude mice | CQ 30 mM/day intracranially for 17 days | ↓Tumor growth ↓Cell viability ↓Number of mitotic cells | [25] | |
| CQ | Melanoma SKMel23 cells xenografts of NOD-SCID mice | CQ 25 mg/kg (IP) twice/week for 3 weeks | ↓Tumor growth ↓Autophagy | [26] | |
| CQ | Immunocompromised mice implanted with patient-resected PDAC cells | CQ 50 mg/kg (IP) for 21 days | ↓CSCs-driven metastases ↓Tumorigenicity | ↓CD133+ cells number ↓ALK4 ↓Nodal/Activin ↓Self-renewal genes | [27] |
| CQ | Liver cancer HepG2-GFP xenograft of nude mice | CQ 80 mg/kg twice daily 3 d on/2 d off (SC) for 25 days | ↓Tumor growth and weight ↓Proliferation | ↓Ki-67 ↑cleaved PARP | [28] |
| CQ | Athymic nude mice with orthotopic MDAMB231 breast cancer tumor | CQ 10 mg/kg daily (IP) for 2 2 weeks | ↓Tumor growth ↓Lung metastasis | ↓CD44+/CD24−/low stem cells number | [32] |
| CQ HCQ | Immunodeficient NOD/Shi-scid/IL-2Rγnull (NOG) mice transplanted with ATLL MT2 or Su9T01 cells | CQ 50 mg/kg/day (IP) or HCQ 6.5–60 mg/kg/day (OR) for 21 days | ↑Survival ↓Tumor growth and weight Degeneration and necrosis of tumor cells | ↑Caspase-3 ↑Condensed hyperchromatic or fragmented nuclei with shrunken cytoplasm | [34] |
| CQ | Female BALB/c mice with MCF-7 xenograft | CQ 50 mg/kg (IP) once/3 days for 43 days | ↓Viability and growth ↑Apoptosis ↓Autophagy | DNA damage Cytochrome C release ↑Bax and p53 ↑Caspases 3 and 9 | [40] |
| CQ + DOX | Female BALB/c mice with MCF-7 xenograft | DOX 2 mg/kg (IP) + CQ 50 mg/kg (IP) once/3 days for 43 days | ↓Tumor growth, ↑Apoptosis ↓Autophagy | DNA damage ↑Autophagosomes Cytochrome C release ↑Bax, p53, caspases 3 and 9, Beclin-1, ATG7, LC3-II, and p62 ↓PI3K, Akt, mTOR, and Bcl-2 | [40] |
| CQ + DOX | Female mice injected with Ehrlich ascites carcinoma (EAC) cells | DOX 1.5 mg/kg and 3 mg/kg + CQ 25 mg/kg and 50 mg/kg (IP) at 2, 7, and 12 days | ↓Disruption of alveolar structure ↓Oxidative stress | ↓MDA, CAT, GPx, SOD, iNOS, and eNOS ↑ NGAL | [53] |
| CQ + PEG-DOX+ pUH | BALB/c mice subcutaneously injected with 4T1 breast tumor cells | PEG-DOX 10 mg/kg (IV) + CQ 50 mg/kg + 15 min on-tumor pUH on day 5 after tumor implantation up to 60 days | ↓Viability ↓Tumor growth ↑ Animal survival | DNA damage ↑LC3-II ↑TUNEL-positive cells | [58,59] |
| CQ + DOX. HCl in DA-DIV/C nanovesicles | Female BALB/c nude mice subcutaneously inoculated with DOX-resistant K562/ADR cells | DOX-HCl 5 mg/kg + CQ 10 mg/kg (IV) at 0, 2, 4, and 6 days | ↓Tumor volume and weight ↓Autophagy ↓Cell density ↑Necrosis DNA damage | ↓Ki67 ↑TUNEL-positive cells ↑LC3-II | [62] |
| CQ + PTX | Athymic nude mice with orthotopic MDAMB231 and SUM159PT tumors | PTX 15–30 mg/kg (IP) weekly + CQ 10 mg/kg daily for 2 weeks or twice/week for 4 weeks | ↑Sensitivity to PTX ↓Tumor growth ↓Lung metastasis ↓Tumor recurrence ↓PTX-induced CSCs population | ↓CD44+/CD24−/low CSCs | [32] |
| CQ + CIS | Nude mice with ovarian cancer SKOV3 xenograft | CIS 5 mg/kg/6 days + CQ 60 mg/kg/day (IP) for 21 days | ↓Tumor volume and weight | ↑Cleaved caspase-3 ↓Ki-67-positive cells | [64] |
| CQ + CIS | Nude BALB/C female mice with gastric cancer SGC7901 xenograft | CIS 5 mg/kg + CQ 45 mg/kg every three days 10 times | ↓Tumor weight | ↓LC3II/I ratio and Beclin-1 ↓MDR1/P-gp ↑caspase-3 | [94] |
| CQ + CIS | BALB/C nude mice with hepatocarcinoma SMMC-7721 xenograft | CQ 60 mg/kg + CIS 3 mg/kg (IP) thrice/week for 2 weeks | ↓Tumor volume and weight ↑Apoptosis ↓Proliferation | DNA damage ↓Ki-67-positive cells | [95] |
| CQ + CPT | Immunodeficient SCID-Beige mice with TNBC SUM159 xenograft | CPT 24 mg/kg weekly + CQ 30 mg/kg every 3 days for 3 weeks | ↓Tumor volume ↓Viability ↑Apoptosis | ↓Mitochondrial metabolic activity ↓Bcl-2, Rad50, Rad51 ↑LC3B-II, and p62 | [33] |
| CQ + CPT | Immunodeficient NSG mice injected with CD45-CD44+ epithelial ovarian tumor cells | CPT 50 mg/kg + CQ 100 mg/kg every 2 days weekly for 16 weeks | ↓Tumor volume | ↓CD44+/CD117+ cells population ↓Ki67 | [38] |
| CQ + OXP | Immunodeficient C/.B.17 SCID mice injected with colon cancer HT29 cells | OXP 5 mg/kg (IP) per week for 2 weeks + CQ 3.5 mg/kg daily for 21 days | ↓Tumor growth and volume ↓Autophagosomal cells | ↓LC3 staining | [67] |
| TH-NP with HCQ + OXP | Nude mice with hepatocellular carcinoma HCCLM3 xenograft | OXP 10 mg/kg + HCQ 20 mg/kg (IV) every three days for 30–49 days | ↓Tumor growth ↓Metastases ↓Autophagy | ↑Cleaved caspase 3 and PARP ↓Ki67 ↓Autophagosomes/ autolysosomess | [68] |
| CQ + GEM | Immunocompromised mice implanted with patient-resected PDAC | GEM 125 mg/kg (IP) for 52 days + CQ 50 mg/kg (IP) for 21 days | ↓Tumor growth ↑Survival rate | ↓ CD133+ CSCs ↓Nodal/Activin pathway | [27] |
| CQ + GEM | Male BALB/c nude mice injected with gallbladder cancer SGC-996 cells | GEM 20 mg/kg (IP) + CQ 60 mg/kg (IP) twice/week for 22 days | ↑Sensitivity to GEM ↓Tumor growth | [69] | |
| CQ-loaded PLGA nanoparticles + GEM | BALB/c AJcl nu/nu female mice orthotopically transplanted with immortalized patient- derived pancreatic stem cells and SUIT-2 cancer cells | GEM 40 mg/kg (IV) on days 10, 17, and 24 + Nano-CQ 30 mg/kg (IV) on days 10, 17, and 24 | ↓Density of activated cancer stem cells ↑Sensitivity to GEM ↓Tumor volume and weight | ↓αSMA | [99] |
| PDGL-GEM@CAP/ CQ | Mice bearing pancreatic cancer Pan 02 xenografts and orthotopic pancreas Pan 02 tumor | GEM 3 mg/kg (IV) + CQ 15 mg/kg (IV) every other day 4 times | ↓Tumor growth ↓Metastases ↑Tumor necrosis ↓Number of activated fibroblasts ↓Fibrosis ↓Autophagy | ↑Autophagosomes ↑LC3II/LC3I ratio and p62 ↓MMP-2, IL-6 ↓Collagen ↑Paxillin ↓αSMA | [100] |
| CQ + IMA | Female athymic nude NMRI nu/nu with heterotopic GIST-T1 xenograft | IMA 50 mg/kg (OR) twice/day + CQ 60 mg/kg (IP) daily for 15 days | ↑Apoptosis No effect on tumor growth | ↑CC-3 staining | [73] |
| CQ + IMA | NOD/SCID male mice implanted with IMA- sensitive and resistant GIST882 cells | IMA 150 mg/kg (OR) twice/day + CQ 60 mg/kg (IP) daily for 28 days | ↓Autophagy No effect on tumor growth | ↑LC3II ↓p-ERK/ERK | [74] |
| CQ + Lenvatinib | Nude mice injected with thyroid cancer K1 cells | Lenvatinib 30 mg/kg + CQ 50 mg/kg for 14 days | ↑Anticancer LEN effect ↓Tumor growth ↓Angiogenesis | ↓VEGFA, CD31, and C-Myc | [75] |
| CQ + Lenvatinib | Nude BALB/c mice injected with hepatocellular carcinoma HCCLM3 cells | Lenvatinib 5–10 mg/kg (IP) + HCQ 50 mg/kg (IP) | ↓Tumor growth ↓Lung metastases ↑Overall survival | [102] | |
| CQ + Apatinib | Male BALB/c nude mice injected with KHM-5M thyroid cancer cells | Apatinib 50 mg/kg (OR) daily + CQ 60 mg/kg (OR) daily for 26 days | ↓Tumor volume and weight ↓Proliferation ↑Apoptosis | ↑Cleaved caspase-3 ↑TUNEL-positive cells ↓Ki67 | [76] |
| CQ + Apatinib | Male BALB/c nude mice injected with esophageal carcinoma ECA-109 cells | Apatinib 60 mg/kg OR) daily + CQ 60 mg/kg (OR) daily for 4 weeks | ↓Tumor volume and weight ↓Proliferation ↑Apoptosis | ↑Cleaved caspase-3 ↑TUNEL-positive cells ↓Ki67-positive cells | [77] |
| CQ + RAPA | Athymic nude mice injected with patient- derived dedifferentiated liposarcoma | RAPA 1 mg/kg/day (IP) + CQ 100 mg/kg/day (IP) for 15 days | ↓Tumor growth ↓Cancer cells density ↑Apoptosis | ↑TUNEL-positive cells | [103] |
| CQ + Salid- roside | Female BALB/c mice subcutaneously injected with HepG2 cells | Salidroside 80 mg/kg (IP) + CQ 5 mg/kg (IP) every other day for 4 weeks | ↓Tumor growth ↓Number of tumor cells | ↑Bax ↓Bcl-2 | [80] |
| CQ + 5-FU | BALB/c nude mice with hepatocarcinoma SMMC-7721 xenograft | 5FU 30 mg/kg (IP) + 60 mg/kg CQ (IP) trice/week for 2 weeks | ↑Sensitivity to 5-FU ↑Apoptosis ↓Proliferation ↓Tumor growth | ↑TUNEL-positive cells ↓Ki67-positive cells | [95] |
| CQ + Tamo- xifen | Athymic nude mice injected with breast cancer MCF7-RR or LCC9 cells | Tamoxifen 32 mg/kg/d + CQ 1–2 mg/mouse/d (OR) for 5 weeks | ↓Tumor growth ↑Angiogenesis ↓Macrophage activation | ↑CD31-positive cells ↑pVEGFR2 ↑CD68-positive cells | [84] |
| CQ + Fas- lodex | Athymic nude mice with breast cancer MCF7-RR or LCC9 xenografts | Faslodex 0.5 mg/mouse/w (SC) + CQ 1–2 mg/mouse/d (OR) for 5 weeks | ↓Tumor growth ↑Angiogenesis | ↑CD31-positive cells ↑pVEGFR2 | [84] |
| CQ + Tase-lisib | Female NOD/SCID athymic mice injected With TNBC MDAMB231 cells | Taselisib 5 mg/kg (OR) 5 days/week + CQ 30 mg/kg (OR) 5 days/week for 2 weeks | ↑Antitumor PTX effect ↓Tumor growth | [85] | |
| CQ + Nelfi- navir + RAPA + Dasatinib + Metformin | Female Nu/nu mice subcutaneously injected with cisplatin-resistant ovarian cancer OVCAR3 cells | CQ 30 mg/kg + Nelfinavir 250 mg/kg + RAPA 2.24 mg/kg + Dasatinib 4 mg/kg + Metformin 150 mg/kg in 50% PEG400 for 7 days | Tumor remission | ↑ LC3B-II and Grp78 | [104] |
| CQ + Apatinib + PTX | Nude BALB/c mice injected with esophageal carcinoma ECA-109 cells | Apatinib 60 mg/kg (OR) daily + CQ 60 mg/kg (OR) daily + PTX 15 mg/kg (IP) twice/week for 4 weeks | ↓Tumor volume and weight ↑apoptosis ↓Proliferation ↑Apoptosis | ↑Cleaved caspase-3 ↑TUNEL-positive cells ↓Ki67 | [77] |
| CQ + Tase- lisib + PTX | Female NOD/SCID athymic mice injected With TNBC MDAMB231 cells | Taselisib 5 mg/kg (OR) 5 days/week + CQ 30 mg/kg (OR) 5 days/week + PTX 10 mg/kg IP once/week for 2 weeks | ↑Antitumor effect of PXT and Taselisib ↓Tumor volume and weight | [85] | |
| CQ + IR | Female NMRI immunodeficient mice injected with GBCs no. 993, no. 1095 and G112SP cells | CQ 14 mg/kg IP IR 2.5 Gy for 6 days | ↑Survival ↑Sensitization to IR | [30] |
4.5. Chloroquine and Tyrosine Kinase Inhibitors
4.6. Chloroquine and PI3K/Akt/mTOR Inhibitors
4.7. Chloroquine and Other Agents
4.8. Chloroquine in Multi-Drug Combinations
5. Conclusions
| Agents | Tumor Type | Concentration | Effects | Reference |
|---|---|---|---|---|
| CQ + PTX, nab-PTX, Docetaxel, or Ixabepilone | Advanced or metastatic anthracycline- refractory breast cancer | CQ 250 mg (OR) daily + PTX 80–175 mg/m2 (IV) every 3 weeks, docetaxel 75–100 mg/m2 (IV) every 3 weeks, nab-PTX 100–260 mg/m2 (IV) every 3 weeks, or Ixabepilone 40 mg/m2 iv every 3 weeks. Maximum 6 cycles. | Increase in ORR | [90] |
| CQ or HCQ + Carboplatin/ Gemcitabine | Phase I trial, refractory advanced solid tumors | CQ 50 mg/day or HCQ 100–150 mg/day (OR) on 7–21 days + CPT 5 AUC (IV) on day 1 + GEM 1000 mg/day (IV) on days 1 and 8 for 21 days, 4 cycles | PR SD PD Improved PFS and OS | [96] |
| HCQ + GEM | Pancreatic carcinoma | Preoperative GEM 1500 mg/m2 + HCQ for 31 days until surgery | ↑OS and PFS Partial histopathological response ↓CA19-9 level | [101] |
| CQ + IMA | Chronic phase CML | IMA 400–800 mg + CQ 400–800 mg (OR) daily for 48 weeks | No significant effect | [107] |
| HCQ + Everolimus | Advanced renal cell carcinoma | Everolimus 10 mg for 1 week + HCQ 600 mg/twice daily for 35–28 days | Partial response and stable disease ↑PFS | [115] |
| HCQ + Temsirolimus | Melanoma, colorectal carcinoma, head and neck cancer, and breast cancer | TEM 25 mg (IV) + HCQ 200–1200 mg/day (OR) daily for 4–6 weeks | Stable disease | [116] |
| CQ + Carmustine + IR | Glioblastoma multiforme (GBM) | Carmustine 200 mg/L once every 6 weeks + CQ 150 mg daily from 1 day after surgery + radiotherapy 6000 Gy | Longer survival Tumor remission | [117] |
| CQ + Carmustine + IR | Glioblastoma multiforme (GBM) | Carmustine 200 mg/L + CQ 150 mg daily from 5 day after surgery for 12 months + 6000 Gy, 4 cycles | Improved mid-term survival | [118] |
| HCQ + Bortezomib | Relapsed/refractory myeloma | 2-week HCQ 100–1200 mg (OR) + Bortezomib 1–1.3 mg/m2 on days 1, 4, 8, and 11 of 21 d cycle | Partial response Minor response Stable disease | [120] |
| HCQ + CPT/PTX+/− Bevacizumab | Untreated metastatic non-small-cell lung cancer | PTX 200 mg/m2 (IV) on day 1 + CPT 6 AUC on day 1 +/− Bevacizumab 15 mg/kg (IV) on day 1 + CQ 200 mg (OR) on days 1–21 for 6 cycles | Modest improvement in RR ↑ORR and PFS in patients with Kras mutations | [122] |
| HCQ + GEM/nab-PTX | Pancreatic carcinoma | Two preoperative cycles of GEM 1000 mg/L + nab-PTX 125 mg/L on days 1, 8, and 15 + HCQ 1200 mg/day from day 1 | Improved OS ↑Evans grade histopathologic tumor response ↑Tumor immune infiltration index | [123] |
| HCQ + GEM or HCQ + GEM + nab-PTX | Pancreatic carcinoma | 1 month of preoperative GEM + HCQ 1200 mg/day or 2 months of GEM/nab-PTX + HCQ 600 mg twice daily | ↑Evans grade histopathological responses in patients with SMAD4 loss. Improvement of biochemical markers | [124] |
| HCQ + GEM/nab-PTX | Metastatic pancreatic cancer | HCQ 600 mg/twice daily (OR) for 28 days + standard chemotherapy | No improvement of OS Partial response | [125] |
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Krafts, K.; Hempelmann, E.; Skórska-Stania, A. From methylene blue to chloroquine: A brief review of the development of an antimalarial therapy. Parasitol. Res. 2012, 111, 1–6. [Google Scholar] [CrossRef] [PubMed]
- Schrezenmeier, E.; Dörner, T. Mechanisms of action of hydroxychloroquine and chloroquine: Implications for rheumatology. Nat. Rev. Rheumatol. 2020, 16, 155–166. [Google Scholar] [CrossRef] [PubMed]
- De Sanctis, J.B.; Charris, J.; Blanco, Z.; Ramírez, H.; Martínez, G.P.; Mijares, M.R. Molecular mechanisms of chloroquine and hydroxychloroquine used in cancer therapy. Anticancer Agents Med. Chem. 2023, 23, 1122–1144. [Google Scholar] [CrossRef] [PubMed]
- Niemann, B.; Puleo, A.; Stout, C.; Markel, J.; Boone, B.A. Biologic functions of hydroxychloroquine in disease: From COVID-19 to cancer. Pharmaceutics 2022, 14, 2551. [Google Scholar] [CrossRef] [PubMed]
- Płusa, T.; Lengier-Krajewska, M.; Baranowska, A.; Krawczyk, J. Chloroquine in controlling biological infections. Pol. Merkur. Lekarski. 2020, 48, 199–203. [Google Scholar]
- Rainsford, K.D.; Parke, A.L.; Clifford-Rashotte, M.; Kean, W.F. Therapy and pharmacological properties of hydroxychloroquine and chloroquine in treatment of systemic lupus erythematosus, rheumatoid arthritis and related diseases. Inflammopharmacology 2015, 23, 231–269. [Google Scholar] [CrossRef]
- Ponticelli, C.; Moroni, G. Hydroxychloroquine in systemic lupus erythematosus (SLE). Expert Opin. Drug Saf. 2017, 16, 411–419. [Google Scholar] [CrossRef]
- El Hussein, M.T.; Wong, C. Systemic lupus erythematosus: An approach to pharmacologic interventions. Nurse Pract. 2023, 48, 37–46. [Google Scholar] [CrossRef]
- Olafuyi, O.; Badhan, R.K.S. Dose optimization of chloroquine by pharmacokinetic modeling during pregnancy for the treatment of Zika virus infection. J. Pharm. Sci. 2019, 108, 661–673. [Google Scholar] [CrossRef]
- Rampini, D.; Prieto, D.C.; Colzi, A.L.; de Araújo, R.V.; Giarolla, J. Future and perspectives of the Zika virus: Drug repurposing as a powerful tool for treatment insights. Mini Rev. Med. Chem. 2020, 20, 1917–1928. [Google Scholar] [CrossRef]
- Savarino, A.; Gennero, L.; Chen, H.C.; Serrano, D.; Malavasi, F.; Boelaert, J.R.; Sperber, K. Anti-HIV effects of chloroquine: Mechanisms of inhibition and spectrum of activity. AIDS 2001, 15, 2221–2229. [Google Scholar] [CrossRef] [PubMed]
- Paton, N.I.; Goodall, R.; Dunn, D.T.; Franzen, S.; Collaco-Moraes, Y.; Gazzard, B.G.; Williams, I.G.; Fisher, M.J.; Winston, A.; Fox, J.; et al. Effects of hydroxychloroquine on immune activation and disease progression among HIV-infected patients not receiving antiretroviral therapy: A randomized controlled trial. JAMA 2012, 308, 353–361. [Google Scholar] [CrossRef] [PubMed]
- Vaz, E.S.; Vassiliades, S.V.; Giarolla, J.; Polli, M.C.; Parise-Filho, R. Drug repositioning in the COVID-19 pandemic: Fundamentals, synthetic routes, and overview of clinical studies. Eur. J. Clin. Pharmacol. 2023, 79, 723–751. [Google Scholar] [CrossRef] [PubMed]
- Manuja, A.; Chhabra, D.; Kumar, B. Chloroquine chaos and COVID-19: Smart delivery perspectives through pH sensitive polymers/micelles and ZnO nanoparticles. Arab. J. Chem. 2023, 16, 104468. [Google Scholar] [CrossRef] [PubMed]
- Fong, W.; To, K.K.W. Repurposing chloroquine analogs as an adjuvant cancer therapy. Recent Pat. Anticancer Drug Discov. 2021, 16, 204–221. [Google Scholar] [CrossRef] [PubMed]
- Low, L.E.; Kong, C.K.; Yap, W.H.; Siva, S.P.; Gan, S.H.; Siew, W.S.; Ming, L.C.; Lai-Foenander, A.S.; Chang, S.K.; Lee, W.L.; et al. Hydroxychloroquine: Key therapeutic advances and emerging nanotechnological landscape for cancer mitigation. Chem. Biol. Interact. 2023, 13, 110750. [Google Scholar] [CrossRef] [PubMed]
- Verbaanderd, C.; Hannelore Maes, H.; Schaaf, M.B.; Sukhatme, V.P.; Pan Pantziarka, P.; Sukhatme, V.; Agostinis, P.; Bouche, G. Repurposing Drugs in Oncology (ReDO)—Chloroquine and hydroxychloroquine as anti-cancer agents. Ecancermedicalscience 2017, 11, 781. [Google Scholar] [CrossRef]
- Mohsen, S.; Sobash, P.T.; Algwaiz, G.F.; Nasef, N.; Al-Zeidaneen, S.A.; Karim, N.A. Autophagy agents in clinical trials for cancer therapy: A brief review. Curr. Oncol. 2022, 29, 1695–1708. [Google Scholar] [CrossRef]
- Hama, Y.; Ogasawara, Y.; Noda, N.N. Autophagy and cancer: Basic mechanisms and inhibitor development. Cancer Sci. 2023, 114(7), 2699–2708. [Google Scholar] [CrossRef]
- Noguchi, M.; Hirata, N.; Tanaka, T.; Suizu, F.; Nakajima, H.; Chiorini, J.A. Autophagy as a modulator of cell death machinery. Cell Death Dis. 2020, 11, 517. [Google Scholar] [CrossRef]
- Russell, R.C.; Guan, K.L. The multifaceted role of autophagy in cancer. EMBO J. 2022, 41, e110031. [Google Scholar] [CrossRef] [PubMed]
- Chern, Y.J.; Tai, I.T. Adaptive response of resistant cancer cells to chemotherapy. Cancer Biol. Med. 2020, 17, 842–863. [Google Scholar] [CrossRef] [PubMed]
- Zhang, T.; Yu, J.; Cheng, S.; Zhang, Y.; Zhou, C.H.; Qin, J.; Luo, H. Research progress on the anticancer molecular mechanism of targets regulating cell autophagy. Pharmacology 2023, 108, 224–237. [Google Scholar] [CrossRef] [PubMed]
- Rahman, M.A.; Saikat, A.S.; Rahman, M.S.; Islam, M.; Parvez, M.A.; Kim, B. Recent update and drug target in molecular and pharmacological insights into autophagy modulation in cancer treatment and future progress. Cells 2023, 12, 458. [Google Scholar] [CrossRef] [PubMed]
- Kim, E.L.; Wüstenberg, R.; Rübsam, A.; Schmitz-Salue, C.; Warnecke, G.; Bücker, E.M.; Pettkus, N.; Speidel, D.; Rohde, V.; Schulz-Schaeffer, W.; et al. Chloroquine activates the p53 pathway and induces apoptosis in human glioma cells. Neuro-Oncology 2010, 12, 389–400. [Google Scholar] [CrossRef] [PubMed]
- Lakhter, A.J.; Sahu, R.P.; Sun, Y.; Kaufmann, W.K.; Androphy, E.J.; Travers, J.B.; Naidu, S.R. Chloroquine promotes apoptosis in melanoma cells by inhibiting BH3 domain-mediated PUMA degradation. J. Investig. Dermatol. 2013, 133, 2247–2254. [Google Scholar] [CrossRef] [PubMed]
- Balic, A.; Sørensen, M.D.; Trabulo, S.M.; Sainz, B., Jr.; Cioffi, M.; Vieira, C.R.; Miranda-Lorenzo, I.; Hidalgo, M.; Kleeff, J.; Erkan, M.; et al. Chloroquine targets pancreatic cancer stem cells via inhibition of CXCR4 and hedgehog signaling. Mol. Cancer Ther. 2014, 13, 1758–1771. [Google Scholar] [CrossRef]
- Hu, T.; Li, P.; Luo, Z.; Chen, X.; Zhang, J.; Wang, C.; Chen, P.; Dong, Z. Chloroquine inhibits hepatocellular carcinoma cell growth in vitro and in vivo. Oncol. Rep. 2016, 35, 43–49. [Google Scholar] [CrossRef]
- Nakano, K.; Masui, T.; Yogo, A.; Uchida, Y.; Sato, A.; Kasai, Y.; Nagai, K.; Anazawa, T.; Kawaguchi, Y.; Uemoto, S. Chloroquine induces apoptosis in pancreatic neuro- endocrine neoplasms via endoplasmic reticulum stress. Endocr. Relat. Cancer 2020, 27, 431–439. [Google Scholar] [CrossRef]
- Müller, A.; Weyerhäuser, P.; Berte, N.; Jonin, F.; Lyubarskyy, B.; Sprang, B.; Kantelhardt, S.R.; Salinas, G.; Opitz, L.; Schulz-Schaeffer, W.; et al. Concurrent Activation of Both Survival-Promoting and Death-Inducing Signaling by Chloroquine in Glioblastoma Stem Cells: Implications for Potential Risks and Benefits of Using Chloroquine as Radiosensitizer. Cells 2023, 12, 1290. [Google Scholar] [CrossRef]
- Mauthe, M.; Orhon, I.; Rocchi, C.; Zhou, X.; Luhr, M.; Hijlkema, K.J.; Coppes, R.P.; Engedal, N.; Mari, M.; Reggiori, F. Chloroquine inhibits autophagic flux by decreasing autophagosome-lysosome fusion. Autophagy 2018, 48, 1435–1455. [Google Scholar] [CrossRef] [PubMed]
- Choi, D.S.; Blanco, E.; Kim, Y.S.; Rodriguez, A.A.; Zhao, H.; Huang, T.H.M.; Chen, C.L.; Jin, G.; Landis, M.D.; Burey, L.A.; et al. Chloroquine eliminates cancer stem cells through deregulation of Jak2 and DNMT1. Stem Cells 2014, 32, 2309–2323. [Google Scholar] [CrossRef] [PubMed]
- Liang, D.H.; Choi, D.S.; Ensor, J.E.; Kaipparettu, B.A.; Bass, B.L.; Chang, J.C. The autophagy inhibitor chloroquine targets cancer stem cells in triple negative breast cancer by inducing mitochondrial damage and impairing DNA break repair. Cancer Lett. 2016, 376, 249–258. [Google Scholar] [CrossRef] [PubMed]
- Fauzi, Y.R.; Nakahata, S.; Chilmi, S.; Ichikawa, T.; Nueangphuet, P.; Yamaguchi, R.; Nakamura, T.; Shimoda, K.; Morishita, K. Antitumor effects of chloroquine/hydroxychloroquine mediated by inhibition of the NF-κB signaling pathway through abrogation of autophagic p47 degradation in adult T-cell leukemia/lymphoma cells. PLoS ONE 2021, 16, e0256320. [Google Scholar] [CrossRef]
- Fukuda, T.; Oda, K.; Wada-Hiraike, O.; Sone, K.; Inaba, K.; Ikeda, Y.; Miyasaka, A.; Kashiyama, T.; Tanikawa, M.; Arimoto, T.; et al. The anti-malarial chloroquine suppresses proliferation and overcomes cisplatin resistance of endometrial cancer cells via autophagy inhibition. Gynecol. Oncol. 2015, 137, 538–545. [Google Scholar] [CrossRef] [PubMed]
- Lin, Y.C.; Lin, J.F.; Wen, S.I.; Yang, S.C.; Tsai, T.F.; Chen, H.E.; Chou, K.Y.; Hwang, T.I. Chloroquine and hydroxychloroquine inhibit bladder cancer cell growth by targeting basal autophagy and enhancing apoptosis. Kaohsiung J. Med. Sci. 2017, 33, 215–223. [Google Scholar] [CrossRef] [PubMed]
- Mulcahy Levy, J.M.; Zahedi, S.; Griesinger, A.M.; Morin, A.; Davies, K.D.; Aisner, D.L.; Kleinschmidt-DeMasters, B.K.; Fitzwalter, B.E.; Goodall, M.L.; Thorburn, J.; et al. Autophagy inhibition overcomes multiple mechanisms of resistance to BRAF inhibition in brain tumors. Elife 2017, 6, e19671. [Google Scholar] [CrossRef] [PubMed]
- Pagotto, A.; Pilotto, G.; Mazzoldi, E.L.; Nicoletto, M.O.; Frezzini, S.; Pastò, A.; Amadori, A. Autophagy inhibition reduces chemoresistance and tumorigenic potential of human ovarian cancer stem cells. Cell Death Dis. 2017, 8, e2943. [Google Scholar] [CrossRef]
- Duarte, D.; Vale, N. New trends for antimalarial drugs: Synergism between antineoplastics and antimalarials on breast cancer cells. Biomolecules 2020, 10, 1623. [Google Scholar] [CrossRef]
- El-Gowily, A.H.; Loutfy, S.A.; Ali, E.M.; Mohamed, T.M.; Mansour, M.A. Tioconazole and chloroquine act synergistically to combat doxorubicin-induced toxicity via inactivation of PI3K/AKT/mTOR signaling mediated ROS-dependent apoptosis and autophagic flux inhibition in MCF-7 breast cancer cells. Pharmaceuticals 2021, 14, 254. [Google Scholar] [CrossRef]
- Kazakova, D.; Shimamura, M.; Kurashige, T.; Hamada, K.; Nagayama, Y. Re-evaluation of the role of autophagy in thyroid cancer treatment. Endocr. J. 2022, 69, 847–862. [Google Scholar] [CrossRef] [PubMed]
- Mattioli, R.; Ilari, A.; Colotti, B.; Mosca, L.; Fazi, F.; Colotti, G. Doxorubicin and other anthracyclines in cancers: Activity, chemoresistance and its overcoming. Mol. Asp. Med. 2023, 93, 101205. [Google Scholar] [CrossRef]
- Kciuk, M.; Gielecińska, A.; Mujwar, S.; Kołat, D.; Kałuzińska-Kołat, Ż.; Celik, I.; Kontek, R. Doxorubicin-an agent with multiple mechanisms of anticancer activity. Cells 2023, 12, 659. [Google Scholar] [CrossRef] [PubMed]
- Christidi, E.; Brunham, L.R. Regulated cell death pathways in doxorubicin-induced cardiotoxicity. Cell Death Dis. 2021, 12, 339. [Google Scholar] [CrossRef] [PubMed]
- Kumar, A.; Singh, U.K.; Chaudhary, A. Targeting autophagy to overcome drug resistance in cancer therapy. Future Med. Chem. 2015, 7, 1535–1542. [Google Scholar] [CrossRef]
- Usman, R.M.; Razzaq, F.; Akbar, A.; Farooqui, A.A.; Iftikhar, A.; Latif, A.; Hassan, H.; Zhao, J.; Carew, J.S.; Nawrocki, S.T.; et al. Role and mechanism of autophagy-regulating factors in tumorigenesis and drug resistance. Asia Pac. J. Clin. Oncol. 2021, 17, 193–208. [Google Scholar] [CrossRef]
- Zhou, Y.; Chen, E.; Tang, Y.; Mao, J.; Shen, J.; Zheng, X.; Xie, S.; Zhang, S.; Wu, Y.; Liu, H.; et al. MiR-223 overexpression inhibits doxorubicin-induced autophagy by targeting FOXO3a and reverses chemoresistance in hepatocellular carcinoma cells. Cell Death Dis. 2019, 10, 843. [Google Scholar] [CrossRef]
- Yu, P.; Wang, H.Y.; Tian, M.; Li, A.X.; Chen, X.S.; Wang, X.L.; Zhang, Y.; Cheng, Y. Eukaryotic elongation factor-2 kinase regulates the cross-talk between autophagy and pyroptosis in doxorubicin-treated human melanoma cells in vitro. Acta Pharmacol. Sin. 2019, 40, 1237–1244. [Google Scholar] [CrossRef]
- Duarte, D.; Nunes, M.; Ricardo, S.; Vale, N. Combination of Antimalarial and CNS Drugs with Antineoplastic Agents in MCF-7 Breast and HT-29 Colon Cancer Cells: Biosafety Evaluation and Mechanism of Action. Biomolecules 2022, 12, 1490. [Google Scholar] [CrossRef]
- Abdel-Mohsen, M.A.; Abdel Malak, C.A.; El-Shafey, E.S. Influence of copper (I) nicotinate complex and autophagy modulation on doxorubicin-induced cytotoxicity in HCC1806 breast cancer cells. Adv. Med. Sci. 2019, 64, 202–209. [Google Scholar] [CrossRef]
- Bano, N.; Ansari, M.I.; Kainat, K.M.; Singh, V.K.; Sharma, P.K. Chloroquine synergizes doxorubicin efficacy in cervical cancer cells through flux impairment and down regulation of proteins involved in the fusion of autophagosomes to lysosomes. Biochem. Biophys. Res. Commun. 2023, 656, 131–138. [Google Scholar] [CrossRef]
- Sato, K.; Ota, N.; Endo, S.; Nakata, A.; Yamashita, H.; Tatsunami, R. Effect of chloroquine on doxorubicin-induced apoptosis in A549 cells. Anticancer Res. 2022, 42, 4025–4035. [Google Scholar] [CrossRef] [PubMed]
- Utkusavas, A.; Gurel Gurevin, E.; Yilmazer, N.; Uvez, A.; Oztay, F.; Bulut, H.; Ustunova, S.; Esener, O.B.B.; Sonmez, K.; Erol Kutucu, D.; et al. Effects of combined administration of doxorubicin and chloroquine on lung pathology in mice with solid Ehrlich ascites carcinoma. Biotech. Histochem. 2022, 97, 555–566. [Google Scholar] [CrossRef] [PubMed]
- Gurel-Gurevin, E.; Kiyan, H.T.; Esener, O.B.B.; Aydinlik, S.; Uvez, A.; Ulukaya, E.; Dimas, K.; Armutak, E.I. Chloroquine used in combination with chemotherapy synergistically suppresses growth and angiogenesis in vitro and in vivo. Anticancer Res. 2018, 38, 4011–4020. [Google Scholar] [CrossRef] [PubMed]
- Zhao, H.; Yu, J.; Zhang, R.; Chen, P.; Jiang, H.; Yu, W. Doxorubicin prodrug-based nanomedicines for the treatment of cancer. Eur. J. Med. Chem. 2023, 258, 115612. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.; Choi, M.K.; Song, I.S. Recent advances in doxorubicin formulation to enhance pharmacokinetics and tumor targeting. Pharmaceuticals 2023, 16, 802. [Google Scholar] [CrossRef] [PubMed]
- Gabizon, A.A. Pegylated liposomal doxorubicin: Metamorphosis of an old drug into a new form of chemotherapy. Cancer Investig. 2001, 19, 424–436. [Google Scholar] [CrossRef]
- Chiang, C.F.; Hsu, Y.H.; Liu, C.C.; Liang, P.C.; Miaw, S.C.; Lin, W.L. Pulsed-wave ultrasound hyperthermia enhanced nanodrug delivery combined with chloroquine exerts effective antitumor response and postpones recurrence. Sci. Rep. 2019, 9, 12448. [Google Scholar] [CrossRef]
- Chiang, C.F.; Wang, Z.Z.; Hsu, Y.H.; Miaw, S.C.; Lin, W.L. Exercise improves the outcome of anticancer treatment with ultrasound-hyperthermia-enhanced nanochemotherapy and autophagy inhibitor. PLoS ONE 2023, 18, e0288380. [Google Scholar] [CrossRef]
- Dos Reis, S.B.; de Oliveira Silva, J.; Garcia-Fossa, F.; Leite, E.A.; Malachias, A.; Pound-Lana, G.; Mosqueira, V.C.F.; Oliveira, M.C.; de Barros, A.L.B.; de Jesus, M.B. Mechanistic insights into the intracellular release of doxorubicin from pH-sensitive liposomes. Biomed. Pharmacother. 2021, 134, 110952. [Google Scholar] [CrossRef]
- Xu, S.; Zhong, Y.; Nie, C.; Pan, Y.; Adeli, M.; Haag, R. Activation co-delivery of doxorubicin and chloroquine by polyglycerol functionalized MoS2 nanosheets for efficient multidrug-resistant cancer therapy. Macromol. Biosci. 2021, 21, e2100233. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Qiu, L. Drug-induced self-assembled nanovesicles for doxorubicin resistance reversal via autophagy inhibition and delivery synchronism. Theranostics 2022, 12, 3977–3994. [Google Scholar] [CrossRef]
- Chen, T.; Zeng, C.; Li, Z.; Wang, J.; Sun, F.; Huang, J.; Lu, S.; Zhu, J.; Zhang, Y.; Sun, X.; et al. Investigation of chemoresistance to first-line chemotherapy and its possible association with autophagy in high-risk neuroblastoma. Front. Oncol. 2022, 12, 1019106. [Google Scholar] [CrossRef] [PubMed]
- Zhu, J.; Zheng, Y.; Zhang, H.; Zhu, J.; Sun, H. Low concentration of chloroquine enhanced efficacy of cisplatin in the treatment of human ovarian cancer dependent on autophagy. Am. J. Transl. Res. 2017, 9, 4046–4058. [Google Scholar] [PubMed]
- Wear, D.; Bhagirath, E.; Balachandar, A.; Vegh, C.; Pandey, S. Autophagy inhibition via hydroxychloroquine or 3-methyladenine enhances chemotherapy-induced apoptosis in neuro-blastoma and glioblastoma. Int. J. Mol. Sci. 2023, 24, 12052. [Google Scholar] [CrossRef] [PubMed]
- Du, H.; Yang, W.; Chen, L.; Shi, M.; Seewoo, V.; Wang, J.; Lin, A.; Liu, Z.; Qiu, W. Role of autophagy in resistance to oxaliplatin in hepatocellular carcinoma cells. Oncol. Rep. 2012, 27, 143–150. [Google Scholar] [CrossRef] [PubMed]
- Selvakumaran, M.; Amaravadi, R.K.; Vasilevskaya, I.A.; O'Dwyer, P.J. Autophagy inhibition sensitizes colon cancer cells to antiangiogenic and cytotoxic therapy. Clin. Cancer Res. 2013, 19, 2995–3007. [Google Scholar] [CrossRef] [PubMed]
- Shi, Y.; Lin, G.; Zheng, H.; Mu, D.; Chen, H.; Lu, Z.; He, P.; Zhang, Y.; Liu, C.; Lin, Z.; et al. Biomimetic nanoparticles blocking autophagy for enhanced chemotherapy and metastasis inhibition via reversing focal adhesion disassembly. J. Nanobiotechnol. 2021, 19, 447. [Google Scholar] [CrossRef]
- Wang, F.-T.; Wang, H.; Wang, Q.-W.; Pan, M.-S.; Li, X.-P.; Sun, W.; Fan, Y.-Z. Inhibition of autophagy by chloroquine enhances the antitumor activity of gemcitabine for gallbladder cancer. Cancer Chemother. Pharmacol. 2020, 86, 221–232. [Google Scholar] [CrossRef]
- Hara, K.; Horikoshi, Y.; Morimoto, M.; Nakaso, K.; Sunaguchi, T.; Kurashiki, T.; Nakayama, Y.; Hanaki, T.; Yamamoto, M.; Sakamoto, T.; et al. TYRO3 promotes chemoresistance via increased LC3 expression in pancreatic cancer. Transl. Oncol. 2023, 28, 101608. [Google Scholar] [CrossRef]
- Mishima, Y.; Terui, Y.; Mishima, Y.; Taniyama, A.; Kuniyoshi, R.; Takizawa, T.; Kimura, S.; Ozawa, K.; Hatake, K. Autophagy and autophagic cell death are next targets for elimination of the resistance to tyrosine kinase inhibitors. Cancer Sci. 2008, 99, 2200–2208. [Google Scholar] [CrossRef] [PubMed]
- Crowley, L.C.; O'Donovan, T.R.; Nyhan, M.J.; McKenna, S.L. Pharmacological agents with inherent anti-autophagic activity improve the cytotoxicity of imatinib. Oncol. Rep. 2013, 29, 2261–2268. [Google Scholar] [CrossRef] [PubMed]
- Gupta, A.; Roy, S.; Lazar, A.J.; Wang, W.L.; McAuliffe, J.C.; Reynoso, D.; McMahon, J.; Taguchi, T.; Floris, G.; Debiec-Rychter, M.; et al. Autophagy inhibition and antimalarials promote cell death in gastrointestinal stromal tumor (GIST). Proc. Natl. Acad. Sci. USA 2010, 107, 14333–14338. [Google Scholar] [CrossRef] [PubMed]
- Zheng, S.; Shu, Y.; Lu, Y.; Sun, Y. Chloroquine combined with imatinib overcomes imatinib resistance in gastrointestinal stromal tumors by inhibiting autophagy via the MAPK/ERK pathway. Onco Targets Ther. 2020, 13, 6433–6441. [Google Scholar] [CrossRef]
- Xue, L.; Gong, Z.; Vlantis, A.C.; Chan, J.Y.; Meehan, K.; van Hasselt, C.A.; Li, D.; Zeng, X.; Wei, M.; Tong, M.C.; et al. Autophagy regulates anti-angiogenic property of lenvatinib in thyroid cancer. Am. J. Cancer Res. 2023, 13, 1457–1470. [Google Scholar] [PubMed]
- Feng, H.; Cheng, X.; Kuang, J.; Chen, L.; Yuen, S.; Shi, M.; Liang, J.; Shen, B.; Jin, Z.; Yan, J.; et al. Apatinib-induced protective autophagy and apoptosis through the AKT-mTOR pathway in anaplastic thyroid cancer. Cell Death Dis. 2018, 9, 1030. [Google Scholar] [CrossRef]
- Wang, Y.M.; Xu, X.; Tang, J.; Sun, Z.Y.; Fu, Y.J.; Zhao, X.J.; Ma, X.M.; Ye, Q. Apatinib induces endoplasmic reticulum stress-mediated apoptosis and autophagy and potentiates cell sensitivity to paclitaxel via the IRE-1α-AKT-mTOR pathway in esophageal squamous cell carcinoma. Cell Biosci. 2021, 11, 124. [Google Scholar] [CrossRef]
- Ishibashi, Y.; Nakamura, O.; Yamagami, Y.; Nishimura, H.; Fukuoka, N.; Yamamoto, T. Chloroquine Enhances Rapamycin-induced Apoptosis in MG63 Cells. Anticancer Res. 2019, 39, 649–654. [Google Scholar] [CrossRef]
- Masaki, N.; Aoki, Y.; Obara, K.; Kubota, Y.; Bouvet, M.; Miyazaki, J.; Hoffman, R.M. Targeting autophagy with the synergistic combination of chloroquine and rapamycin as a novel effective treatment for well-differentiated liposarcoma. Cancer Genom. Proteom. 2023, 20, 317–322. [Google Scholar] [CrossRef]
- Jiang, B.; Cui, Y.; Ma, X.; Zhang, Y.; Feng, X.; Yang, T.; Feng, L.; Guo, W.; Li, Y.; Wang, T.; et al. Crosstalk between autophagy inhibitor and salidroside-induced apoptosis: A novel strategy for autophagy-based treatment of hepatocellular cancer. Int. Immunopharmacol. 2023, 124 Pt B, 111040. [Google Scholar] [CrossRef]
- Abdelwahab, M.; Saeed, H.; Elnikhely, N.; Nematalla, H. Synergistic effect of Dactolisib/Lys05 combination on autophagy in A549 cells. Acta Biochim. Pol. 2023, 70, 615–622. [Google Scholar] [CrossRef] [PubMed]
- Grimaldi, A.; Santini, D.; Zappavigna, S.; Lombardi, A.; Misso, G.; Boccellino, M.; Desiderio, V.; Vitiello, P.P.; Di Lorenzo, G.; Zoccoli, A.; et al. Antagonistic effects of chloroquine on autophagy occurrence potentiate the anticancer effects of everolimus on renal cancer cells. Cancer Biol. Ther. 2015, 16, 567–579. [Google Scholar] [CrossRef] [PubMed]
- Erkisa, M.; Aydinlik, S.; Cevatemre, B.; Aztopal, N.; Akar, R.O.; Celikler, S.; Yilmaz, V.T.; Ari, F.; Ulukaya, E. A promising therapeutic combination for metastatic prostate cancer: Chloroquine as autophagy inhibitor and palladium(II) barbiturate complex. Biochimie 2020, 175, 159–172. [Google Scholar] [CrossRef] [PubMed]
- Cook, K.; Wärri, A.; Soto-Pantoja, D.R.; Clarke, P.A.; Cruz, M.I.; Zwart, A.; Clarke, R. Chloroquine inhibits autophagy to potentiate antiestrogen responsiveness in ER+ breast cancer. Clin. Cancer Res. 2014, 20, 3222–3232. [Google Scholar] [CrossRef] [PubMed]
- Cocco, S.; Leone, A.; Roca, M.S.; Lombardi, R.; Piezzo, M.; Caputo, R.; Ciardiello, C.; Costantini, S.; Bruzzese, F.; Sisalli, M.J.; et al. Inhibition of autophagy by chloroquine prevents resistance to PI3K/AKT inhibitors and potentiates their antitumor effect in combination with paclitaxel in triple negative breast cancer models. J. Transl. Med. 2022, 20, 290. [Google Scholar] [CrossRef]
- Sousa-Pimenta, M.; Estevinho, L.M.; Szopa, A.; Basit, M.; Khan, K.; Armaghan, M.; Ibrayeva, M.; Sönmez Gürer, E.; Calina, D.; Hano, C.; et al. Chemotherapeutic properties and side-effects associated with the clinical practice of tepene alkaloids: Paclitaxel, docetaxel, and cabazitaxel. Front. Pharmacol. 2023, 14, 1157306. [Google Scholar] [CrossRef]
- Liu, Y.; Zhao, F.; Wang, Q.; Zhao, Q.; Hou, G.; Meng, Q. Current perspectives on paclitaxel: Focus on its production, delivery and combination therapy. Mini Rev. Med. Chem. 2023, 23, 1780–1796. [Google Scholar] [CrossRef]
- Škubník, J.; Svobodová Pavlíčková, V.; Ruml, T.; Rimpelová, S. Autophagy in cancer resistance to paclitaxel: Development of combination strategies. Biomed. Pharmacother. 2023, 161, 114458. [Google Scholar] [CrossRef]
- Yuan, Z.; Cai, J.; Du, Q.; Ma, Q.; Xu, L.; Cai, Y.; Zhong, X.; Guo, X. chloroquine sensitizes esophageal carcinoma EC109 cells to paclitaxel by inhibiting autophagy. Crit. Rev. Eukaryot. Gene Expr. 2023, 33, 43–53. [Google Scholar] [CrossRef]
- Anand, K.; Niravath, P.; Patel, T.; Ensor, J.; Rodriguez, A.; Boone, T.; Wong, S.T.; Chang, J.C. A Phase II Study of the Efficacy and Safety of Chloroquine in Combination with Taxanes in the Treatment of Patients with Advanced or Metastatic Anthracycline-refractory Breast Cancer. Clin. Breast Cancer 2021, 21, 199–204. [Google Scholar] [CrossRef]
- Forgie, B.N.; Prakash, R.; Telleria, C.M. Revisiting the anti-cancer toxicity of clinically approved platinating derivatives. Int. J. Mol. Sci. 2022, 23, 15410. [Google Scholar] [CrossRef] [PubMed]
- Mason, S.R.; Willson, M.L.; Egger, S.J.; Beith, J.; Dear, R.F.; Goodwin, A. Platinum-based chemotherapy for early triple-negative breast cancer. Cochrane Database Syst. Rev. 2023, 9, CD014805. [Google Scholar] [CrossRef] [PubMed]
- Lu, E.; Gareev, I.; Yuan, C.; Liang, Y.; Sun, J.; Chen, X.; Beylerli, O.; Sufianov, A.; Zhao, S.; Yang, G. The mechanisms of current platinum anticancer drug resistance in the glioma. Curr. Pharm. Des. 2022, 28, 1863–1869. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.Q.; Fang, N.; Liu, X.M.; Xiong, S.P.; Liao, Y.Q.; Jin, W.J.; Song, R.F.; Wan, Y.Y. Antitumor activity of chloroquine in combination with cisplatin in human gastric cancer xenografts. Asian Pac. J. Cancer Prev. 2015, 16, 3907–3912. [Google Scholar] [CrossRef]
- Guo, X.L.; Li, D.; Hu, F.; Song, J.R.; Zhang, S.S.; Deng, W.J.; Sun, K.; Zhao, Q.D.; Xie, X.Q.; Song, Y.J.; et al. Targeting autophagy potentiates chemotherapy-induced apoptosis and proliferation inhibition in hepatocarcinoma cells. Cancer Lett. 2012, 320, 171–179. [Google Scholar] [CrossRef]
- Karim, N.A.; Ullah, A.; Ahmad, I.; Bahassi, E.; Olowokure, O.; Khaled, A.; Davis, H.; Morris, J.C. A phase I trial to determine the safety and tolerability of autophagy inhibition using chloroquine or hydroxychloroquine in combination with carboplatin and gemcitabine in patients with advanced solid tumors. Front. Oncol. 2022, 12, 811411. [Google Scholar] [CrossRef] [PubMed]
- Abdel-Karim, N.; Gaber, O.; Aljohani, H.M.; Eldessouki, I.; Bahassi, E.M.; Morris, J. Exosomes as a surrogate marker for autophagy in peripheral blood, correlative data from phase I study of chloroquine in combination with carboplatin/gemcitabine in advanced solid tumors. Asian Pac. J. Cancer Prev. 2019, 20, 3789–3796. [Google Scholar] [CrossRef][Green Version]
- Beutel, A.K.; Halbrook, C.J. Barriers and opportunities for gemcitabine in pancreatic cancer therapy. Am. J. Physiol. Cell Physiol. 2023, 324, C540–C552. [Google Scholar] [CrossRef]
- Matsumoto, S.; Nakata, K.; Sagara, A.; Guan, W.; Ikenaga, N.; Ohuchida, K.; Nakamura, M. Efficient pre-treatment for pancreatic cancer using chloroquine-loaded nanoparticles targeting pancreatic stellate cells. Oncol. Lett. 2021, 22, 633. [Google Scholar] [CrossRef]
- Chen, X.; Tao, Y.; He, M.; Deng, M.; Guo, R.; Sheng, Q.; Wang, X.; Ren, K.; Li, T.; He, X.; et al. Co-delivery of autophagy inhibitor and gemcitabine using a pH-activatable core-shell nanobomb inhibits pancreatic cancer progression and metastasis. Theranostics 2021, 11, 8692–8705. [Google Scholar] [CrossRef]
- AlMasri, S.S.; Zenati, M.S.; Desilva, A.; Nassour, I.; Boone, B.A.; Singhi, A.D.; Bartlett, D.L.; Liotta, L.A.; Espina, V.; Loughran, P.; et al. Encouraging long-term survival following autophagy inhibition using neoadjuvant hydroxychloroquine and gemcitabine for high-risk patients with resectable pancreatic carcinoma. Cancer Med. 2021, 10, 7233–7241. [Google Scholar] [CrossRef] [PubMed]
- Zheng, Y.; Huang, C.; Lu, L.; Yu, K.; Zhao, J.; Chen, M.; Liu, L.; Sun, Q.; Lin, Z.; Zheng, J.; et al. STOML2 potentiates metastasis of hepatocellular carcinoma by promoting PINK1-mediated mitophagy and regulates sensitivity to lenvatinib. J. Hematol. Oncol. 2021, 14, 16. [Google Scholar] [CrossRef] [PubMed]
- Masaki, N.; Aoki, Y.; Kubota, Y.; Obara, K.; Miyazaki, J.; Hoffman, R.M. Chloroquine combined with rapamycin arrests tumor growth in a patient-derived orthotopic xenograft (PDOX) mouse model of dedifferentiated liposarcoma. In Vivo 2022, 36, 2630–2637. [Google Scholar] [CrossRef] [PubMed]
- Delaney, J.R.; Patel, C.B.; Willis, K.M.; Haghighiabyaneh, M.; Axelrod, J.; Tancioni, I.; Lu, D.; Bapat, J.; Young, S.; Cadassou, O.; et al. Haploinsufficiency Networks Identify Targetable Patterns of Allelic Deficiency in Low Mutation Ovarian Cancer. Nat. Commun. 2017, 8, 14423. [Google Scholar] [CrossRef]
- Kronick, O.; Chen, X.; Mehra, N.; Varmeziar, A.; Fisher, R.; Kartchner, D.; Kota, V.; Mitchell, C.S. Hematological adverse events with tyrosine kinase inhibitors for chronic myeloid leukemia: A systematic review with meta-analysis. Cancers 2023, 15, 4354. [Google Scholar] [CrossRef] [PubMed]
- Golčić, M.; Jones, R.L.; Huang, P.; Napolitano, A. Evaluation of systemic treatment options for gastrointestinal stromal tumours. Cancers 2023, 15, 4081. [Google Scholar] [CrossRef] [PubMed]
- Horne, G.A.; Stobo, J.; Kelly, C.; Mukhopadhyay, A.; Latif, A.L.; Dixon-Hughes, J.; McMahon, L.; Cony-Makhoul, P.; Byrne, J.; Smith, G.; et al. A randomised phase II trial of hydroxychloroquine and imatinib versus imatinib alone for patients with chronic myeloid leukaemia in major cytogenetic response with residual disease. Leukemia 2020, 34, 1775–1786. [Google Scholar] [CrossRef]
- Hua, X.; Yin, Z.; Liang, J.; Chen, W.; Gong, H. Efficacy and safety comparison between Lenvatinib and Sorafenib in hepatocellular carcinoma treatment: A systematic review and meta-analysis of real-world study. Eur. J. Gastroenterol. Hepatol. 2024, 36(1), 120–128. [Google Scholar] [CrossRef]
- Buttell, A.; Qiu, W. The action and resistance mechanisms of Lenvatinib in liver cancer. Mol. Carcinog. 2023, 62, 1918–1934. [Google Scholar] [CrossRef]
- Peng, D.; Cai, Y.; Chen, G.; Hou, M.; Luo, X.; Dongzhi, Z.; Xie, H.; Liu, Y. Efficacy and safety of apatinib versus sorafenib/placebo in first-line treatment for intermediate and advanced primary liver cancer: A systematic review and meta-analysis. Front. Pharmacol. 2023, 14, 1101063. [Google Scholar] [CrossRef]
- Glaviano, A.; Foo, A.S.C.; Lam, H.Y.; Yap, K.C.H.; Jacot, W.; Jones, R.H.; Eng, H.; Nair, M.G.; Makvandi, P.; Geoerger, B.; et al. PI3K/AKT/mTOR signaling transduction pathway and targeted therapies in cancer. Mol. Cancer 2023, 22, 138. [Google Scholar] [CrossRef] [PubMed]
- Wylaź, M.; Kaczmarska, A.; Pajor, D.; Hryniewicki, M.; Gil, D.; Dulińska-Litewka, J. Exploring the role of PI3K/AKT/mTOR inhibitors in hormone-related cancers: A focus on breast and prostate cancer. Biomed. Pharmacother. 2023, 168, 115676. [Google Scholar] [CrossRef] [PubMed]
- Singh, S.; Barik, D.; Lawrie, K.; Mohapatra, I.; Prasad, S.; Naqvi, A.R.; Singh, A.; Singh, G. Unveiling novel avenues in mtor-targeted therapeutics: Advancements in glioblastoma treatment. Int. J. Mol. Sci. 2023, 24, 14960. [Google Scholar] [CrossRef] [PubMed]
- Jiang, B.; Feng, L.; Yang, T.; Guo, W.; Li, Y.; Wang, T.; Liu, C.; Su, H. Combination of chloroquine diphosphate and salidroside induces human liver cell apoptosis via regulation of mitochondrial dysfunction and autophagy. Mol. Med. Rep. 2023, 27, 37. [Google Scholar] [CrossRef] [PubMed]
- Haas, N.; Appleman, L.J.; Stein, M.; Redlinger, M.; Wilks, M.; Xu, X.; Onorati, A.; Kalavacharla, A.; Kim, T.; Zhen, C.J.; et al. Autophagy Inhibition to Augment mTOR Inhibition: A Phase I/II Trial of Everolimus and Hydroxychloroquine in Patients with Previously Treated Renal Cell Carcinoma. Clin. Cancer Res. 2019, 25, 2080–2087. [Google Scholar] [CrossRef]
- Rangwala, R.; Chang, Y.C.; Hu, J.; Algazy, K.M.; Evans, T.L.; Fecher, L.A.; Schuchter, L.M.; Torigian, D.A.; Panosian, J.T.; Troxel, A.B.; et al. Combined MTOR and autophagy inhibition: Phase I trial of hydroxychloroquine and temsirolimus in patients with advanced solid tumors and melanoma. Autophagy 2014, 10, 1391–1402. [Google Scholar] [CrossRef] [PubMed]
- Briceño, E.; Reyes, S.; Sotelo, J. Therapy of glioblastoma multiforme improved by the antimutagenic chloroquine. Neurosurg. Focus 2003, 14, e3. [Google Scholar] [CrossRef]
- Sotelo, J.; Briceño, E.; López-González, M.A. Adding chloroquine to conventional treatment for glioblastoma multiforme: A randomized, double-blind, placebo-controlled trial. Ann. Intern. Med. 2006, 144, 337–343. [Google Scholar] [CrossRef]
- Deng, Q.; Tao, S.; Huang, H.; Lv, Q.; Wang, W. Chloroquine Supplementation for the Treatment of Glioblastoma: A Meta-analysis of Randomized Controlled Studies. Clin. Neuropharmacol. 2023, 46, 1–5. [Google Scholar] [CrossRef]
- Vogl, D.T.; Stadtmauer, E.A.; Tan, K.-S.; Heitjan, D.F.; Davis, L.E.; Pontiggia, L.; Rangwala, R.; Piao, S.; Chang, Y.C.; Scott, E.C.; et al. Combined Autophagy and Proteasome Inhibition: A Phase 1 Trial of Hydroxychloroquine and Bortezomib in Patients With Relapsed/Refractory Myeloma. Autophagy 2014, 10, 1380–1390. [Google Scholar] [CrossRef]
- Loh, J.S.; Rahim, N.A.; Tor, Y.S.; Foo, J.B. Simultaneous proteasome and autophagy inhibition synergistically enhances cytotoxicity of doxorubicin in breast cancer cells. Cell Biochem. Funct. 2022, 40, 403–416. [Google Scholar] [CrossRef] [PubMed]
- Malhotra, J.; Jabbour, S.; Orlick, M.; Riedlinger, G.; Guo, Y.; White, E.; Aisner, J. Phase Ib/II study of hydroxychloroquine in combination with chemotherapy in patients with metastatic non-small cell lung cancer (NSCLC). Cancer Treat. Res. Commun. 2019, 21, 100158. [Google Scholar] [CrossRef] [PubMed]
- Zeh, H.J.; Bahary, N.; Boone, B.A.; Singhi, A.D.; Miller-Ocuin, J.L.; Normolle, D.P.; Zureikat, A.H.; Hogg, M.E.; Bartlett, D.L.; Lee, K.K.; et al. A randomized phase II preoperative study of autophagy inhibition with high-dose hydroxychloroquine and gemcitabine/nab-paclitaxel in pancreatic cancer patients. Clin. Cancer Res. 2020, 26, 3126–3134. [Google Scholar] [CrossRef] [PubMed]
- Fei, N.; Wen, S.; Ramanathan, R.; Hogg, M.E.; Zureikat, A.H.; Lotze, M.T.; Bahary, N.; Singhi, A.D.; Zeh, H.J.; Boone, B.A. SMAD4 loss is associated with response to neoadjuvant chemotherapy plus hydroxychloroquine in patients with pancreatic adenocarcinoma. Clin. Transl. Sci. 2021, 14, 1822–1829. [Google Scholar] [CrossRef]
- Karasic, T.; O'Hara, M.H.; Loaiza-Bonilla, A.; Reiss, K.A.; Teitelbaum, U.R.; Borazanci, E.; De Jesus-Acosta, A.; Redlinger, C.; Burrell, J.A.; Laheru, D.A.; et al. Effect of Gemcitabine and nab-Paclitaxel with or Without Hydroxychloroquine on Patients with Advanced Pancreatic Cancer: A Phase 2 Randomized Clinical Trial. JAMA Oncol. 2019, 5, 993–998. [Google Scholar] [CrossRef]
- Stokkermans, T.J.; Falkowitz, D.M.; Georgios Trichonas, G. Chloroquine and Hydroxychloroquine Toxicity. In StatPearls; StatPearls Publishing: Treasure Island, FL, USA, 2023. [Google Scholar]



Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Agalakova, N.I. Chloroquine and Chemotherapeutic Compounds in Experimental Cancer Treatment. Int. J. Mol. Sci. 2024, 25, 945. https://doi.org/10.3390/ijms25020945
Agalakova NI. Chloroquine and Chemotherapeutic Compounds in Experimental Cancer Treatment. International Journal of Molecular Sciences. 2024; 25(2):945. https://doi.org/10.3390/ijms25020945
Chicago/Turabian StyleAgalakova, Natalia I. 2024. "Chloroquine and Chemotherapeutic Compounds in Experimental Cancer Treatment" International Journal of Molecular Sciences 25, no. 2: 945. https://doi.org/10.3390/ijms25020945
APA StyleAgalakova, N. I. (2024). Chloroquine and Chemotherapeutic Compounds in Experimental Cancer Treatment. International Journal of Molecular Sciences, 25(2), 945. https://doi.org/10.3390/ijms25020945