
Response to Fingolimod in Multiple Sclerosis Patients Is Associated with a Differential Transcriptomic Regulation
 , ,
, ,
Abstract
1. Introduction
2. Results
2.1. Clinical Response to Fingolimod
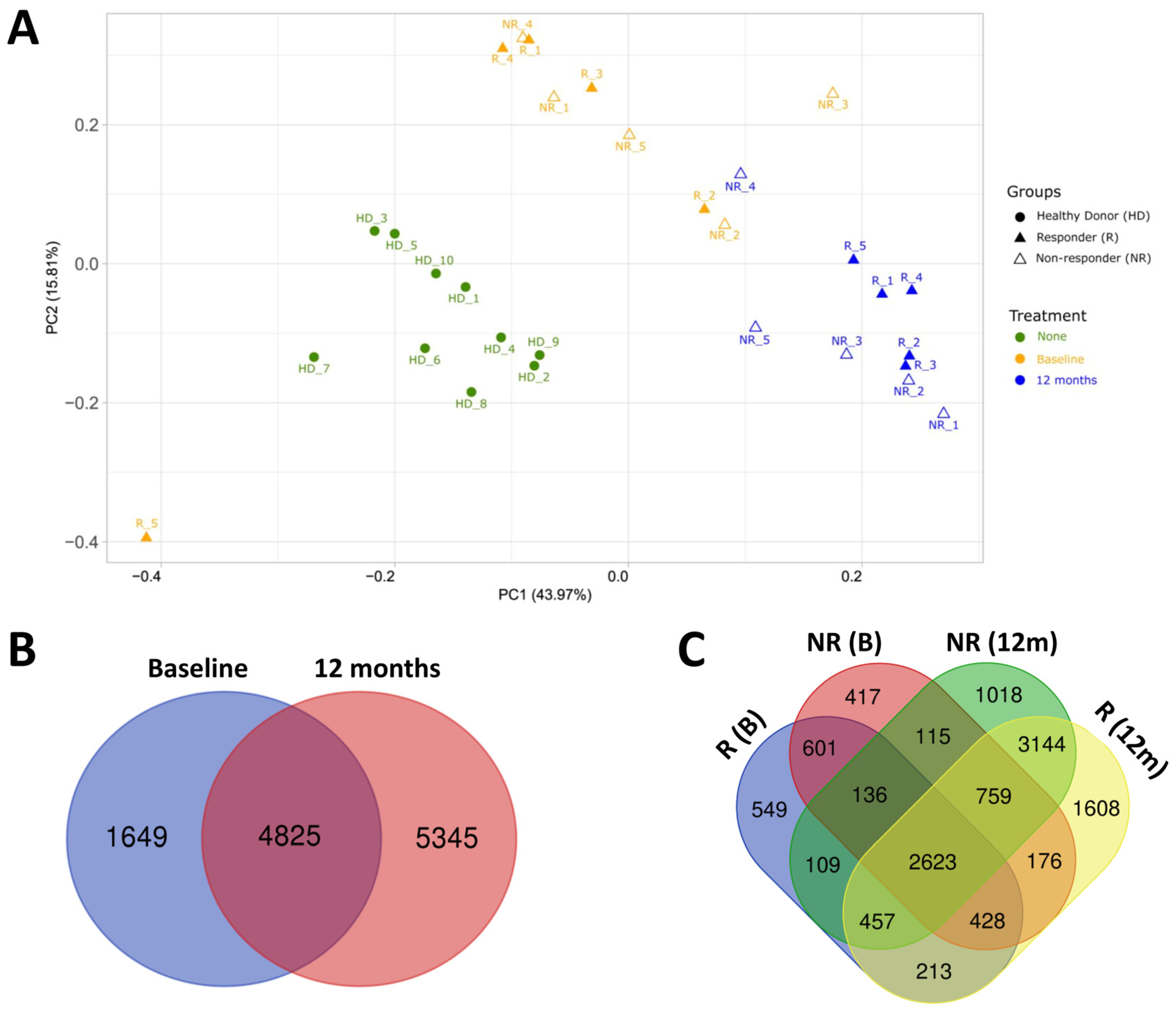
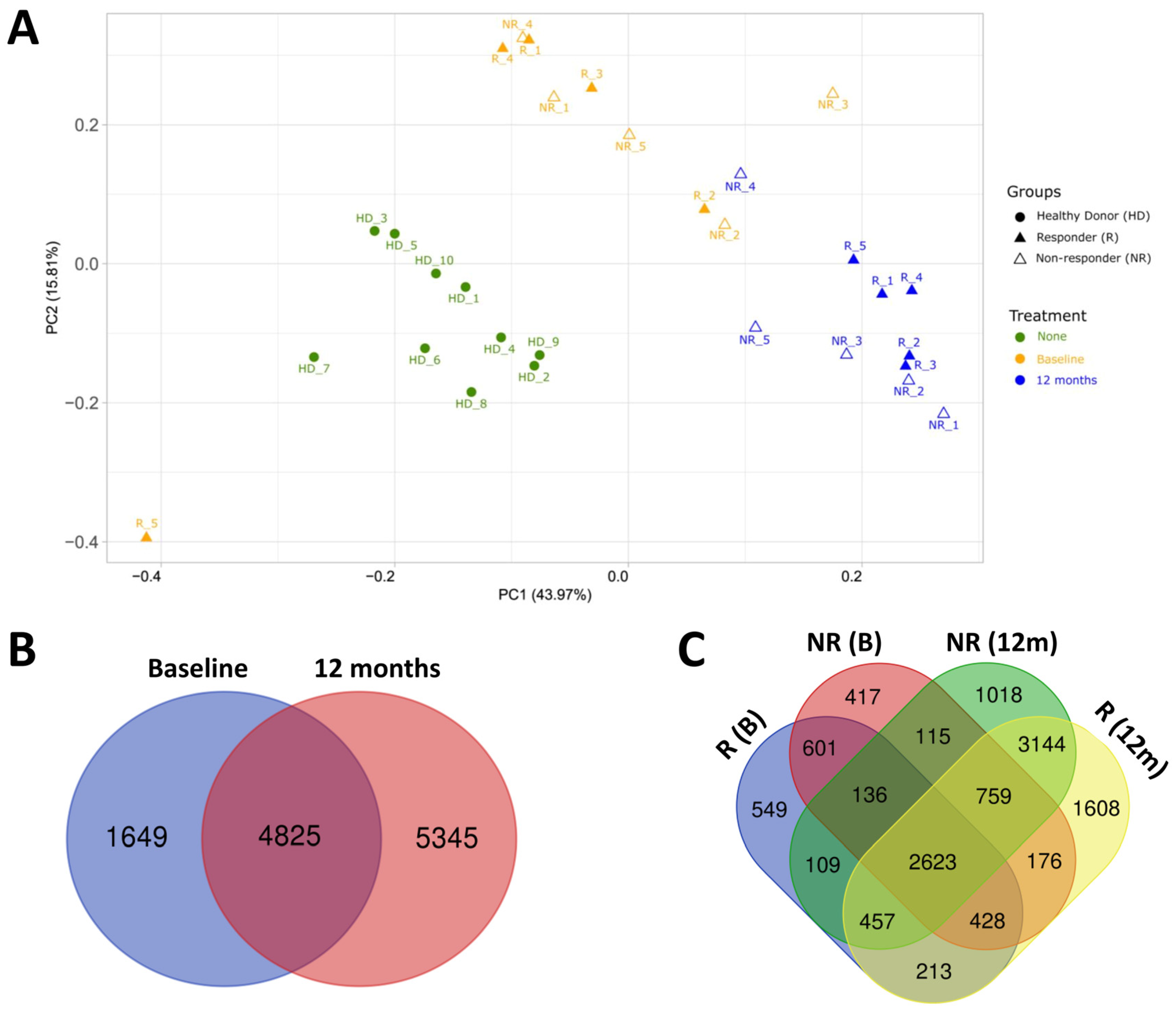
2.2. Transcriptomic Profile of HD and MS Patients
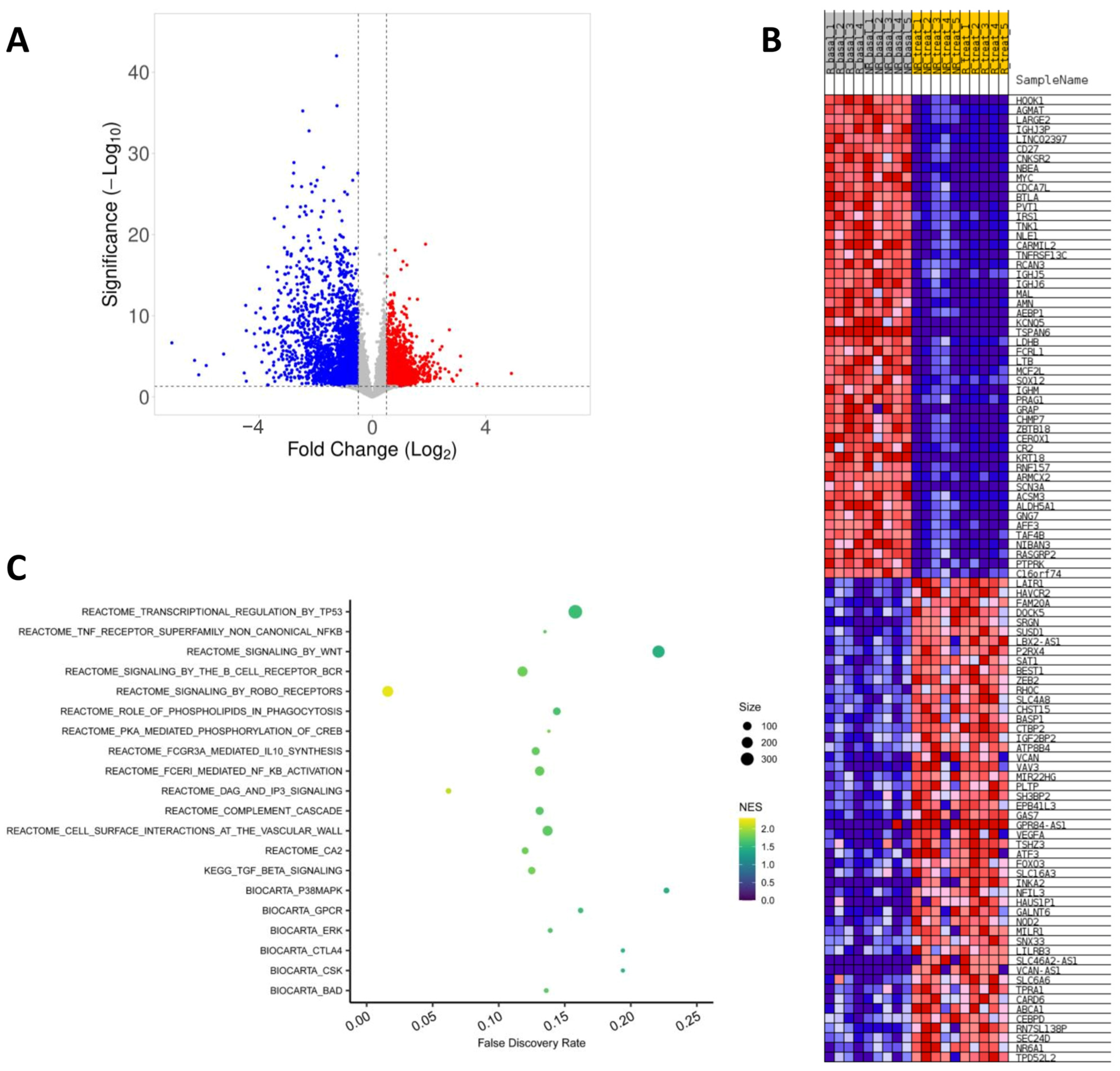
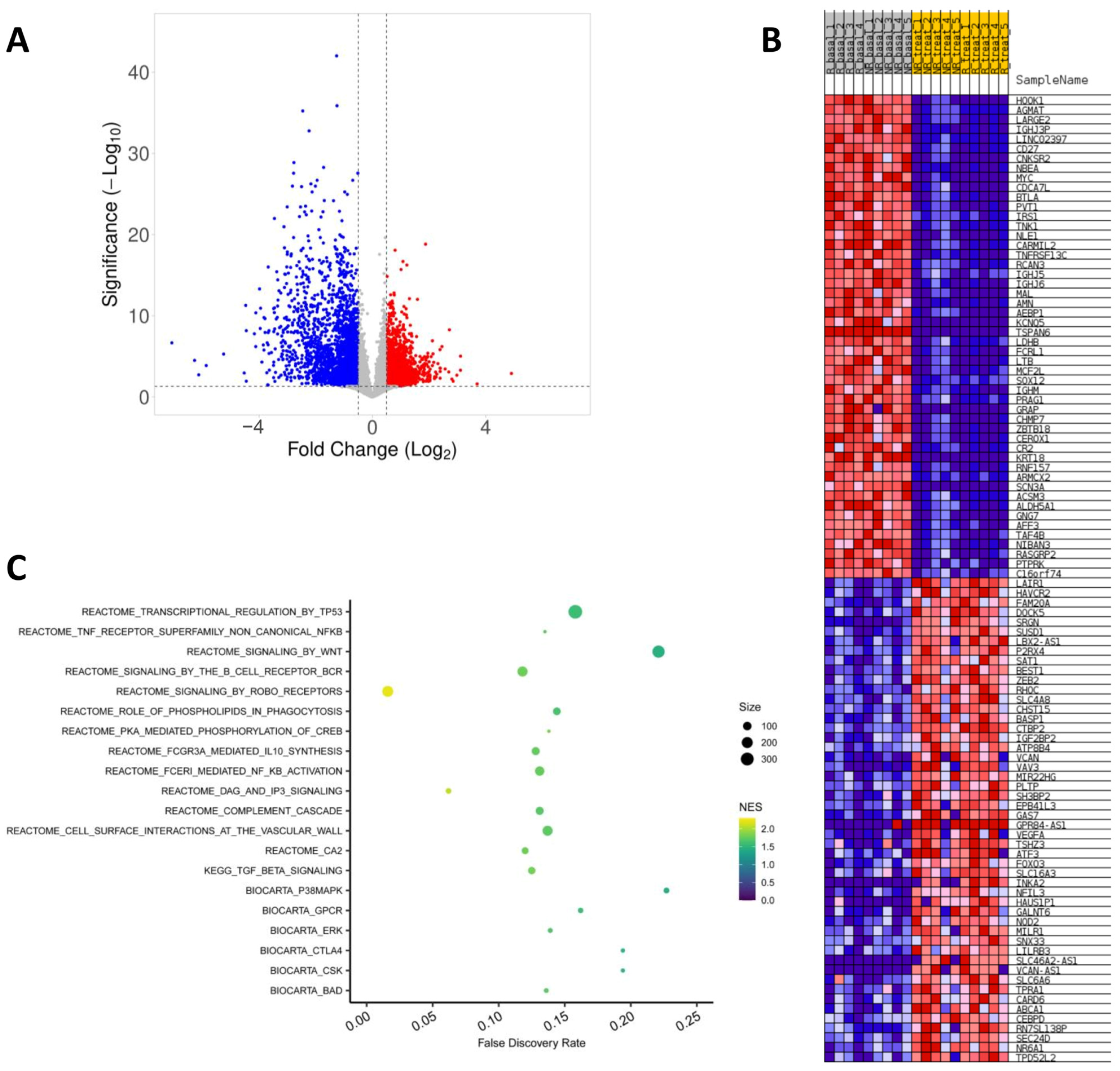
2.3. Effect of Fingolimod Treatment on Gene Expression
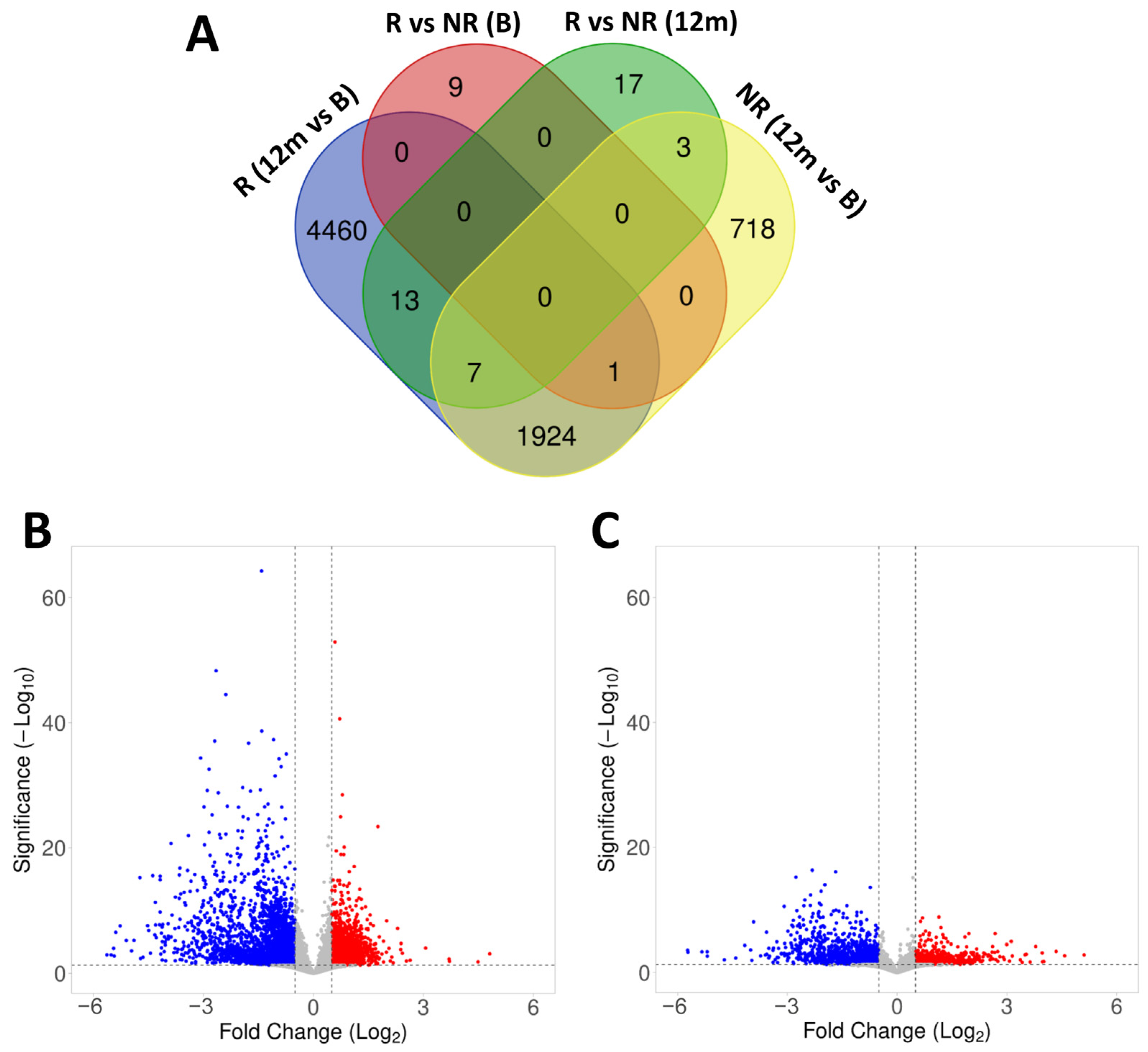
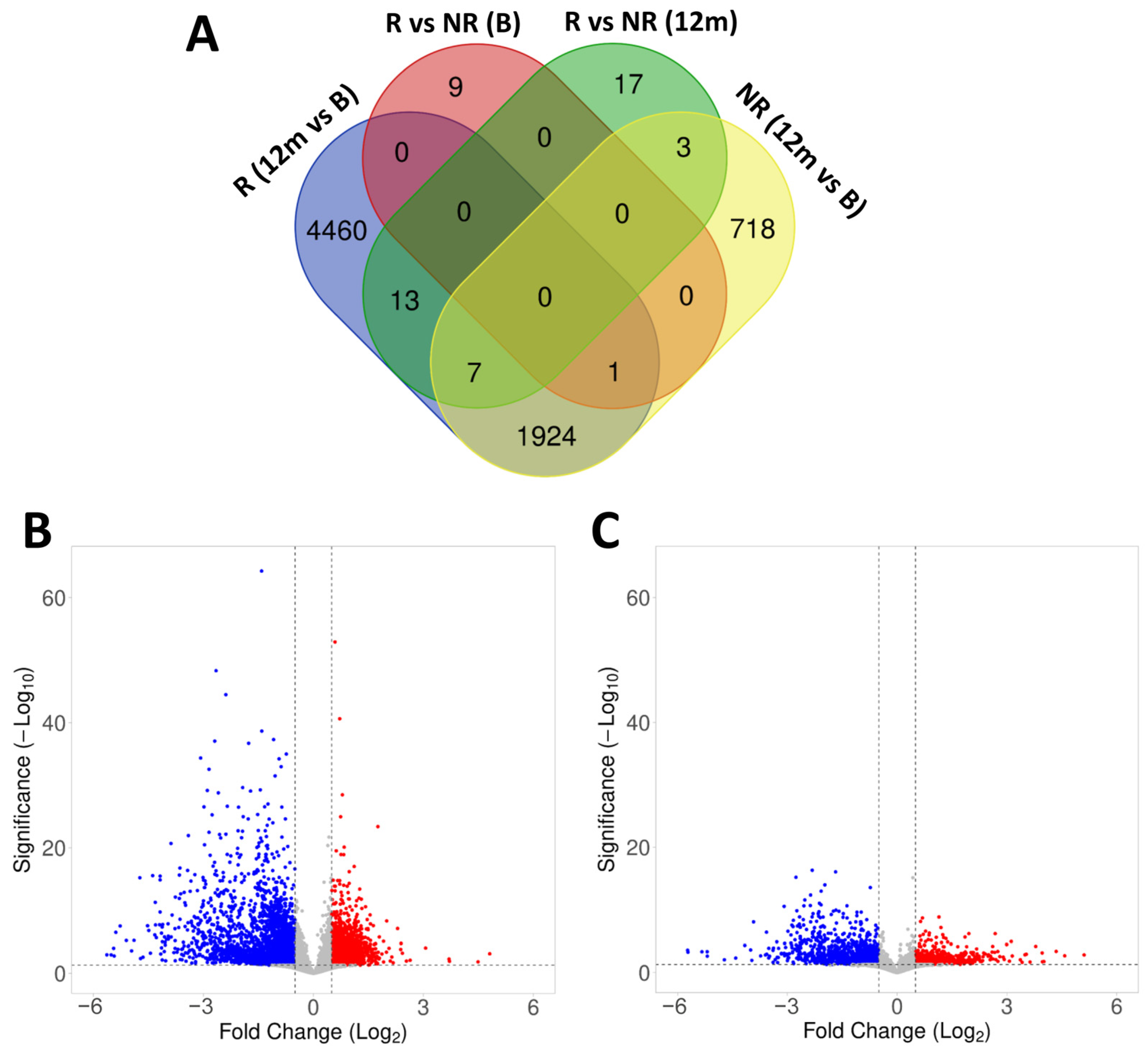
2.4. Gene Expression in Responder and Non-Responder Patients to Fingolimod
3. Discussion
4. Materials and Methods
4.1. Patients and Healthy Donors
4.2. Clinical and MRI Measures
4.3. RNA Isolation and Sequencing
4.4. Bioinformatics Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Dendrou, C.A.; Fugger, L.; Friese, M.A. Immunopathology of multiple sclerosis. Nat. Rev. Immunol. 2015, 15, 545–558. [Google Scholar] [CrossRef] [PubMed]
- Attfield, K.E.; Jensen, L.T.; Kaufmann, M.; Friese, M.A.; Fugger, L. The immunology of multiple sclerosis. Nat. Rev. Immunol. 2022, 22, 734–750. [Google Scholar] [CrossRef] [PubMed]
- Comi, G.; Radaelli, M.; Sørensen, P.S. Evolving concepts in the treatment of relapsing multiple sclerosis. Lancet 2017, 389, 1347–1356. [Google Scholar] [CrossRef] [PubMed]
- Engelhardt, B.; Comabella, M.; Chan, A. Multiple sclerosis: Immunopathological heterogeneity and its implications. Eur. J. Immunol. 2022, 52, 869–881. [Google Scholar] [CrossRef] [PubMed]
- Roy, R.; Alotaibi, A.A.; Freedman, M.S. Sphingosine 1-Phosphate Receptor Modulators for Multiple Sclerosis. CNS Drugs 2021, 35, 385–402. [Google Scholar] [CrossRef] [PubMed]
- Kappos, L.; Radue, E.-W.; O’Connor, P.; Polman, C.; Hohlfeld, R.; Calabresi, P.; Selmaj, K.; Agoropoulou, C.; Leyk, M.; Zhang-Auberson, L.; et al. A placebo-controlled trial of oral fingolimod in relapsing multiple sclerosis. N. Engl. J. Med. 2010, 362, 387–401. [Google Scholar] [CrossRef] [PubMed]
- Calabresi, P.A.; Radue, E.W.; Goodin, D.; Jeffery, D.; Rammohan, K.W.; Reder, A.T.; Vollmer, T.; Agius, M.A.; Kappos, L.; Stites, T.; et al. Safety and efficacy of fingolimod in patients with relapsing-remitting multiple sclerosis (FREEDOMS II): A double-blind, randomised, placebo-controlled, phase 3 trial. Lancet Neurol. 2014, 13, 545–556. [Google Scholar] [CrossRef]
- Hjorth, M.; Dandu, N.; Mellergård, J. Treatment effects of fingolimod in multiple sclerosis: Selective changes in peripheral blood lymphocyte subsets. PLoS ONE 2020, 15, e0228380. [Google Scholar] [CrossRef]
- Song, Z.Y.; Yamasaki, R.; Kawano, Y.; Sato, S.; Masaki, K.; Yoshimura, S.; Matsuse, D.; Murai, H.; Matsushita, T.; Kira, J.I. Peripheral blood T cell dynamics predict relapse in multiple sclerosis patients on fingolimod. PLoS ONE 2015, 10, e0124923. [Google Scholar] [CrossRef]
- Mao-Draayer, Y.; Cohen, J.A.; Bar-Or, A.; Han, M.H.; Singer, B.; Williams, I.M.; Meng, X.; Elam, C.; Weiss, J.L.; Cox, G.M.; et al. Immune cell subset profiling in multiple sclerosis after fingolimod initiation and continued treatment: The FLUENT study. Mult. Scler. J.—Exp. Transl. Clin. 2022, 8, 20552173221115023. [Google Scholar] [CrossRef]
- Blumenfeld, S.; Staun-Ram, E.; Miller, A. Fingolimod therapy modulates circulating B cell composition, increases B regulatory subsets and production of IL-10 and TGFβ in patients with Multiple Sclerosis. J. Autoimmun. 2016, 70, 40–51. [Google Scholar] [CrossRef] [PubMed]
- Kemmerer, C.L.; Pernpeintner, V.; Ruschil, C.; Abdelhak, A.; Scholl, M.; Ziemann, U.; Krumbholz, M.; Hemmer, B.; Kowarik, M.C. Differential effects of disease modifying drugs on peripheral blood B cell subsets: A cross sectional study in multiple sclerosis patients treated with interferon-β, glatiramer acetate, dimethyl fumarate, fingolimod or natalizumab. PLoS ONE 2020, 15, e0235449. [Google Scholar] [CrossRef] [PubMed]
- Claes, N.; Dhaeze, T.; Fraussen, J.; Broux, B.; Van Wijmeersch, B.; Stinissen, P.; Hupperts, R.; Hellings, N.; Somers, V. Compositional changes of B and T cell subtypes during fingolimod treatment in multiple sclerosis patients: A 12-month follow-up study. PLoS ONE 2014, 9, e111115. [Google Scholar] [CrossRef] [PubMed]
- Nakamura, M.; Matsuoka, T.; Chihara, N.; Miyake, S.; Sato, W.; Araki, M.; Okamoto, T.; Lin, Y.; Ogawa, M.; Murata, M.; et al. Differential effects of fingolimod on B-cell populations in multiple sclerosis. Mult. Scler. 2014, 20, 1371–1380. [Google Scholar] [CrossRef] [PubMed]
- Grützke, B.; Hucke, S.; Gross, C.C.; Herold, M.V.B.; Posevitz-Fejfar, A.; Wildemann, B.T.; Kieseier, B.; Dehmel, T.; Wiendl, H.; Klotz, L. Fingolimod treatment promotes regulatory phenotype and function of B cells. Ann. Clin. Transl. Neurol. 2015, 2, 119–130. [Google Scholar] [CrossRef] [PubMed]
- Teniente-Serra, A.; Hervás, J.V.; Quirant-Sánchez, B.; Mansilla, M.J.; Grau-López, L.; Ramo-Tello, C.; Martínez-Cáceres, E.M. Baseline Differences in Minor Lymphocyte Subpopulations may Predict Response to Fingolimod in Relapsing–Remitting Multiple Sclerosis Patients. CNS Neurosci. Ther. 2016, 22, 584–592. [Google Scholar] [CrossRef] [PubMed]
- Ghadiri, M.; Rezk, A.; Li, R.; Evans, A.; Giacomini, P.S.; Barnett, M.H.; Antel, J.; Bar-Or, A. Pre-treatment T-cell subsets associate with fingolimod treatment responsiveness in multiple sclerosis. Sci. Rep. 2020, 10, 356. [Google Scholar] [CrossRef]
- Quirant-Sánchez, B.; Presas-Rodriguez, S.; Mansilla, M.J.; Teniente-Serra, A.; Hervás-García, J.V.; Brieva, L.; Moral-Torres, E.; Cano, A.; Munteis, E.; Navarro-Barriuso, J.; et al. Th1Th17CM lymphocyte subpopulation as a predictive biomarker of disease activity in multiple sclerosis patients under dimethyl fumarate or fingolimod treatment. Mediat. Inflamm. 2019, 2019, 8147803. [Google Scholar] [CrossRef]
- Camacho-Toledano, C.; Machín-Díaz, I.; Calahorra, L.; Cabañas-Cotillas, M.; Otaegui, D.; Castillo-Triviño, T.; Villar, L.M.; Costa-Frossard, L.; Comabella, M.; Midaglia, L.; et al. Peripheral myeloid-derived suppressor cells are good biomarkers of the efficacy of fingolimod in multiple sclerosis. J. Neuroinflamm. 2022, 19, 277. [Google Scholar] [CrossRef]
- Quirant-Sánchez, B.; Hervás-García, J.V.; Teniente-Serra, A.; Brieva, L.; Moral-Torres, E.; Cano, A.; Munteis, E.; Mansilla, M.J.; Presas-Rodríguez, S.; Navarro-Barriuso, J.; et al. Predicting therapeutic response to fingolimod treatment in multiple sclerosis patients. CNS Neurosci. Ther. 2018, 24, 1175–1184. [Google Scholar] [CrossRef]
- Ferraro, D.; De Biasi, S.; Simone, A.M.; Orlandi, R.; Nasi, M.; Vitetta, F.; Pinti, M.; Fogliani, M.; Meletti, S.; Cossarizza, A.; et al. Modulation of Tregs and iNKT by Fingolimod in Multiple Sclerosis Patients. Cells 2021, 10, 3324. [Google Scholar] [CrossRef] [PubMed]
- Moreno-Torres, I.; González-García, C.; Marconi, M.; García-Grande, A.; Rodríguez-Esparragoza, L.; Elvira, V.; Ramil, E.; Campos-Ruíz, L.; García-Hernández, R.; Al-Shahrour, F.; et al. Immunophenotype and transcriptome profile of patients with multiple sclerosis treated with fingolimod: Setting up a model for prediction of response in a 2-year translational study. Front. Immunol. 2018, 9, 1693. [Google Scholar] [CrossRef] [PubMed]
- Brinkmann, V.; Davis, M.D.; Heise, C.E.; Albert, R.; Cottens, S.; Hof, R.; Bruns, C.; Prieschl, E.; Baumruker, T.; Hiestand, P.; et al. The immune modulator FTY720 targets sphingosine 1-phosphate receptors. J. Biol. Chem. 2002, 277, 21453–21457. [Google Scholar] [CrossRef] [PubMed]
- Honig, S.M.; Fu, S.; Mao, X.; Yopp, A.; Gunn, M.D.; Randolph, G.J.; Bromberg, J.S. FTY720 stimulates multidrug transporter– and cysteinyl leukotriene–dependent T cell chemotaxis to lymph nodes. J. Clin. Investig. 2003, 111, 627–637. [Google Scholar] [CrossRef] [PubMed]
- Dadyar, M.; Hussen, B.M.; Eslami, S.; Taheri, M.; Emadi, F.; Ghafouri-Fard, S.; Sayad, A. Expression of T cell-related lncRNAs in multiple sclerosis. Front. Genet. 2022, 13, 967157. [Google Scholar] [CrossRef] [PubMed]
- Li, M.; Chen, H.; Yin, P.; Song, J.; Jiang, F.; Tang, Z.; Fan, X.; Xu, C.; Wang, Y.; Xuw, Y.; et al. Identification and Clinical Validation of Key Extracellular Proteins as the Potential Biomarkers in Relapsing-Remitting Multiple Sclerosis. Front. Immunol. 2021, 12, 753929. [Google Scholar] [CrossRef] [PubMed]
- Parchami Barjui, S.; Reiisi, S.; Bayati, A. Human glutathione s-transferase enzyme gene variations and risk of multiple sclerosis in Iranian population cohort. Mult. Scler. Relat. Disord. 2017, 17, 41–46. [Google Scholar] [CrossRef] [PubMed]
- Gregory, S.G.; Schmidt, S.; Seth, P.; Oksenberg, J.R.; Hart, J.; Prokop, A.; Caillier, S.J.; Ban, M.; Goris, A.; Barcellos, L.F.; et al. Interleukin 7 receptor α chain (IL7R) shows allelic and functional association with multiple sclerosis. Nat. Genet. 2007, 39, 1083–1091. [Google Scholar] [CrossRef]
- Sferruzza, G.; Clarelli, F.; Mascia, E.; Ferrè, L.; Ottoboni, L.; Sorosina, M.; Santoro, S.; Filippi, M.; Provero, P.; Esposito, F. Transcriptional effects of fingolimod treatment on peripheral T cells in relapsing remitting multiple sclerosis patients. Pharmacogenomics 2022, 23, 161–171. [Google Scholar] [CrossRef]
- Sferruzza, G.; Clarelli, F.; Mascia, E.; Ferrè, L.; Ottoboni, L.; Sorosina, M.; Santoro, S.; Moiola, L.; Martinelli, V.; Comi, G.; et al. Transcriptomic Analysis of Peripheral Monocytes upon Fingolimod Treatment in Relapsing Remitting Multiple Sclerosis Patients. Mol. Neurobiol. 2021, 58, 4816–4827. [Google Scholar] [CrossRef]
- Angerer, I.C.; Hecker, M.; Koczan, D.; Roch, L.; Friess, J.; Rüge, A.; Fitzner, B.; Boxberger, N.; Schröder, I.; Flechtner, K.; et al. Transcriptome profiling of peripheral blood immune cell populations in multiple sclerosis patients before and during treatment with a sphingosine-1-phosphate receptor modulator. CNS Neurosci. Ther. 2018, 24, 193–201. [Google Scholar] [CrossRef] [PubMed]
- Mandala, S.; Hajdu, R.; Bergstrom, J.; Quackenbush, E.; Xie, J.; Milligan, J.; Thornton, R.; Shei, G.J.; Card, D.; Keohane, C.A.; et al. Alteration of Lymphocyte Trafficking by Sphingosine-1-Phosphate Receptor Agonists. Science 2016, 296, 346–349. [Google Scholar] [CrossRef]
- Mehling, M.; Brinkmann, V.; Antel, J.; Bar-Or, A.; Goebels, N.; Vedrine, C.; Kristofic, C.; Kuhle, J.; Lindberg, R.L.P.; Kappos, L. FTY720 therapy exerts differential effects on T cell subsets in multiple sclerosis. Neurology 2008, 71, 1261–1267. [Google Scholar] [CrossRef] [PubMed]
- Suryawanshi, A.; Manoharan, I.; Hong, Y.; Swafford, D.; Majumdar, T.; Taketo, M.M.; Manicassamy, B.; Koni, P.A.; Thangaraju, M.; Sun, Z.; et al. Canonical Wnt Signaling in Dendritic Cells Regulates Th1/Th17 Responses and Suppresses Autoimmune Neuroinflammation. J. Immunol. 2015, 194, 3295–3304. [Google Scholar] [CrossRef] [PubMed]
- Friess, J.; Hecker, M.; Roch, L.; Koczan, D.; Fitzner, B.; Angerer, I.C.; Schröder, I.; Flechtner, K.; Thiesen, H.J.; Winkelmann, A.; et al. Fingolimod alters the transcriptome profile of circulating CD4+ cells in multiple sclerosis. Sci. Rep. 2017, 7, 42087. [Google Scholar] [CrossRef]
- Roch, L.; Hecker, M.; Friess, J.; Angerer, I.C.; Koczan, D.; Fitzner, B.; Schröder, I.; Flechtner, K.; Thiesen, H.J.; Meister, S.; et al. High-Resolution Expression Profiling of Peripheral Blood CD8+ Cells in Patients with Multiple Sclerosis Displays Fingolimod-Induced Immune Cell Redistribution. Mol. Neurobiol. 2017, 54, 5511–5525. [Google Scholar] [CrossRef] [PubMed]
- Sánchez-Sanz, A.; García-Martín, S.; Sabín-Muñoz, J.; Moreno-Torres, I.; Elvira, V.; Al-Shahrour, F.; García-Grande, A.; Ramil, E.; Rodríguez-De la Fuente, O.; Brea-Álvarez, B.; et al. Dimethyl fumarate-related immune and transcriptional signature is associated with clinical response in multiple sclerosis-treated patients. Front. Immunol. 2023, 14, 1209923. [Google Scholar] [CrossRef]
- Gafson, A.R.; Kim, K.; Cencioni, M.T.; Van Hecke, W.; Nicholas, R.; Baranzini, S.E.; Matthews, P.M. Mononuclear cell transcriptome changes associated with dimethyl fumarate in MS. Neurol. Neuroimmunol. Neuroinflamm. 2018, 5, 1–11. [Google Scholar] [CrossRef]
- Zhou, Y.; Cui, C.; Ma, X.; Luo, W.; Zheng, S.G.; Qiu, W. Nuclear Factor κB (NF-κB)–Mediated Inflammation in Multiple Sclerosis. Front. Immunol. 2020, 11, 391. [Google Scholar] [CrossRef]
- Ghosh, S.; Dass, J.F.P. Study of pathway cross-talk interactions with NF-κB leading to its activation via ubiquitination or phosphorylation: A brief review. Gene 2016, 584, 97–109. [Google Scholar] [CrossRef]
- Thompson, A.J.; Banwell, B.L.; Barkhof, F.; Carroll, W.M.; Coetzee, T.; Comi, G.; Correale, J.; Fazekas, F.; Filippi, M.; Freedman, M.S.; et al. Diagnosis of multiple sclerosis: 2017 revisions of the McDonald criteria. Lancet Neurol. 2018, 17, 162–173. [Google Scholar] [CrossRef]
- European Medicines Agency. Gilenya [Internet]. Available online: https://www.ema.europa.eu/en/documents/product-information/gilenya-epar-product-information_en.pdf (accessed on 22 December 2023).
- Cohen, M.; Maillart, E.; Tourbah, A.; De Sèze, J.; Vukusic, S.; Brassat, D.; Anne, O.; Wiertlewski, S.; Camu, W.; Courtois, S.; et al. Switching from natalizumab to fingolimod in multiple sclerosis: A French prospective study. JAMA Neurol. 2014, 71, 436–441. [Google Scholar] [CrossRef] [PubMed]
- Martin, M. Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnet J. 2011, 17, 10–12. [Google Scholar] [CrossRef]
- Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: A flexible trimmer for Illumina sequence data. Bioinformatics 2014, 30, 2114–2120. [Google Scholar] [CrossRef]
- Kim, D.; Paggi, J.M.; Park, C.; Bennett, C.; Salzberg, S.L. Graph-based genome alignment and genotyping with HISAT2 and HISAT-genotype. Nat. Biotechnol. 2019, 37, 907–915. [Google Scholar] [CrossRef] [PubMed]
- Love, M.I.; Huber, W.; Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014, 15, 550. [Google Scholar] [CrossRef] [PubMed]
- Subramanian, A.; Tamayo, P.; Mootha, V.K.; Mukherjee, S.; Ebert, B.L.; Gillette, M.A.; Paulovich, A.; Pomeroy, S.L.; Golub, T.R.; Lander, E.S.; et al. Gene set enrichment analysis: A knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl. Acad. Sci. USA 2005, 102, 15545–15550. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
{kind=link}
{kind=link}
| Groups | MS Subgroups | |||
|---|---|---|---|---|
| HD (n = 10) | MS (n = 10) | Responders (n = 5) | Non-Responders (n = 5) | |
| Age (years) 1 | 43.30 ± 10.60 | 36.70 ± 11.03 | 40.00 ± 13.32 | 33.40 ± 8.29 |
| Sex (% of female) 2 | 70% | 60% | 40% | 80% |
| Disease duration (years) 1,3 | 6.35 ± 4.94 | 6.32 ± 5.02 | 6.38 ± 5.45 | |
| Time since DMT onset (years) 1 | 3.38 ± 3.73 | 2.55 ± 1.84 | 4.20 ± 5.11 | |
| Nº of previous DMTs 1 | 1.40 ± 0.70 | 1.20 ± 0.45 | 1.60 ± 0.89 | |
| Immediately previous DMT | ||||
| Natalizumab | 60% | 60% | 60% | |
| Interferon beta | 20% | 20% | 20% | |
| Dimethyl fumarate | 20% | 20% | 20% | |
| EDSS 1 | 1.60 ± 1.84 | 2.10 ± 2.38 | 1.10 ± 1.14 | |
| EDA-3 Patients | MRI Activity | Nº of Relapses | CDP |
|---|---|---|---|
| NonResponder_1 | No | 1 | No |
| NonResponder_2 | No | 0 | Yes |
| NonResponder_3 | Yes | 1 | No |
| NonResponder_4 | Yes | 1 | Yes |
| NonResponder_5 | No | 1 | No |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sánchez-Sanz, A.; Muñoz-Viana, R.; Sabín-Muñoz, J.; Moreno-Torres, I.; Brea-Álvarez, B.; Rodríguez-De la Fuente, O.; García-Merino, A.; Sánchez-López, A.J. Response to Fingolimod in Multiple Sclerosis Patients Is Associated with a Differential Transcriptomic Regulation. Int. J. Mol. Sci. 2024, 25, 1372. https://doi.org/10.3390/ijms25031372
Sánchez-Sanz A, Muñoz-Viana R, Sabín-Muñoz J, Moreno-Torres I, Brea-Álvarez B, Rodríguez-De la Fuente O, García-Merino A, Sánchez-López AJ. Response to Fingolimod in Multiple Sclerosis Patients Is Associated with a Differential Transcriptomic Regulation. International Journal of Molecular Sciences. 2024; 25(3):1372. https://doi.org/10.3390/ijms25031372
Chicago/Turabian StyleSánchez-Sanz, Alicia, Rafael Muñoz-Viana, Julia Sabín-Muñoz, Irene Moreno-Torres, Beatriz Brea-Álvarez, Ofir Rodríguez-De la Fuente, Antonio García-Merino, and Antonio J. Sánchez-López. 2024. "Response to Fingolimod in Multiple Sclerosis Patients Is Associated with a Differential Transcriptomic Regulation" International Journal of Molecular Sciences 25, no. 3: 1372. https://doi.org/10.3390/ijms25031372
APA StyleSánchez-Sanz, A., Muñoz-Viana, R., Sabín-Muñoz, J., Moreno-Torres, I., Brea-Álvarez, B., Rodríguez-De la Fuente, O., García-Merino, A., & Sánchez-López, A. J. (2024). Response to Fingolimod in Multiple Sclerosis Patients Is Associated with a Differential Transcriptomic Regulation. International Journal of Molecular Sciences, 25(3), 1372. https://doi.org/10.3390/ijms25031372