
Hepatic Amyloid Beta-42-Metabolizing Proteins in Liver Steatosis and Metabolic Dysfunction-Associated Steatohepatitis

 ,
,  ,
,  ,
,  and
and 
Abstract

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results
2.1. Saturated Palmitic Acid Induces the Gene Expression of APP and Its Metabolizing Proteins In Vitro, Which Is Attenuated by Mono-Unsaturated Oleic Acid
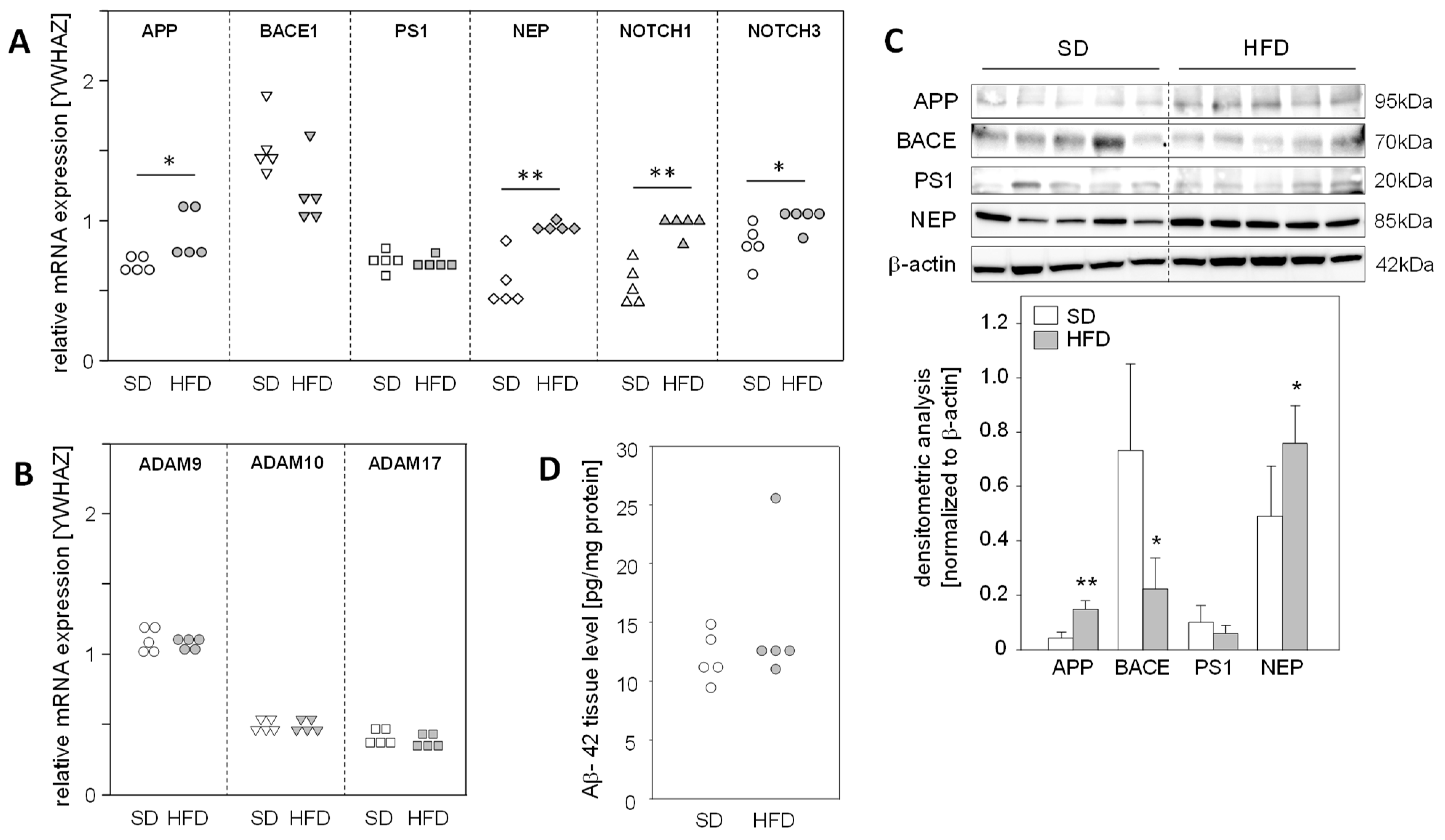
2.2. Hepatic Expression of APP and Its Metabolizing Protein, but Not Aβ-42, Levels Are Altered in a Fatty Liver Mouse Model
2.3. Hepatic Aβ-42 Levels Are Reduced in Patients with MASH
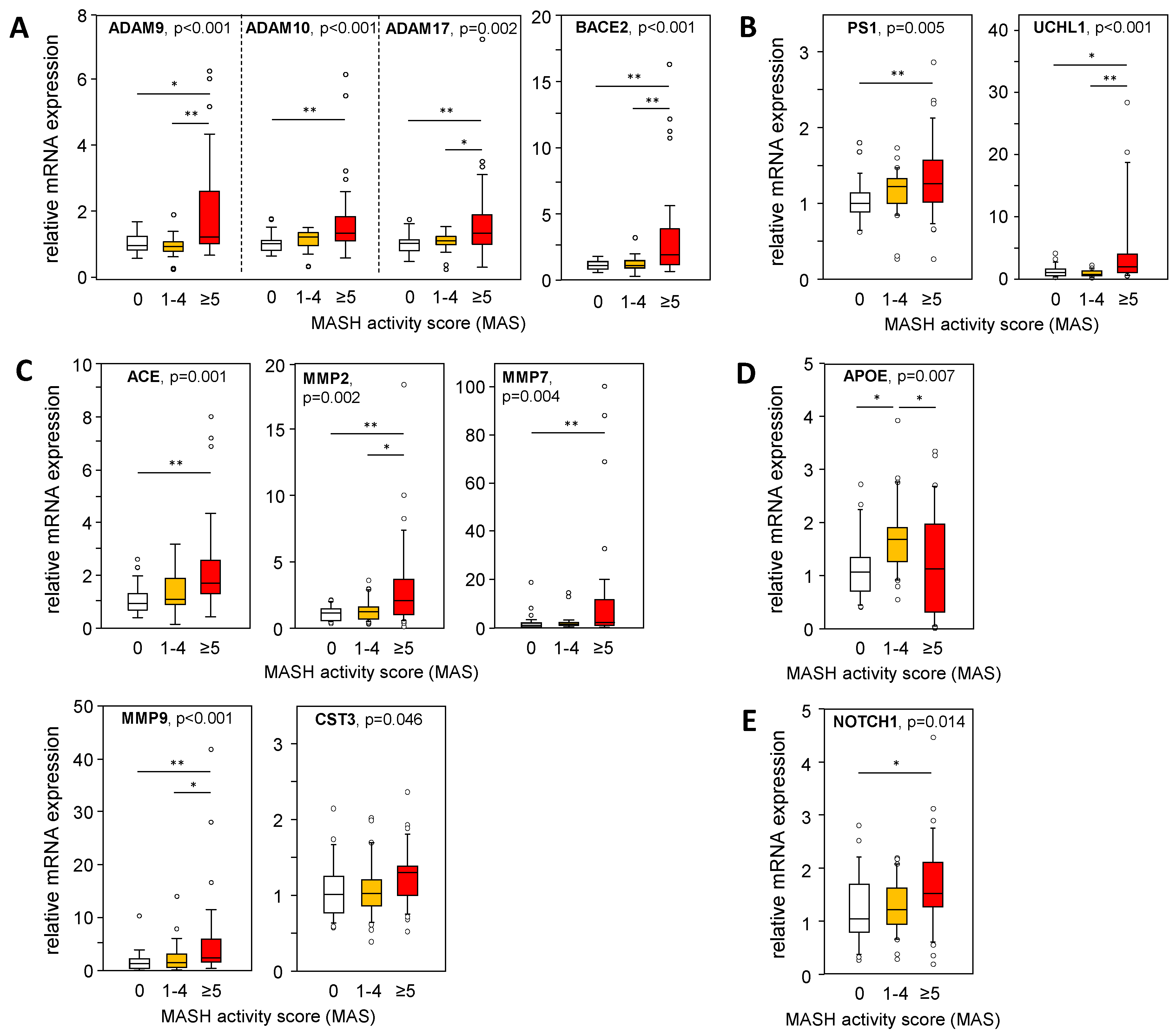
2.4. mRNA Expression of APP, APP Degradation, and Aβ-42-Processing Proteins Is Altered in Liver Tissue from Patients with MASH
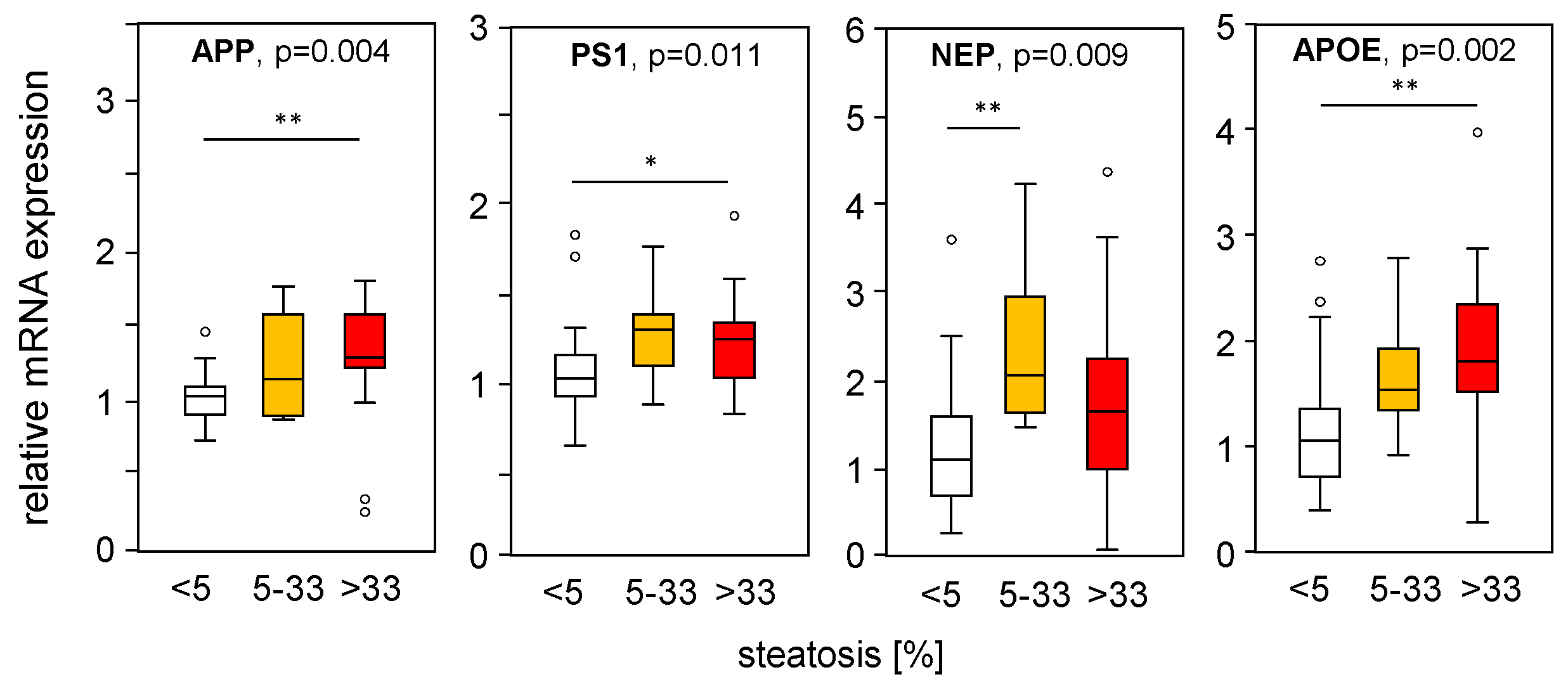
2.5. Fibrotic Rather Than Steatotic Liver Tissue Conditions Are Responsible for Differentially Expressed APP- and Aβ-42-Processing Genes

3. Discussion
4. Materials and Methods
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Knopman, D.S.; Amieva, H.; Petersen, R.C.; Chetelat, G.; Holtzman, D.M.; Hyman, B.T.; Nixon, R.A.; Jones, D.T. Alzheimer disease. Nat. Rev. Dis. Primers 2021, 7, 33. [Google Scholar] [CrossRef] [PubMed]
- Bassendine, M.F.; Taylor-Robinson, S.D.; Fertleman, M.; Khan, M.; Neely, D. Is Alzheimer’s Disease a Liver Disease of the Brain? J. Alzheimers Dis. 2020, 75, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Estrada, L.D.; Ahumada, P.; Cabrera, D.; Arab, J.P. Liver Dysfunction as a Novel Player in Alzheimer’s Progression: Looking Outside the Brain. Front. Aging Neurosci. 2019, 11, 174. [Google Scholar] [CrossRef] [PubMed]
- Nalivaeva, N.N.; Beckett, C.; Belyaev, N.D.; Turner, A.J. Are amyloid-degrading enzymes viable therapeutic targets in Alzheimer’s disease? J. Neurochem. 2012, 120, 167–185. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Wen, D.X.; Zhao, Y.H.; Hang, Y.N.; Mandell, M.S. Increase of beta-amyloid and C-reactive protein in liver transplant recipients with postoperative cognitive dysfunction. Hepatobiliary Pancreat. Dis. Int. 2013, 12, 370–376. [Google Scholar] [CrossRef]
- Chen, T.B.; Yiao, S.Y.; Sun, Y.; Lee, H.J.; Yang, S.C.; Chiu, M.J.; Chen, T.F.; Lin, K.N.; Tang, L.Y.; Lin, C.C.; et al. Comorbidity and dementia: A nationwide survey in Taiwan. PLoS ONE 2017, 12, e0175475. [Google Scholar] [CrossRef] [PubMed]
- Wiest, R.; Weiss, T.S.; Danielyan, L.; Buechler, C. Serum Amyloid Beta42 Is Not Eliminated by the Cirrhotic Liver: A Pilot Study. J. Clin. Med. 2021, 10, 2669. [Google Scholar] [CrossRef]
- Wang, Y.R.; Wang, Q.H.; Zhang, T.; Liu, Y.H.; Yao, X.Q.; Zeng, F.; Li, J.; Zhou, F.Y.; Wang, L.; Yan, J.C.; et al. Associations Between Hepatic Functions and Plasma Amyloid-Beta Levels-Implications for the Capacity of Liver in Peripheral Amyloid-Beta Clearance. Mol. Neurobiol. 2017, 54, 2338–2344. [Google Scholar] [CrossRef]
- Buniatian, G.H.; Weiskirchen, R.; Weiss, T.S.; Schwinghammer, U.; Fritz, M.; Seferyan, T.; Proksch, B.; Glaser, M.; Lourhmati, A.; Buadze, M.; et al. Antifibrotic Effects of Amyloid-Beta and Its Loss in Cirrhotic Liver. Cells 2020, 9, 452. [Google Scholar] [CrossRef]
- Buniatian, G.H.; Schwinghammer, U.; Tremmel, R.; Cynis, H.; Weiss, T.S.; Weiskirchen, R.; Lauschke, V.M.; Youhanna, S.; Ramos, I.; Valcarcel, M.; et al. Consequences of Amyloid-β Deficiency for the Liver. Adv. Sci. 2024, 11, e2307734. [Google Scholar] [CrossRef]
- Chalasani, N.; Younossi, Z.; Lavine, J.E.; Charlton, M.; Cusi, K.; Rinella, M.; Harrison, S.A.; Brunt, E.M.; Sanyal, A.J. The diagnosis and management of nonalcoholic fatty liver disease: Practice guidance from the American Association for the Study of Liver Diseases. Hepatology 2018, 67, 328–357. [Google Scholar] [CrossRef] [PubMed]
- Celikbilek, A.; Celikbilek, M.; Bozkurt, G. Cognitive assessment of patients with nonalcoholic fatty liver disease. Eur. J. Gastroenterol. Hepat. 2018, 30, 944–950. [Google Scholar] [CrossRef] [PubMed]
- Weinstein, G.; Zelber-Sagi, S.; Preis, S.R.; Beiser, A.S.; DeCarli, C.; Speliotes, E.K.; Satizabal, C.L.; Vasan, R.S.; Seshadri, S. Association of Nonalcoholic Fatty Liver Disease with Lower Brain Volume in Healthy Middle-aged Adults in the Framingham Study. JAMA Neurol. 2018, 75, 97–104. [Google Scholar] [CrossRef] [PubMed]
- Gomez-Lechon, M.J.; Donato, M.T.; Martinez-Romero, A.; Jimenez, N.; Castell, J.V.; O’Connor, J.E. A human hepatocellular in vitro model to investigate steatosis. Chem-Biol. Interact. 2007, 165, 106–116. [Google Scholar] [CrossRef] [PubMed]
- Ricchi, M.; Odoardi, M.R.; Carulli, L.; Anzivino, C.; Ballestri, S.; Pinetti, A.; Fantoni, L.I.; Marra, F.; Bertolotti, M.; Banni, S.; et al. Differential effect of oleic and palmitic acid on lipid accumulation and apoptosis in cultured hepatocytes. J. Gastroenterol. Hepatol. 2009, 24, 830–840. [Google Scholar] [CrossRef]
- Weiss, T.S.; Lupke, M.; Ibrahim, S.; Buechler, C.; Lorenz, J.; Ruemmele, P.; Hofmann, U.; Melter, M.; Dayoub, R. Attenuated lipotoxicity and apoptosis is linked to exogenous and endogenous augmenter of liver regeneration by different pathways. PLoS ONE 2017, 12, e0184282. [Google Scholar] [CrossRef] [PubMed]
- Eisinger, K.; Rein-Fischboeck, L.; Neumeier, M.; Schmidhofer, S.; Pohl, R.; Haberl, E.M.; Liebisch, G.; Kopp, A.; Schmid, A.; Krautbauer, S.; et al. Alpha-syntrophin deficient mice are protected from adipocyte hypertrophy and ectopic triglyceride deposition in obesity. Exp. Mol. Pathol. 2018, 104, 212–221. [Google Scholar] [CrossRef]
- Kim, D.G.; Krenz, A.; Toussaint, L.E.; Maurer, K.J.; Robinson, S.A.; Yan, A.; Torres, L.; Bynoe, M.S. Non-alcoholic fatty liver disease induces signs of Alzheimer’s disease (AD) in wild-type mice and accelerates pathological signs of AD in an AD model. J. Neuroinflam. 2016, 13, 1. [Google Scholar] [CrossRef]
- Pincon, A.; De Montgolfier, O.; Akkoyunlu, N.; Daneault, C.; Pouliot, P.; Villeneuve, L.; Lesage, F.; Levy, B.I.; Thorin-Trescases, N.; Thorin, E.; et al. Non-Alcoholic Fatty Liver Disease, and the Underlying Altered Fatty Acid Metabolism, Reveals Brain Hypoperfusion and Contributes to the Cognitive Decline in APP/PS1 Mice. Metabolites 2019, 9, 104. [Google Scholar] [CrossRef]
- Zeltser, N.; Meyer, I.; Hernandez, G.V.; Trahan, M.J.; Fanter, R.K.; Abo-Ismail, M.; Glanz, H.; Strand, C.R.; Burrin, D.G.; La Frano, M.R.; et al. Neurodegeneration in juvenile Iberian pigs with diet-induced nonalcoholic fatty liver disease. Am. J. Physiol. Endocrinol. Metab. 2020, 319, E592–E606. [Google Scholar] [CrossRef]
- Ricciarelli, R.; Canepa, E.; Marengo, B.; Marinari, U.M.; Poli, G.; Pronzato, M.A.; Domenicotti, C. Cholesterol and Alzheimer’s disease: A still poorly understood correlation. IUBMB Life 2012, 64, 931–935. [Google Scholar] [CrossRef] [PubMed]
- Cheignon, C.; Tomas, M.; Bonnefont-Rousselot, D.; Faller, P.; Hureau, C.; Collin, F. Oxidative stress and the amyloid beta peptide in Alzheimer’s disease. Redox Biol. 2018, 14, 450–464. [Google Scholar] [CrossRef] [PubMed]
- Schilcher, K.; Dayoub, R.; Kubitza, M.; Riepl, J.; Klein, K.; Buechler, C.; Melter, M.; Weiss, T.S. Saturated Fat-Mediated Upregulation of IL-32 and CCL20 in Hepatocytes Contributes to Higher Expression of These Fibrosis-Driving Molecules in MASLD. Int. J. Mol. Sci. 2023, 24, 13222. [Google Scholar] [CrossRef] [PubMed]
- Serviddio, G.; Bellanti, F.; Vendemiale, G. Free radical biology for medicine: Learning from nonalcoholic fatty liver disease. Free Radic. Biol. Med. 2013, 65, 952–968. [Google Scholar] [CrossRef] [PubMed]
- Brill, A.; Chauhan, A.K.; Canault, M.; Walsh, M.T.; Bergmeier, W.; Wagner, D.D. Oxidative stress activates ADAM17/TACE and induces its target receptor shedding in platelets in a p38-dependent fashion. Cardiovasc. Res. 2009, 84, 137–144. [Google Scholar] [CrossRef] [PubMed]
- Botteri, G.; Salvado, L.; Guma, A.; Hamilton, D.L.; Meakin, P.J.; Montagut, G.; Ashford, M.L.J.; Ceperuelo-Mallafre, V.; Fernandez-Veledo, S.; Vendrell, J.; et al. The BACE1 product sAPP beta induces ER stress and inflammation and impairs insulin signaling. Metab. -Clin. Exp. 2018, 85, 59–75. [Google Scholar] [CrossRef] [PubMed]
- Satoh, J.; Kuroda, Y. Amyloid precursor protein β-secretase (BACE) mRNA expression in human neural cell lines following induction of neuronal differentiation and exposure to cytokines and growth factors. Neuropathology 2000, 20, 289–296. [Google Scholar] [CrossRef] [PubMed]
- Gamba, P.; Guglielmotto, M.; Testa, G.; Monteleone, D.; Zerbinati, C.; Gargiulo, S.; Biasi, F.; Iuliano, L.; Giaccone, G.; Mauro, A.; et al. Up-regulation of -amyloidogenesis in neuron-like human cells by both 24-and 27-hydroxycholesterol: Protective effect of N-acetyl-cysteine. Aging Cell 2014, 13, 561–572. [Google Scholar] [CrossRef] [PubMed]
- Garcia, J.; Chang, R.; Steinberg, R.A.; Arce, A.; Yang, J.S.; van der Eb, P.; Abdullah, T.; Chandrashekar, D.V.; Eck, S.M.; Meza, P.; et al. Modulation of hepatic amyloid precursor protein and lipoprotein receptor-related protein 1 by chronic alcohol intake: Potential link between liver steatosis and amyloid-β. Front. Physiol. 2022, 13, 930402. [Google Scholar] [CrossRef]
- Kjeldsen, S.A.S.; Gluud, L.L.; Werge, M.P.; Pedersen, J.S.; Bendtsen, F.; Alexiadou, K.; Tan, T.C.; Torekov, S.S.; Iepsen, E.W.; Jensen, N.J.; et al. Neprilysin activity is increased in metabolic dysfunction-associated steatotic liver disease and normalizes after bariatric surgery or GLP-1 therapy. Iscience 2023, 26, 108190. [Google Scholar] [CrossRef]
- Bahn, G.; Park, J.S.; Yun, U.J.; Lee, Y.J.; Choi, Y.; Park, J.S.; Baek, S.H.; Choi, B.Y.; Cho, Y.S.; Kim, H.K.; et al. NRF2/ARE pathway negatively regulates BACE1 expression and ameliorates cognitive deficits in mouse Alzheimer’s models. Proc. Natl. Acad. Sci. USA 2019, 116, 12516–12523. [Google Scholar] [CrossRef] [PubMed]
- Qiu, S.; Liang, Z.R.; Wu, Q.A.; Wang, M.; Yang, M.L.; Chen, C.; Zheng, H.T.; Zhu, Z.M.; Li, L.; Yang, G.Y. Hepatic lipid accumulation induced by a high-fat diet is regulated by Nrf2 through multiple pathways. Faseb J. 2022, 36, e22280. [Google Scholar] [CrossRef] [PubMed]
- Chen, G.F.; Xu, T.H.; Yan, Y.; Zhou, Y.R.; Jiang, Y.; Melcher, K.; Xu, H.E. Amyloid beta: Structure, biology and structure-based therapeutic development. Acta Pharmacol. Sin. 2017, 38, 1205–1235. [Google Scholar] [CrossRef] [PubMed]
- Qi, S.; Wang, C.; Li, C.; Wang, P.; Liu, M. Candidate genes investigation for severe nonalcoholic fatty liver disease based on bioinformatics analysis. Medicine 2017, 96, e7743. [Google Scholar] [CrossRef] [PubMed]
- Tutusaus, A.; de Gregorio, E.; Cucarull, B.; Cristobal, H.; Areste, C.; Graupera, I.; Coll, M.; Colell, A.; Gausdal, G.; Lorens, J.B.; et al. A Functional Role of GAS6/TAM in Nonalcoholic Steatohepatitis Progression Implicates AXL as Therapeutic Target. Cell Mol. Gastroenterol. 2020, 9, 349–368. [Google Scholar] [CrossRef] [PubMed]
- Casagrande, V.; Mauriello, A.; Bischetti, S.; Mavilio, M.; Federici, M.; Menghini, R. Hepatocyte specific TIMP3 expression prevents diet dependent fatty liver disease and hepatocellular carcinoma. Sci. Rep. 2017, 7, 6747. [Google Scholar] [CrossRef]
- Palladini, G.; Di Pasqua, L.G.; Croce, A.C.; Ferrigno, A.; Vairetti, M. Recent Updates on the Therapeutic Prospects of Reversion-Inducing Cysteine-Rich Protein with Kazal Motifs (RECK) in Liver Injuries. Int. J. Mol. Sci. 2023, 24, 17407. [Google Scholar] [CrossRef]
- Dashek, R.J.; Cunningham, R.P.; Taylor, C.L.; Alessi, I.; Diaz, C.; Meers, G.M.; Wheeler, A.A.; Ibdah, J.A.; Parks, E.J.; Yoshida, T.; et al. Hepatocellular RECK as a Critical Regulator of Metabolic Dysfunction-Associated Steatohepatitis Development. Cell Mol. Gastroenterol. 2024, 18, 101365. [Google Scholar] [CrossRef] [PubMed]
- Vassar, R. BACE1—The β-secretase enzyme in Alzheimer’s disease. J. Mol. Neurosci. 2004, 23, 105–113. [Google Scholar] [CrossRef]
- Guglielmotto, M.; Monteleone, D.; Vasciaveo, V.; Repetto, I.E.; Manassero, G.; Tabaton, M.; Tamagno, E. The Decrease of Uch-L1 Activity Is a Common Mechanism Responsible for Abeta 42 Accumulation in Alzheimer’s and Vascular Disease. Front. Aging Neurosci. 2017, 9, 320. [Google Scholar] [CrossRef]
- Wilson, C.L.; Murphy, L.B.; Leslie, J.; Kendrick, S.; French, J.; Fox, C.R.; Sheerin, N.S.; Fisher, A.; Robinson, J.H.; Tiniakos, D.G.; et al. Ubiquitin C-terminal hydrolase 1: A novel functional marker for liver myofibroblasts and a therapeutic target in chronic liver disease. J. Hepatol. 2015, 63, 1421–1428. [Google Scholar] [CrossRef] [PubMed]
- Endres, K. Apolipoprotein A1, the neglected relative of Apolipoprotein E and its potential role in Alzheimer’s disease. Neural Regen. Res. 2021, 16, 2141–2148. [Google Scholar] [CrossRef]
- Davies, J.; Zachariades, E.; Rogers-Broadway, K.R.; Karteris, E. Elucidating the role of DEPTOR in Alzheimer’s disease. Int. J. Mol. Med. 2014, 34, 1195–1200. [Google Scholar] [CrossRef]
- Chu, Y.P.; Jin, L.W.; Wang, L.C.; Ho, P.C.; Wei, W.Y.; Tsai, K.J. Transthyretin attenuates TDP-43 proteinopathy by autophagy activation via ATF4 in FTLD-TDP. Brain 2023, 146, 2089–2106. [Google Scholar] [CrossRef]
- Toyoda, H.; Kumada, T.; Kiriyama, S.; Tanikawa, M.; Hisanaga, Y.; Kanamori, A.; Tada, T.; Murakami, Y. Higher hepatic gene expression and serum levels of matrix metalloproteinase-2 are associated with steatohepatitis in non-alcoholic fatty liver diseases. Biomark. Biochem. Indic. Expo. Response Susceptibility Chem. 2013, 18, 82–87. [Google Scholar] [CrossRef] [PubMed]
- Weiss, T.S.; Dayoub, R. Thy-1 (CD90)-Positive Hepatic Progenitor Cells, Hepatoctyes, and Non-parenchymal Liver Cells Isolated from Human Livers. Methods Mol. Biol. 2017, 1506, 75–89. [Google Scholar] [CrossRef]
- Huang, H.; Bihaqi, S.W.; Cui, L.; Zawia, N.H. In vitro Pb exposure disturbs the balance between Abeta production and elimination: The role of AbetaPP and neprilysin. Neurotoxicology 2011, 32, 300–306. [Google Scholar] [CrossRef] [PubMed]
- Zhu, L.; Zhao, Y.; Yu, L.; He, X.; Wang, Y.; Jiang, P.; Yu, R.; Li, W.; Dong, B.; Wang, X.; et al. Overexpression of ADAM9 decreases radiosensitivity of hepatocellular carcinoma cell by activating autophagy. Bioengineered 2021, 12, 5516–5528. [Google Scholar] [CrossRef]
- Pan, B.; Huo, T.; Cao, M.; Jing, L.; Luo, X.; Qu, Z.; Feng, H.; Yuan, F.; Guo, K. ADAM10 promotes the proliferation of ligamentum flavum cells by activating the PI3K/AKT pathway. Int. J. Mol. Med. 2021, 47, 688–698. [Google Scholar] [CrossRef]
- Arcidiacono, P.; Webb, C.M.; Brooke, M.A.; Zhou, H.; Delaney, P.J.; Ng, K.-E.; Blaydon, D.C.; Tinker, A.; Kelsell, D.P.; Chikh, A. p63 is a key regulator of iRHOM2 signalling in the keratinocyte stress response. Nat. Commun. 2018, 9, 1021. [Google Scholar] [CrossRef]
- Wang, T.; Shi, F.; Jin, Y.; Jiang, W.; Shen, D.; Xiao, S. Abnormal Changes of Brain Cortical Anatomy and the Association with Plasma MicroRNA107 Level in Amnestic Mild Cognitive Impairment. Front Aging Neurosci. 2016, 8, 112. [Google Scholar] [CrossRef]
- Fischer, M.; Skowron, M.; Berthold, F. Reliable transcript quantification by real-time reverse transcriptase-polymerase chain reaction in primary neuroblastoma using normalization to averaged expression levels of the control genes HPRT1 and SDHA. J. Mol. Diagn. 2005, 7, 89–96. [Google Scholar] [CrossRef] [PubMed]
- Augustin, S.; Rimbach, G.; Augustin, K.; Schliebs, R.; Wolffram, S.; Cermak, R. Effect of a short- and long-term treatment with Ginkgo biloba extract on amyloid precursor protein levels in a transgenic mouse model relevant to Alzheimer’s disease. Arch. Biochem. Biophys. 2009, 481, 177–182. [Google Scholar] [CrossRef] [PubMed]
- Kim, D.Y.; Lee, M.; Kim, E.J. Involvement of Klotho, TNF-alpha and ADAMs in radiation-induced senescence of renal epithelial cells. Mol. Med. Rep. 2021, 23, 22. [Google Scholar] [CrossRef] [PubMed]
- Wen, Y.; Yu, W.H.; Maloney, B.; Bailey, J.; Ma, J.; Marié, I.; Maurin, T.; Wang, L.; Figueroa, H.; Herman, M.; et al. Transcriptional regulation of beta-secretase by p25/cdk5 leads to enhanced amyloidogenic processing. Neuron 2008, 57, 680–690. [Google Scholar] [CrossRef] [PubMed]
- Kalinin, S.; Gavrilyuk, V.; Polak, P.E.; Vasser, R.; Zhao, J.; Heneka, M.T.; Feinstein, D.L. Noradrenaline deficiency in brain increases beta-amyloid plaque burden in an animal model of Alzheimer’s disease. Neurobiol Aging 2007, 28, 1206–1214. [Google Scholar] [CrossRef] [PubMed]
- Hajdu, M.; Luttun, A.; Pelacho, B.; Burns, T.C.; Chase, L.; Gutiérrez-Pérez, M.; Jiang, Y.; Lenvik, T.; Vas, V.; Uher, F.; et al. Transcriptional characterization of the Notch signaling pathway in rodent multipotent adult progenitor cells. Pathol. Oncol. Res. POR 2007, 13, 302–310. [Google Scholar] [CrossRef]
- Robinson, S.C.; Klobucar, K.; Pierre, C.C.; Ansari, A.; Zhenilo, S.; Prokhortchouk, E.; Daniel, J.M. Kaiso differentially regulates components of the Notch signaling pathway in intestinal cells. Cell Commun. Signal 2017, 15, 24. [Google Scholar] [CrossRef]
- Baeten, J.T.; Lilly, B. Differential Regulation of NOTCH2 and NOTCH3 Contribute to Their Unique Functions in Vascular Smooth Muscle Cells. J. Biol. Chem. 2015, 290, 16226–16237. [Google Scholar] [CrossRef]


Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gross, S.; Danielyan, L.; Buechler, C.; Kubitza, M.; Klein, K.; Schwab, M.; Melter, M.; Weiss, T.S. Hepatic Amyloid Beta-42-Metabolizing Proteins in Liver Steatosis and Metabolic Dysfunction-Associated Steatohepatitis. Int. J. Mol. Sci. 2024, 25, 8768. https://doi.org/10.3390/ijms25168768
Gross S, Danielyan L, Buechler C, Kubitza M, Klein K, Schwab M, Melter M, Weiss TS. Hepatic Amyloid Beta-42-Metabolizing Proteins in Liver Steatosis and Metabolic Dysfunction-Associated Steatohepatitis. International Journal of Molecular Sciences. 2024; 25(16):8768. https://doi.org/10.3390/ijms25168768
Chicago/Turabian StyleGross, Simon, Lusine Danielyan, Christa Buechler, Marion Kubitza, Kathrin Klein, Matthias Schwab, Michael Melter, and Thomas S. Weiss. 2024. "Hepatic Amyloid Beta-42-Metabolizing Proteins in Liver Steatosis and Metabolic Dysfunction-Associated Steatohepatitis" International Journal of Molecular Sciences 25, no. 16: 8768. https://doi.org/10.3390/ijms25168768
APA StyleGross, S., Danielyan, L., Buechler, C., Kubitza, M., Klein, K., Schwab, M., Melter, M., & Weiss, T. S. (2024). Hepatic Amyloid Beta-42-Metabolizing Proteins in Liver Steatosis and Metabolic Dysfunction-Associated Steatohepatitis. International Journal of Molecular Sciences, 25(16), 8768. https://doi.org/10.3390/ijms25168768