
Molecular Mechanisms of Deregulation of Muscle Contractility Caused by the R168H Mutation in TPM3 and Its Attenuation by Therapeutic Agents

 ,
, 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results and Discussion
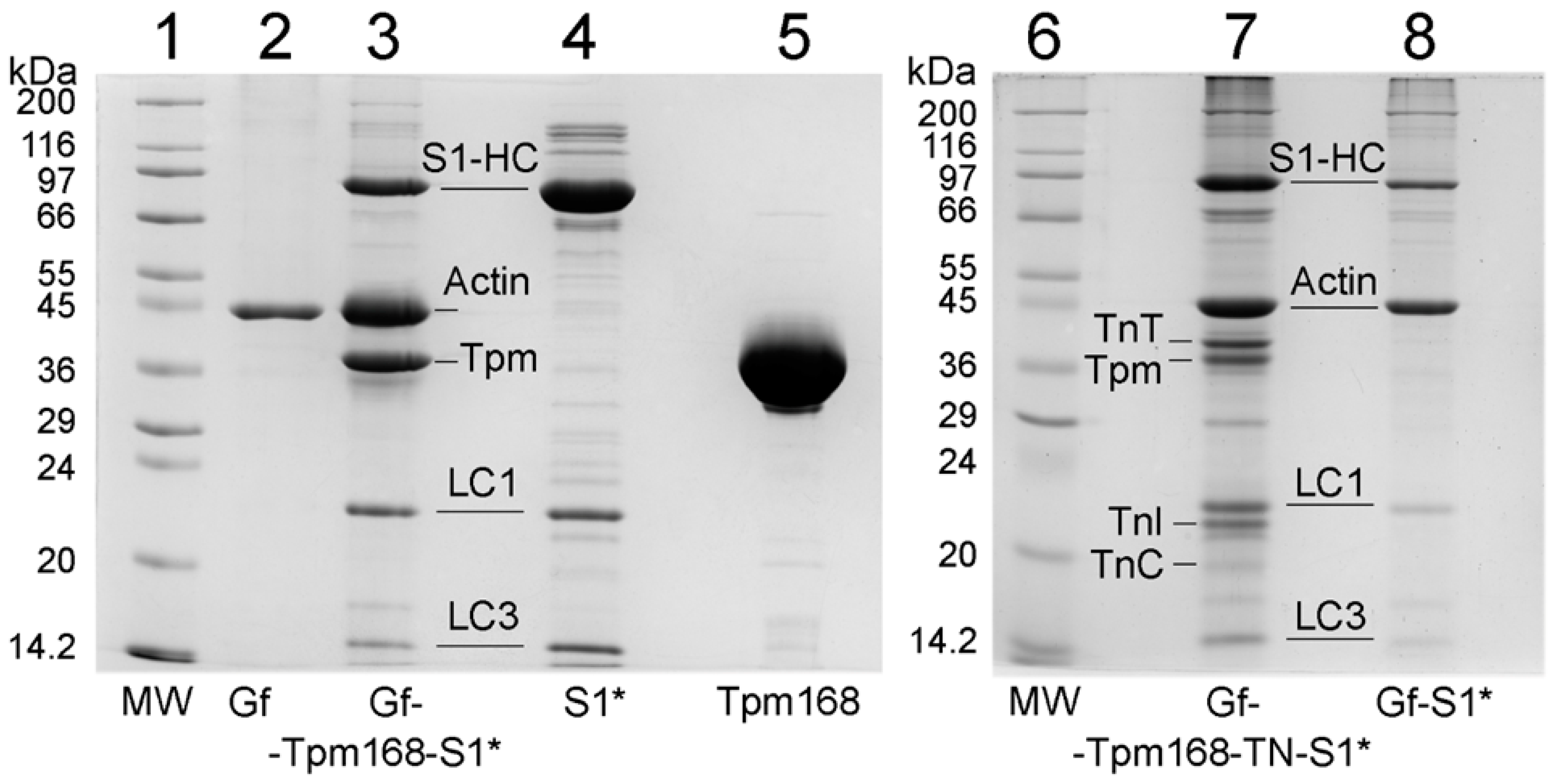
2.1. Ghost Muscle Fibers with Incorporated Labeled Protein as a Model for Studying the Conformational Changes in Proteins during Muscle Contraction
2.2. Troponin (±Ca2+) Can Change Concerted Interdependent Conformational Rearrangements of Major Proteins in the Contractile System during the ATPase Cycle
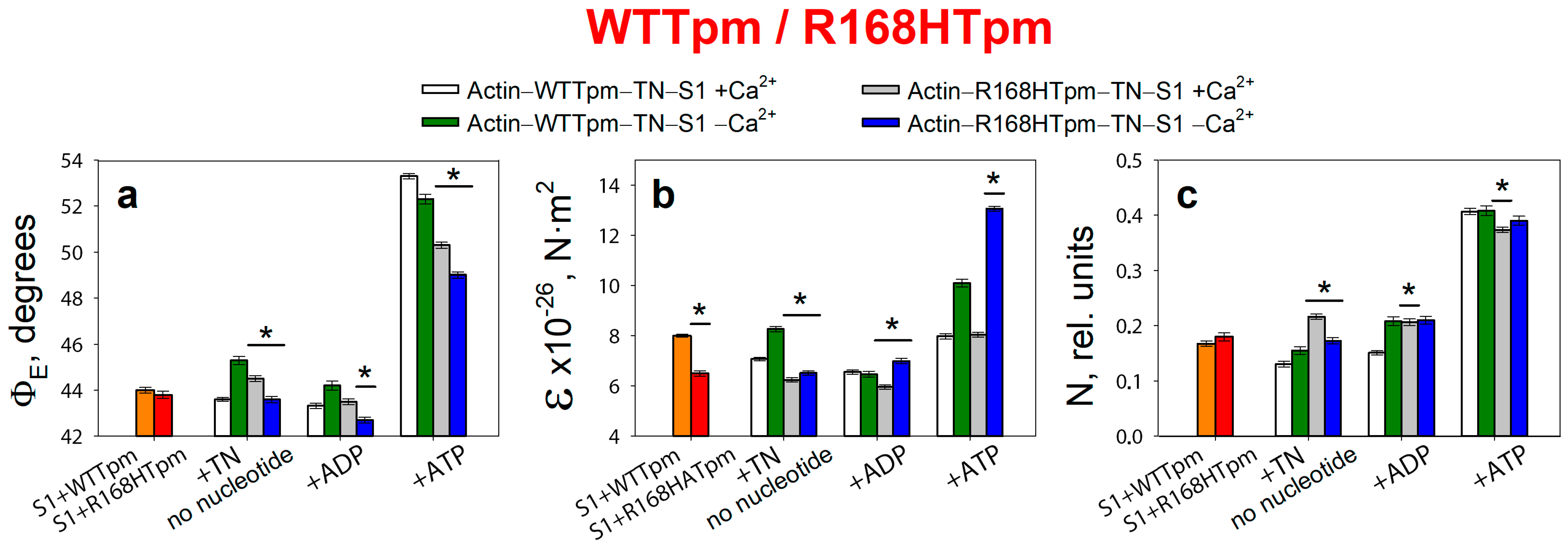
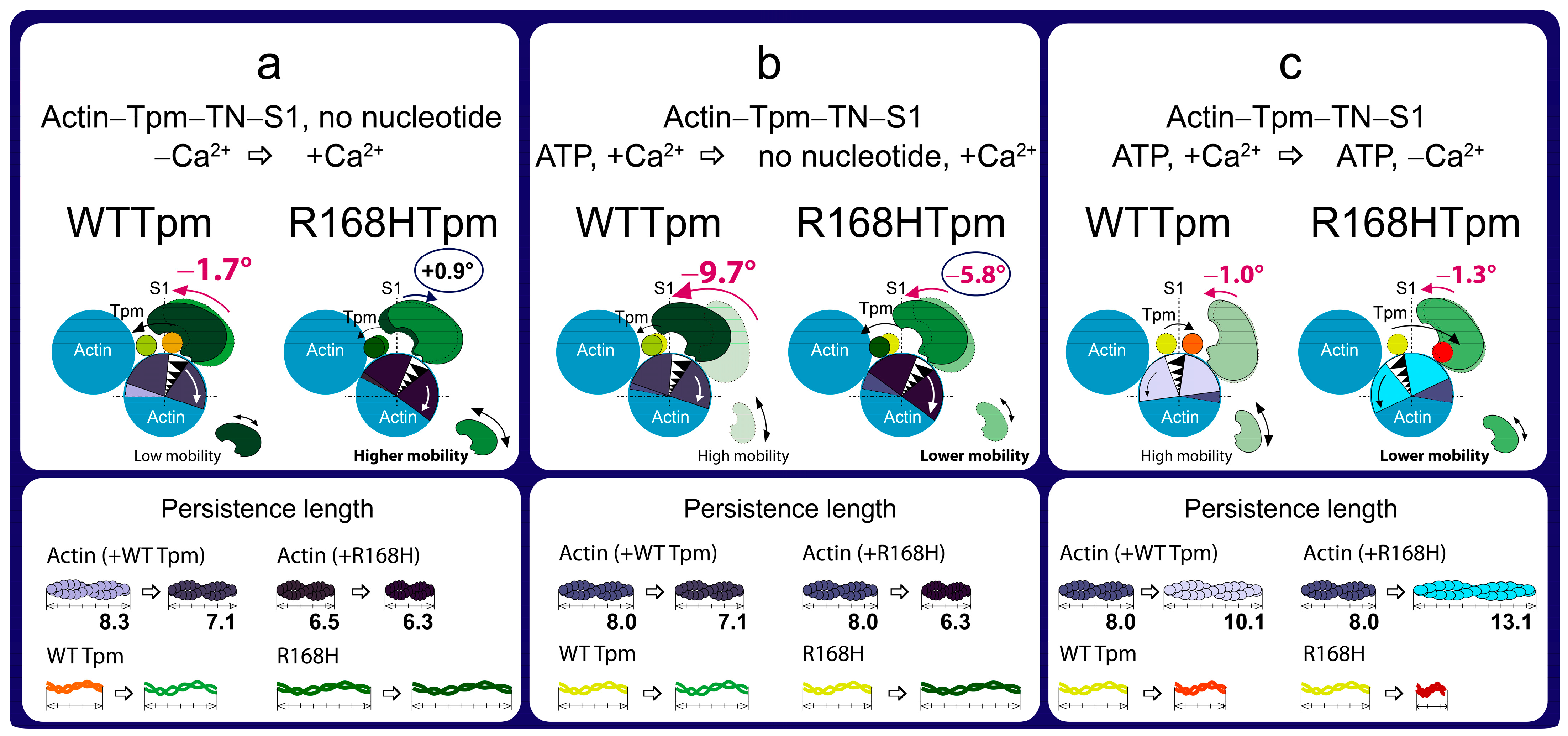
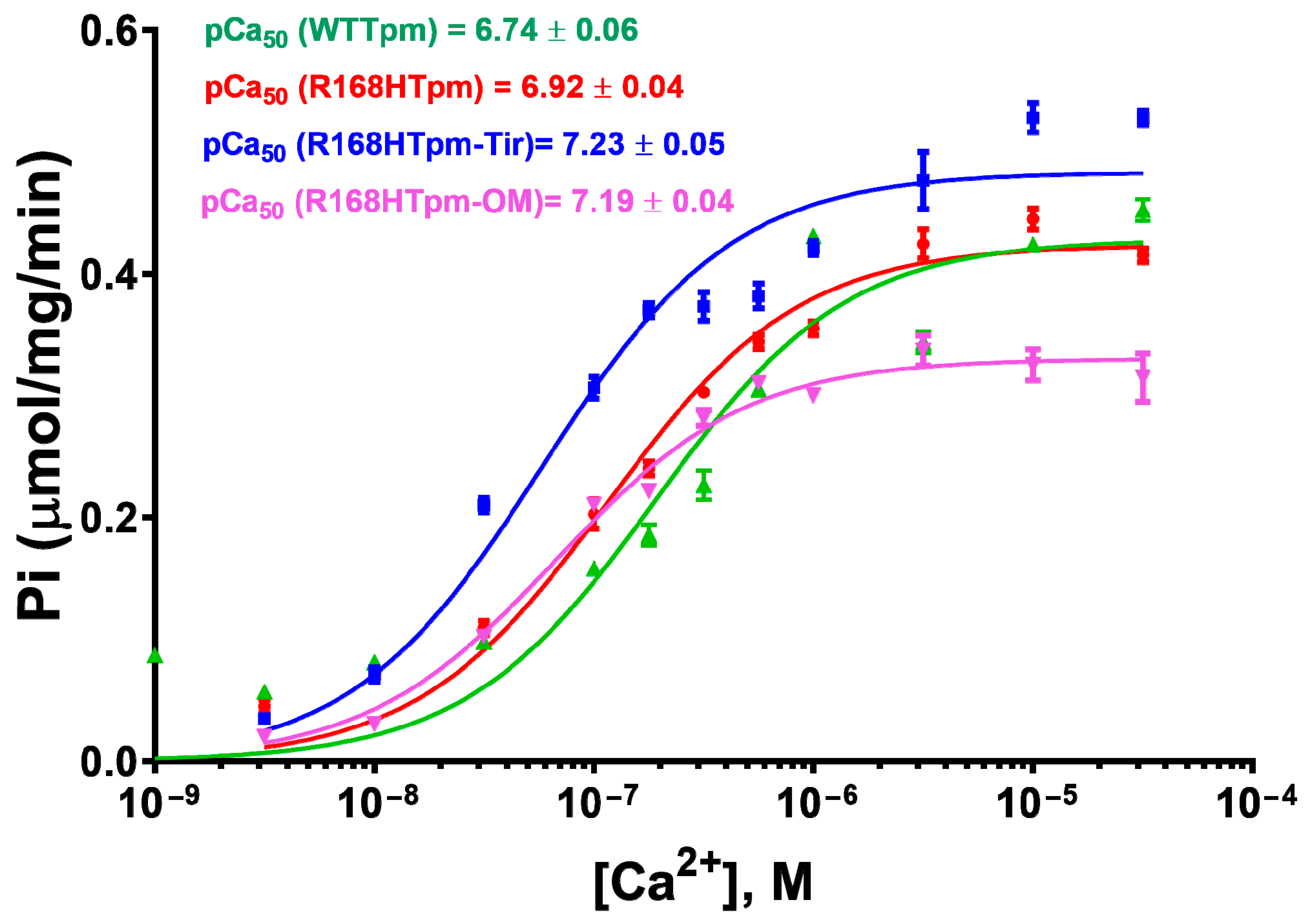
2.3. The R168H Mutation Allows Strong Binding of Myosin Heads to F-Actin at Low Ca2+ and Reduces It at High Ca2+
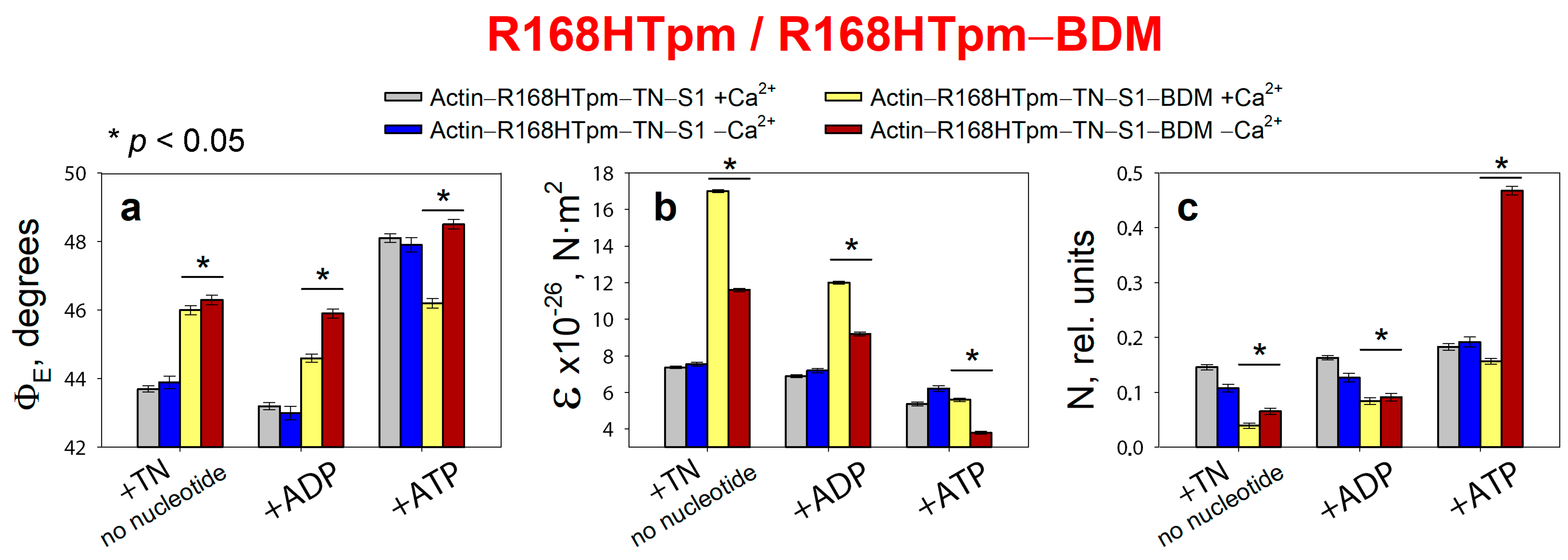
2.4. The Myosin Inhibitor, BDM, Attenuates the Effect of the Mutation on Troponin Function at Low Ca2+, but Is Unable to Repair the Damage Caused by the Arg168His Mutation
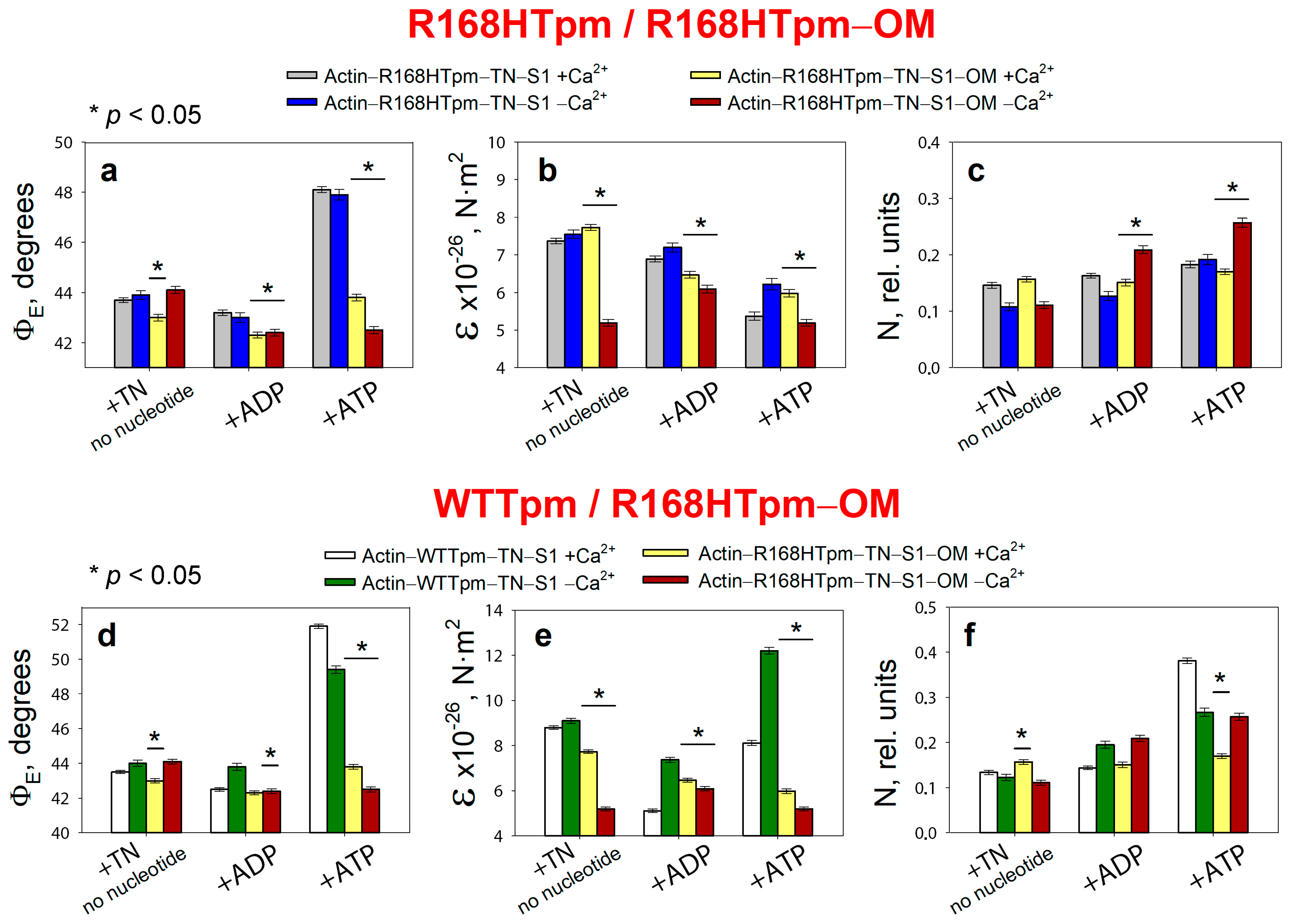
2.5. The Myosin Activator, Omecamtiv Mecarbil, Weakens the Effect of the R168H Mutation on Troponin Function at High Ca2+, but Does Not Reverse It, Provoking Strong Actin–Myosin Binding throughout the ATPase Cycle
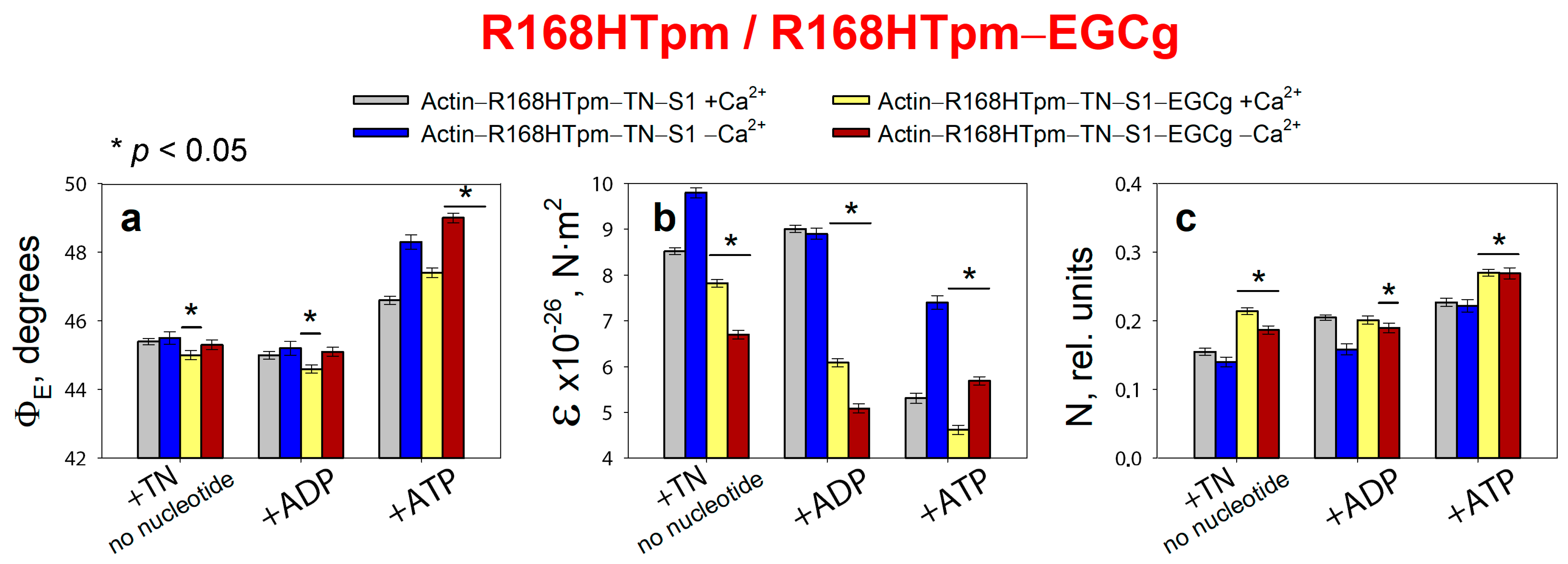
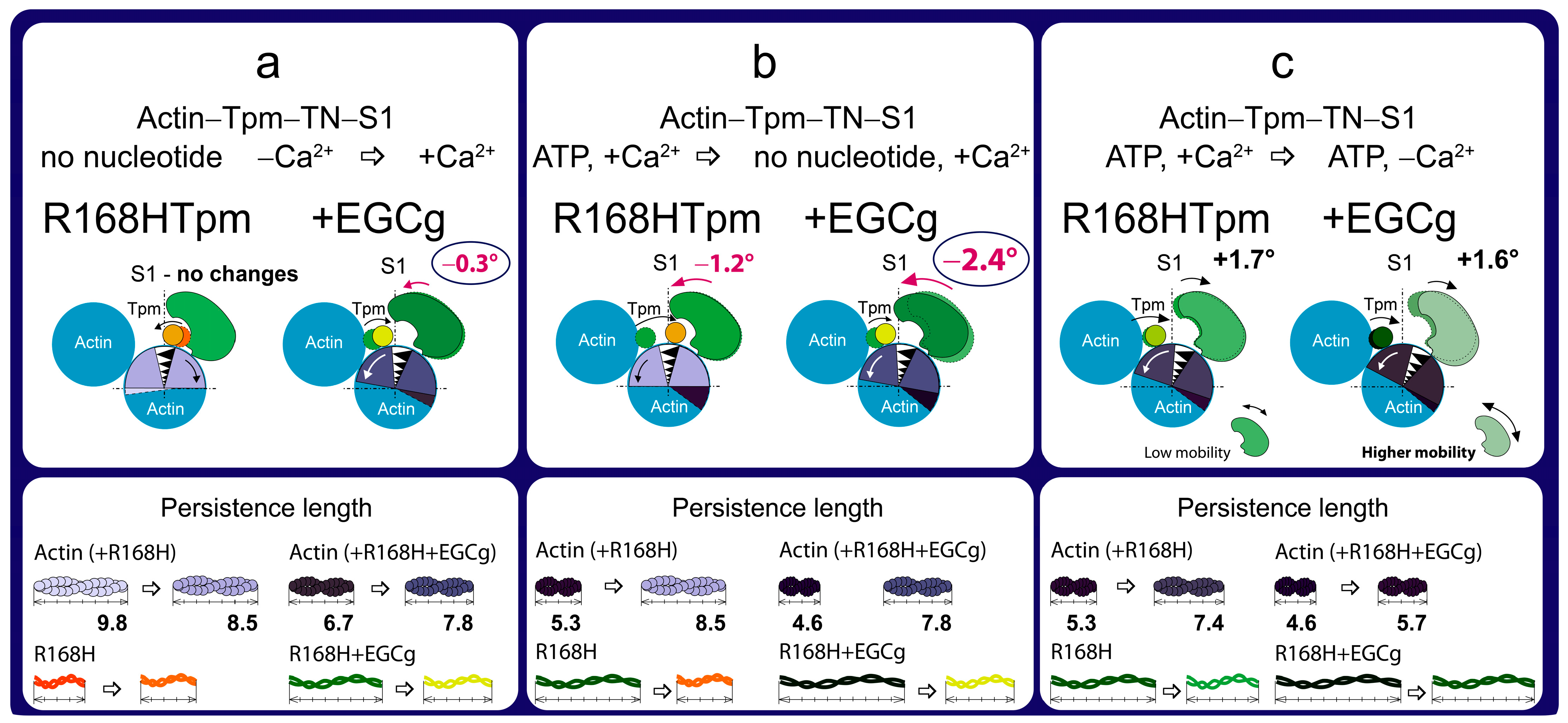
2.6. The Troponin Inhibitor, EGCg, Restores Troponin Function at High Ca2+, the Ability of Muscle Tissue to Relaxation, and Weakly Restore the Ability of Troponin to Switch Actin Monomers off at Low Ca2+
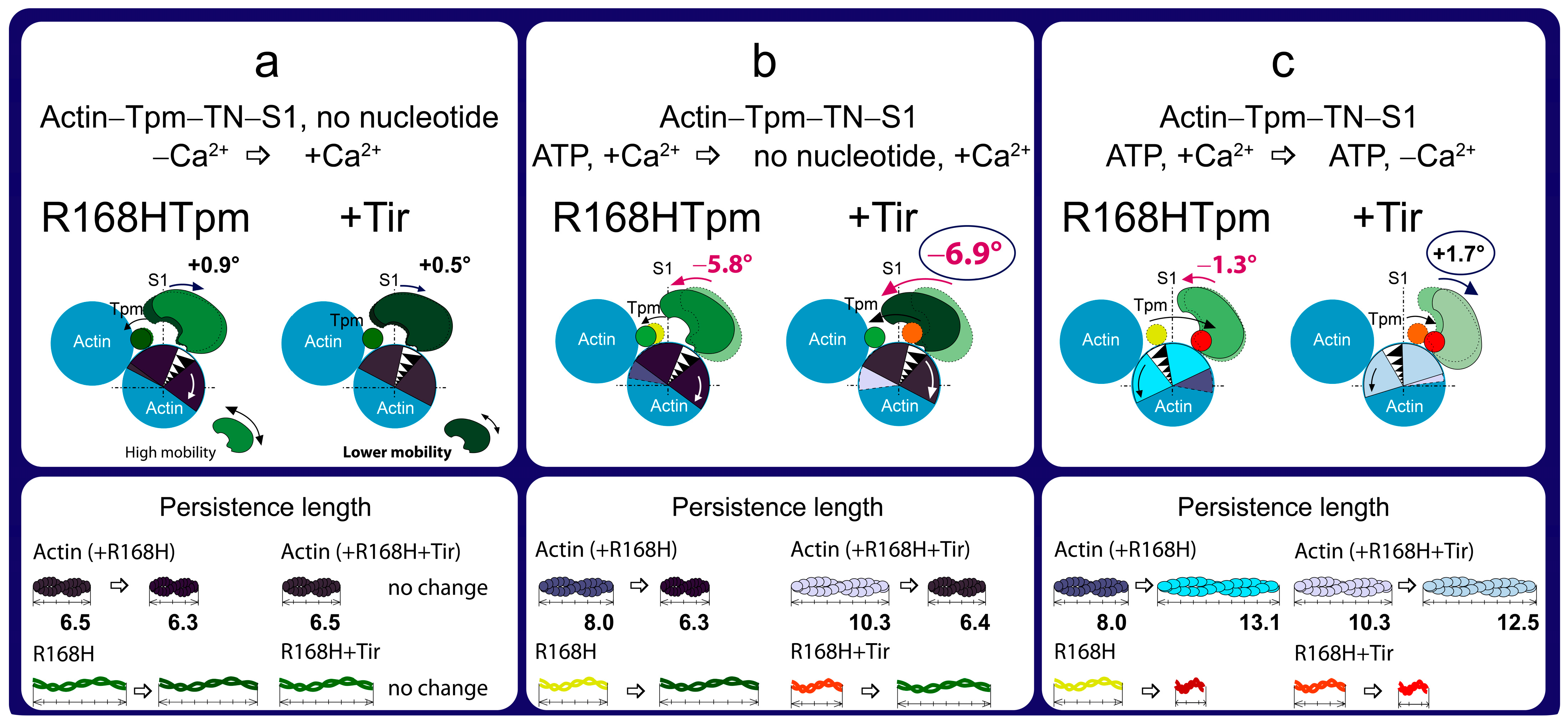
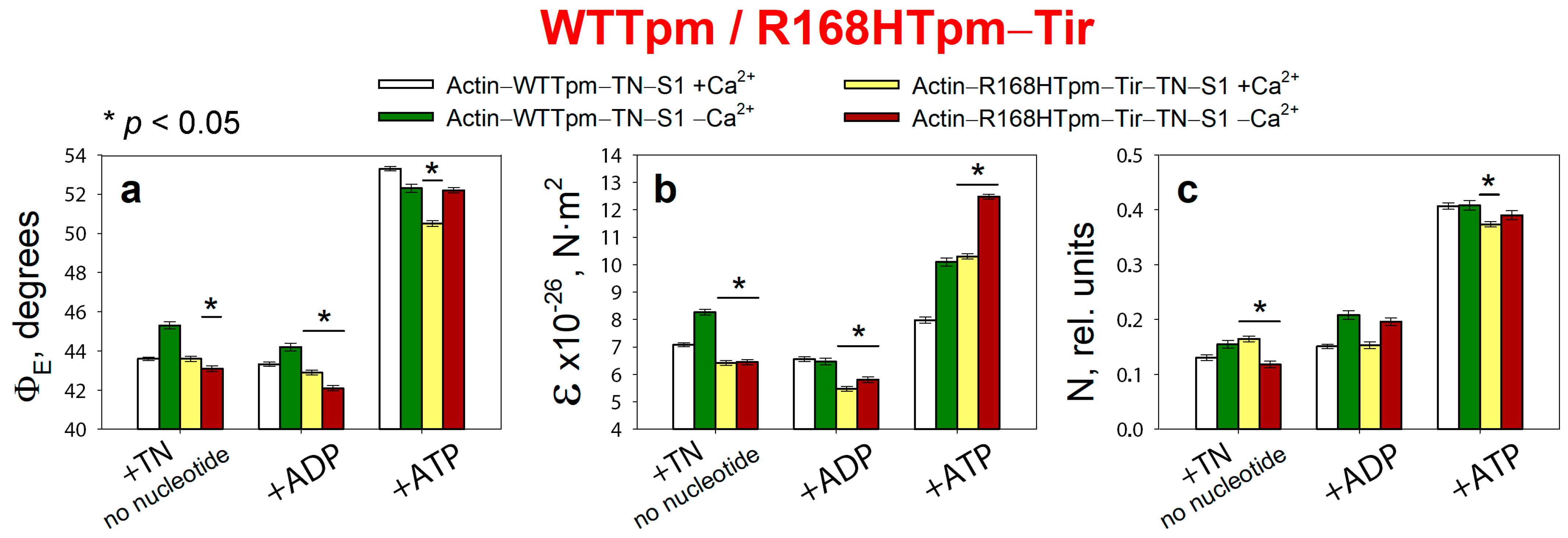
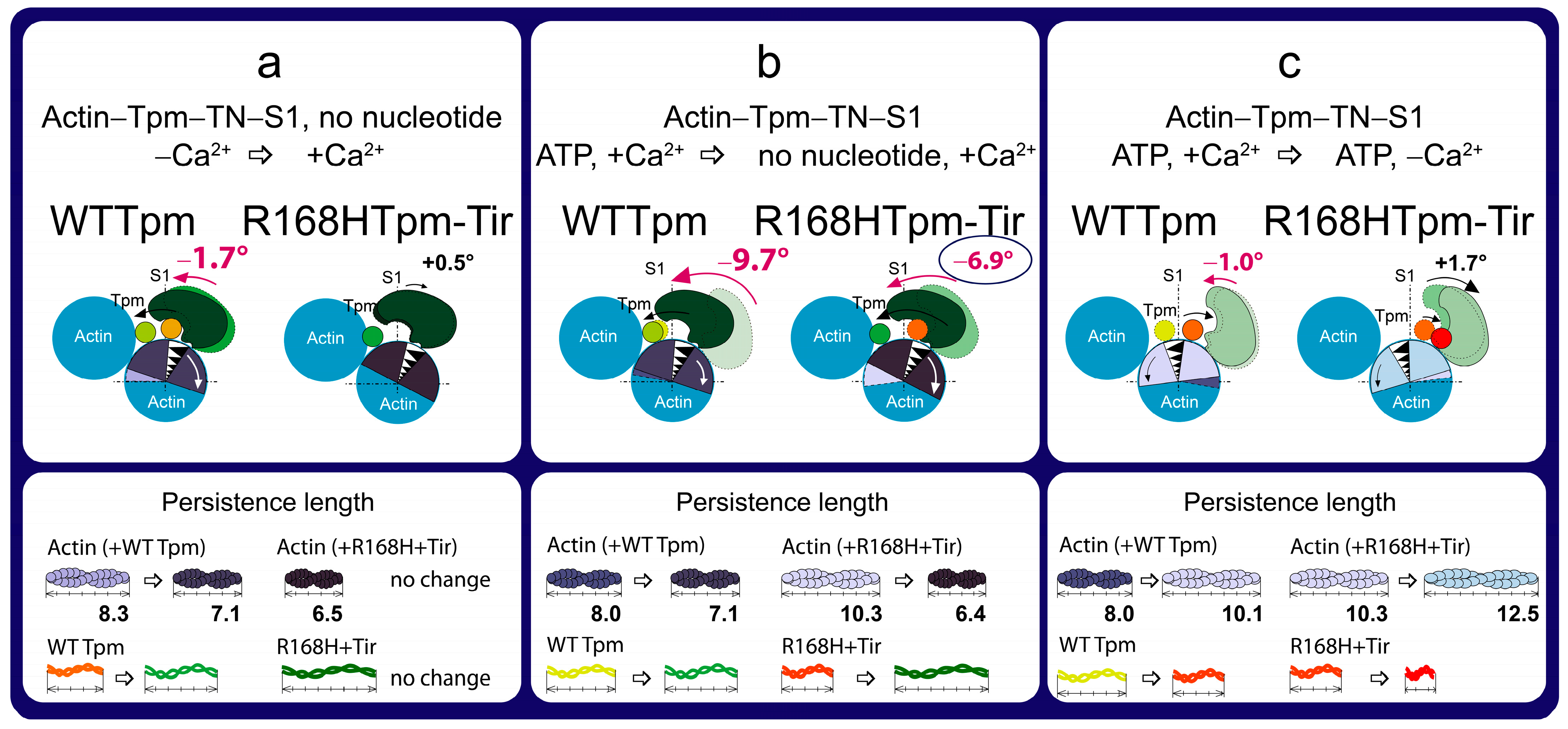
2.7. The Troponin Ca2+-Sensitive Activator, Tirasemtiv, Can Correct the Dysregulation Induced by the R168H Mutation in Tpm
3. Materials and Methods
3.1. Using Experimental Animals
3.2. Preparation of Proteins and Their Labeling by Fluorescent Probes
3.3. Determination of Actin-Activated ATPase of Subfragment-1
3.4. Preparation and Labeling of Ghost Fibers
3.5. Polarized Fluorescence Measurements
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| Tpm | Tropomyosin |
| WTTpm | Wild-type tropomyosin |
| R168HTpm | Tropomyosin with the Arg168His substitution |
| TN | Troponin |
| S1 | Myosin subfragment-1 |
| 1,5-IAEDANS | N-(iodoacetaminoethyl)-1-naphthyl-amine-5-sulfonic acid |
| EGCg | Epigallocatechin-3-gallate |
| BDM | 2,3-Butanedione monoxime |
| OM | Omecamtiv mecarbil |
| Tir | Tirasemtiv |
References
- Gordon, A.M.; Homsher, E.; Regnier, M. Regulation of contraction in striated muscle. Physiol. Rev. 2000, 80, 853–924. [Google Scholar] [CrossRef]
- Houdusse, A.; Sweeney, H.L. How myosin generates force on actin filaments. Trends Biochem. Sci. 2016, 41, 989–997. [Google Scholar] [CrossRef] [PubMed]
- Rynkiewicz, M.J.; Schott, V.; Orzechowski, M.; Lehman, W.; Fischer, S. Electrostatic interaction map reveals a new binding position for tropomyosin on F-actin. J. Muscle Res. Cell Motil. 2015, 36, 525–533. [Google Scholar] [CrossRef]
- Tobacman, L.S. Troponin Revealed: Uncovering the Structure of the Thin Filament On-Off Switch in Striated Muscle. Biophys. J. 2021, 120, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Craig, R.; Lehman, W. Crossbridge and tropomyosin positions observed in native, interacting thick and thin filaments. J. Mol. Biol. 2001, 311, 1027–1036. [Google Scholar] [CrossRef]
- Li, X.E.; Lehman, W.; Fischer, S. The relationship between curvature, flexibility and persistence length in the tropomyosin coiled-coil. J. Struct. Biol. 2010, 170, 313–318. [Google Scholar] [CrossRef] [PubMed]
- Lehman, W.; Li, X.; Kiani, F.A.; Moore, J.R.; Campbell, S.G.; Fischer, S.; Rynkiewicz, M.J. Precise Binding of Tropomyosin on Actin Involves Sequence-Dependent Variance in Coiled-Coil Twisting. Biophys. J. 2018, 115, 1082–1092. [Google Scholar] [CrossRef]
- Borovikov, Y.S.; Karpicheva, O.E.; Avrova, S.V.; Redwood, C.S. Modulation of the effects of tropomyosin on actin and myosin conformational changes by troponin and Ca2+. Biochim. Biophys. Acta 2009, 1794, 985–994. [Google Scholar] [CrossRef]
- Borovikov, Y.S.; Rysev, N.A.; Karpicheva, O.E.; Sirenko, V.V.; Avrova, S.V.; Piers, A.; Redwood, C.S. Molecular mechanisms of dysfunction of muscle fibers associated with Glu139 deletion in TPM2 gene. Sci. Rep. 2017, 7, 16797. [Google Scholar] [CrossRef]
- Borovikov, Y.S.; Karpicheva, O.E.; Simonyan, A.O.; Avrova, S.V.; Rogozovets, E.A.; Sirenko, V.V.; Redwood, C.S. The Primary Causes of Muscle Dysfunction Associated with the Point Mutations in Tpm3.12; Conformational Analysis of Mutant Proteins as a Tool for Classification of Myopathies. Int. J. Mol. Sci. 2018, 19, 3975. [Google Scholar] [CrossRef]
- Borovikov, Y.S.; Simonyan, A.O.; Avrova, S.V.; Sirenko, V.V.; Redwood, C.S.; Karpicheva, O.E. Molecular Mechanisms of Muscle Weakness Associated with E173A Mutation in Tpm3.12. Troponin Ca2+ Sensitivity Inhibitor W7 Can Reduce the Damaging Effect of This Mutation. Int. J. Mol. Sci. 2020, 21, 4421. [Google Scholar] [CrossRef] [PubMed]
- Li, X.E.; Tobacman, L.S.; Mun, J.Y.; Craig, R.; Fischer, S.; Lehman, W. Tropomyosin position on F-actin revealed by EM reconstruction and computational chemistry. Biophys. J. 2010, 100, 1005–1013. [Google Scholar] [CrossRef]
- Yamada, Y.; Namba, K.; Fujii, T. Cardiac Muscle Thin Filament Structures Reveal Calcium Regulatory Mechanism. Nat. Commun. 2020, 11, 153. [Google Scholar] [CrossRef] [PubMed]
- Barua, B. Periodicities designed in the tropomyosin sequence and structure define its functions. Bioarchitecture 2013, 3, 51–56. [Google Scholar] [CrossRef] [PubMed]
- Behrmann, E.; Müller, M.; Penczek, P.A.; Mannherz, H.G.; Manstein, D.J.; Raunser, S. Structure of the rigor actin-tropomyosin-myosin complex. Cell 2012, 150, 327–338. [Google Scholar] [CrossRef] [PubMed]
- Doran, M.H.; Pavadai, E.; Rynkiewicz, M.J.; Walklate, J.; Bullitt, E.; Moore, J.R.; Regnier, M.; Geeves, M.A.; Lehman, W. Cryo-EM and Molecular Docking Shows Myosin Loop 4 Contacts Actin and Tropomyosin on Thin Filaments. Biophys. J. 2020, 119, 821–830. [Google Scholar] [CrossRef]
- Perry, S.V. Vertebrate TM: Distribution, properties and function. J. Muscle Res. Cell. Motil. 2001, 22, 5–49. [Google Scholar] [CrossRef]
- Jin, Y.; Peng, Y.; Lin, Z.; Chen, Y.; Wei, L.; Hacker, T.A.; Larsson, L.; Ge, Y. Comprehensive analysis of tropomyosin isoforms in skeletal muscles by top-down proteomics. J. Muscle Res. Cell Motil. 2016, 37, 41–52. [Google Scholar] [CrossRef]
- Moraczewska, J. Thin filament dysfunctions caused by mutations in tropomyosin Tpm3.12 and Tpm1.1. J. Muscle Res. Cell Motil. 2020, 41, 39–53. [Google Scholar] [CrossRef]
- Geeves, M.A.; Hitchcock-DeGregori, S.E.; Gunning, P.W. A systematic nomenclature for mammalian tropomyosin isoforms. J. Muscle Res. Cell Motil. 2015, 36, 147–153. [Google Scholar] [CrossRef]
- Billeter, R.; Heizmann, C.W.; Reist, U.; Howald, H.; Jenny, E. Alpha- and beta-tropomyosin in typed single fibers of human skeletal muscle. FEBS Lett. 1981, 132, 133–136. [Google Scholar] [CrossRef]
- Pieples, K.; Wieczorek, D.F. Tropomyosin 3 increases striated muscle isoform diversity. Biochemistry 2000, 39, 8291–8297. [Google Scholar] [CrossRef]
- McLachlan, A.D.; Stewart, M. Tropomyosin coiled-coil interactions: Evidence for an unstaggered structure. J. Mol. Biol. 1975, 98, 293–304. [Google Scholar] [CrossRef]
- Ottenheijm, C.A.; Lawlor, M.W.; Stienen, G.J.; Granzier, H.; Beggs, A.H. Changes in cross-bridge cycling underlie muscle weakness in patients with tropomyosin 3-based myopathy. Hum. Mol. Genet. 2011, 20, 2015–2025. [Google Scholar] [CrossRef]
- Ochala, J.; Gokhin, D.S.; Penisson-Besnier, I.; Quijano-Roy, S.; Monnier, N.; Lunardi, J.; Romero, N.B.; Fowler, V.M. Congenital myopathy-causing tropomyosin mutations induce thin filament dysfunction via distinct physiological mechanisms. Hum. Mol. Genet. 2012, 21, 4473–4485. [Google Scholar] [CrossRef]
- Marttila, M.; Lehtokari, V.-L.; Marston, S.; Nyman, T.A.; Barnerias, C.; Beggs, A.H.; Bertini, E.; Ceyhan-Birsoy, O.; Cintas, P.; Gerard, M.; et al. Mutation update and genotype-phenotype correlations of novel and previously described mutations in TPM2 and TPM3 causing congenital myopathies. Hum. Mutat. 2014, 35, 779–790. [Google Scholar] [CrossRef]
- Marston, S.; Memo, M.; Messer, A.; Papadaki, M.; Nowak, K.; McNamara, E.; Ong, R.; El-Mezgueldi, M.; Li, X.; Lehman, W. Mutations in repeating structural motifs of TM cause gain of function in skeletal muscle myopathy patients. Hum. Mol. Genet. 2013, 22, 4978–4987. [Google Scholar] [CrossRef] [PubMed]
- Moraczewska, J.; Robaszkiewicz, K.; Śliwinska, M.; Czajkowska, M.; Ly, T.; Kostyukova, A.; Wen, H.; Zheng, W. Congenital myopathy-related mutations in tropomyosin disrupt regulatory function through altered actin affinity and tropomodulin binding. FEBS J. 2019, 286, 1877–1893. [Google Scholar] [CrossRef]
- Lawlor, M.W.; Dechene, E.T.; Roumm, E.; Geggel, A.S.; Moghadaszadeh, B.; Beggs, A.H. Mutations of tropomyosin 3 (TPM3) are common and associated with type 1 myofiber hypotrophy in congenital fiber type disproportion. Hum. Mutat. 2010, 31, 176–183. [Google Scholar] [CrossRef] [PubMed]
- Borovikov, Y.S. Conformational changes of contractile proteins and their role in muscle contraction. Int. Rev. Cytol. 1999, 189, 267–301. [Google Scholar] [CrossRef] [PubMed]
- Borovikov, Y.S.; Rysev, N.A.; Avrova, S.V.; Karpicheva, O.E.; Borys, D.; Moraczewska, J. Molecular mechanisms of deregulation of the thin filament associated with the R167H and K168E substitutions in tropomyosin Tpm1.1. Arch. Biochem. Biophys. 2017, 614, 28–40. [Google Scholar] [CrossRef]
- Borovikov, Y.S.; Dedova, I.V.; dos Remedios, C.G.; Vikhoreva, N.N.; Vikhorev, P.G.; Avrova, S.V.; Hazlett, T.L.; Van Der Meer, B.W. Fluorescence depolarization of actin filaments in reconstructed myofibers: The effect of S1 or pPDM-S1 on movements of distinct areas of actin. Biophys. J. 2004, 86, 3020–3029. [Google Scholar] [CrossRef]
- Robaszkiewicz, K.; Dudek, E.; Kasprzak, A.A.; Moraczewska, J. Functional effects of congenital myopathy-related mutations in gamma-tropomyosin gene. Biochim. Biophys. Acta 2012, 1822, 1562–1569. [Google Scholar] [CrossRef] [PubMed]
- Fischer, S.; Rynkiewicz, M.J.; Moore, J.R.; Lehman, W. Tropomyosin diffusion over actin subunits facilitates thin filament assembly. Struct. Dyn. 2016, 3, 012002. [Google Scholar] [CrossRef] [PubMed]
- Galazzo, L.; Nogara, L.; LoVerso, F.; Polimeno, A.; Blaauw, B.; Sandri, M.; Reggiani, C.; Carbonera, D. Changes in the fraction of strongly attached cross bridges in mouse atrophic and hypertrophic muscles as revealed by continuous wave electron paramagnetic resonance. Am. J. Physiol. Cell Physiol. 2019, 316, 722–730. [Google Scholar] [CrossRef]
- Onishi, H.; Nitanai, Y. Thiol reactivity as a sensor of rotation of the converter in myosin. Biochem. Biophys. Res. Commun. 2008, 369, 115–123. [Google Scholar] [CrossRef]
- Borovikov, Y.S.; Avrova, S.V.; Rysev, N.A.; Sirenko, V.V.; Simonyan, A.O.; Chernev, A.A.; Karpicheva, O.E.; Piers, A.; Redwood, C.S. Aberrant movement of β-tropomyosin associated with congenital myopathy causes defective response of myosin heads and actin during the ATPase cycle. Arch. Biochem. Biophys. 2015, 577–578, 13–23. [Google Scholar] [CrossRef]
- Borovikov, Y.S.; Andreeva, D.D.; Avrova, S.V.; Sirenko, V.V.; Simonyan, A.O.; Redwood, C.S.; Karpicheva, O.E. Molecular Mechanisms of the Deregulation of Muscle Contraction Induced by the R90P Mutation in Tpm3.12 and the Weakening of This Effect by BDM and W7. Int. J. Mol. Sci. 2021, 22, 6318. [Google Scholar] [CrossRef]
- Karpicheva, O.E.; Simonyan, A.O.; Rysev, N.A.; Redwood, C.S.; Borovikov, Y.S. Looking for Targets to Restore the Contractile Function in Congenital Myopathy Caused by Gln147Pro Tropomyosin. Int. J. Mol. Sci. 2020, 21, 7590. [Google Scholar] [CrossRef] [PubMed]
- Karpicheva, O.E.; Sirenko, V.V.; Rysev, N.A.; Simonyan, A.O.; Borys, D.; Moraczewska, J.; Borovikov, Y.S. Deviations in conformational rearrangements of thin filaments and myosin caused by the Ala155Thr substitution in hydrophobic core of tropomyosin. Biochim. Biophys. Acta 2017, 1865, 1790–1799. [Google Scholar] [CrossRef]
- Borovikov, Y.S.; Avrova, S.V.; Karpicheva, O.E.; Robinson, P.; Redwood, C.S. The effect of the dilated cardiomyopathy-causing Glu40Lys TPM1 mutation on actin-myosin interactions during the ATPase cycle, Biochem. Biophys. Res. Commun. 2011, 411, 496–500. [Google Scholar] [CrossRef]
- Rysev, N.A.; Nevzorov, I.A.; Avrova, S.V.; Karpicheva, O.E.; Redwood, C.S.; Levitsky, D.I.; Borovikov, Y.S. Gly126Arg substitution causes anomalous behaviour of α-skeletal and β-smooth tropomyosins during the ATPase cycle. Arch. Biochem. Biophys. 2014, 543, 57–66. [Google Scholar] [CrossRef]
- Borovikov, Y.S.; Simonyan, A.O.; Karpicheva, O.E.; Avrova, S.V.; Rysev, N.A.; Sirenko, V.V.; Piers, A.; Redwood, C.S. The reason for a high Ca(2+)-sensitivity associated with Arg91Gly substitution in TPM2 gene is the abnormal behavior and high flexibility of tropomyosin during the ATPase cycle. Biochem. Biophys. Res. Commun. 2017, 494, 681–686. [Google Scholar] [CrossRef]
- Crick, F.H.C. The packing of α-helices. Simple coiled-coils. Acta Crystallogr. 1953, 6, 689–697. [Google Scholar] [CrossRef]
- Mudalige, W.A.; Tao, T.C.; Lehrer, S.S. Ca2+-dependent photocrosslinking of tropomyosin residue 146 to residues 157–163 in the C-terminal domain of troponin I in reconstituted skeletal muscle thin filaments. J. Mol. Biol. 2009, 389, 575–583. [Google Scholar] [CrossRef] [PubMed]
- McKillop, D.F.; Fortune, N.S.; Ranatunga, K.W.; Geeves, M.A. The influence of 2,3-butanedione 2-monoxime (BDM) on the interaction between actin and myosin in solution and in skinned muscle fibres. J. Muscle Res. Cell Motil. 1994, 15, 309. [Google Scholar] [CrossRef]
- Lee, B.K.; Jeung, K.W.; Choi, S.S.; Park, S.W.; Yun, S.W.; Lee, S.M.; Kim, N.Y.; Heo, T.; Min, Y.I. Effects of the administration of 2,3-butanedione monoxime during conventional cardiopulmonary resuscitation on ischaemic contracture and resuscitability in a pig model of out-of-hospital cardiac arrest. Resuscitation 2015, 87, 26. [Google Scholar] [CrossRef]
- Herrmann, C.; Wray, J.; Travers, F.; Barman, T. Effect of 2,3-butanedione monoxime on myosin and myofibrillar ATPases. An example of an uncompetitive inhibitor. Biochemistry 1992, 31, 12227. [Google Scholar] [CrossRef] [PubMed]
- Planelles-Herrero, V.J.; Hartman, J.J.; Robert-Paganin, J.; Malik, F.I.; Houdusse, A. Mechanistic and structural basis for activation of cardiac myosin force production by omecamtiv mecarbil. Nat. Commun. 2017, 8, 190. [Google Scholar] [CrossRef]
- Liu, Y.; White, H.D.; Belknap, B.; Winkelmann, D.A.; Forgacs, E. Omecamtiv Mecarbil modulates the kinetic and motile properties of porcine β-cardiac myosin. Biochemistry 2015, 54, 1963–1975. [Google Scholar] [CrossRef]
- Li, Y.; Karim, M.R.; Wang, B.; Peng, J. Effects of Green Tea (-)-Epigallocatechin-3-Gallate (EGCG) on Cardiac Function—A Review of the Therapeutic Mechanism and Potentials. Mini Rev. Med. Chem. 2022, 22, 2371–2382. [Google Scholar] [CrossRef] [PubMed]
- Hotta, Y.; Huang, L.; Muto, T.; Yajima, M.; Miyazeki, K.; Ishikawa, N.; Fukuzawa, Y.; Wakida, Y.; Tushima, H.; Ando, H.; et al. Positive inotropic effect of purified green tea catechin derivative in guinea pig hearts: The measurements of cellular Ca2+ and nitric oxide release. Eur. J. Pharmacol. 2006, 552, 123–130. [Google Scholar] [CrossRef] [PubMed]
- Liou, Y.M.; Kuo, S.C.; Hsieh, S.R. Differential effects of a green tea-derived polyphenol (-)-epigallocatechin-3-gallate on the acidosis-induced decrease in the Ca(2+) sensitivity of cardiac and skeletal muscle. Pflügers Arch.-Eur. J. Physiol. 2008, 456, 787–800. [Google Scholar] [CrossRef] [PubMed]
- Warren, C.M.; Karam, C.N.; Wolska, B.M.; Kobayashi, T.; de Tombe, P.P.; Arteaga, G.M.; Bos, J.M.; Ackerman, M.J.; Solaro, R.J. Green Tea Catechin Normalizes the Enhanced Ca2+ Sensitivity of Myofilaments Regulated by a Hypertrophic Cardiomyopathy-Associated Mutation in Human Cardiac Troponin I (K206I). Circ. Cardiovasc. Genet. 2015, 8, 765–773. [Google Scholar] [CrossRef] [PubMed]
- Collibee, S.E.; Bergnes, G.; Muci, A.; Browne, W.F.; Garard, M.; Hinken, A.C.; Russell, A.J.; Suehiro, I.; Hartman, J.; Kawas, R.; et al. Discovery of Tirasemtiv, the First Direct Fast Skeletal Muscle Troponin Activator. ACS Med. Chem. Lett. 2018, 9, 354–358. [Google Scholar] [CrossRef]
- de Winter, J.M.; Gineste, C.; Minardi, E.; Brocca, L.; Rossi, M.; Borsboom, T.; Beggs, A.H.; Bernard, M.; Bendahan, D.; Hwee, D.T.; et al. Acute and chronic tirasemtiv treatment improves in vivo and in vitro muscle performance in actin-based nemaline myopathy mice. Hum. Mol. Genet. 2021, 30, 1305–1320. [Google Scholar] [CrossRef]
- Hwee, D.T.; Kennedy, A.; Ryans, J.; Russell, A.J.; Jia, Z.; Hinken, A.C.; Morgans, D.J.; Malik, F.I.; Jasper, J.R. Fast Skeletal Muscle Troponin Activator Tirasemtiv Increases Muscle Function and Performance in the B6SJL-SOD1G93A ALS Mouse Model. PLoS ONE 2014, 9, e96921. [Google Scholar] [CrossRef]
- Hansen, R.; Saikali, K.G.; Chou, W.; Russell, A.J.; Chen, M.M.; Vijayakumar, V.; Stoltz, R.R.; Baudry, S.; Enoka, R.M.; Morgans, D.J.; et al. Tirasemtiv amplifies skeletal muscle response to nerve activation in humans. Muscle Nerve 2014, 50, 925–931. [Google Scholar] [CrossRef]
- Hwee, D.T.; Cheng, A.J.; Hartman, J.J.; Hinken, A.C.; Lee, K.; Durham, N.; Russell, A.J.; Malik, F.I.; Westerblad, H.; Jasper, L.R. The Ca2+ sensitizer CK-2066260 increases myofibrillar Ca2+ sensitivity and submaximal force selectively in fast skeletal muscle. J. Physiol. 2017, 595, 1657–1670. [Google Scholar] [CrossRef]
- Margossian, S.; Lowey, S. Preparation of myosin and its subfragments from rabbit skeletal muscle. Methods Enzym. 1982, 85, 55–71. [Google Scholar] [CrossRef]
- Spudich, J.A.; Watt, S. The regulation of rabbit skeletal muscle contraction. J. Biol. Chem. 1971, 246, 4866. [Google Scholar] [CrossRef]
- Okamoto, Y.; Sekine, T. A streamlined method of subfragment one preparation from myosin. J. Biochem. 1985, 98, 1143–1145. [Google Scholar] [CrossRef] [PubMed]
- Borejdo, J.; Putnam, S. Polarization of fluorescence from single skinned glycerinated rabbit psoas fibers in rigor and relaxation. Biochim. Biophys. Acta 1977, 459, 578–595. [Google Scholar] [CrossRef] [PubMed]
- Robinson, P.; Lipscomb, S.; Preston, L.C.; Altin, E.; Watkins, H.; Ashley, C.C.; Redwood, C.S. Mutations in fast skeletal troponin I, troponin T, and beta-tropomyosin that cause distal arthrogryposis all increase contractile function. FASEB J. 2007, 21, 896–905. [Google Scholar] [CrossRef]
- Fiske, C.H.; Subbarow, Y. Determination of inorganic phosphate. J. Biol. Chem. 1925, 66, 375–400. [Google Scholar] [CrossRef]
- Yanagida, T.; Oosawa, F. Polarized fluorescence from epsilon-ADP incorporated into F-actin in a myosin-free single fiber: Conformation of F-actin and changes induced in it by heavy meromyosin. J. Mol. Biol. 1978, 126, 507–524. [Google Scholar] [CrossRef]
- Kakol, I.; Borovikov, Y.S.; Szczesna, D.; Kirillina, V.P.; Levitsky, D.I. Conformational changes of F-actin in myosin-free ghost single fiber induced by either phosphorylated or dephosphorylated heavy meromyosin. Biochim. Biophys. Acta 1987, 913, 1–9. [Google Scholar] [CrossRef]
- Goody, R.S.; Hofmann, W. Stereochemical aspects of the interaction of myosin and actomyosin with nucleotides. J. Muscle Res. Cell Motil. 1980, 1, 101–115. [Google Scholar] [CrossRef]












Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Karpicheva, O.E.; Avrova, S.V.; Bogdanov, A.L.; Sirenko, V.V.; Redwood, C.S.; Borovikov, Y.S. Molecular Mechanisms of Deregulation of Muscle Contractility Caused by the R168H Mutation in TPM3 and Its Attenuation by Therapeutic Agents. Int. J. Mol. Sci. 2023, 24, 5829. https://doi.org/10.3390/ijms24065829
Karpicheva OE, Avrova SV, Bogdanov AL, Sirenko VV, Redwood CS, Borovikov YS. Molecular Mechanisms of Deregulation of Muscle Contractility Caused by the R168H Mutation in TPM3 and Its Attenuation by Therapeutic Agents. International Journal of Molecular Sciences. 2023; 24(6):5829. https://doi.org/10.3390/ijms24065829
Chicago/Turabian StyleKarpicheva, Olga E., Stanislava V. Avrova, Andrey L. Bogdanov, Vladimir V. Sirenko, Charles S. Redwood, and Yurii S. Borovikov. 2023. "Molecular Mechanisms of Deregulation of Muscle Contractility Caused by the R168H Mutation in TPM3 and Its Attenuation by Therapeutic Agents" International Journal of Molecular Sciences 24, no. 6: 5829. https://doi.org/10.3390/ijms24065829
APA StyleKarpicheva, O. E., Avrova, S. V., Bogdanov, A. L., Sirenko, V. V., Redwood, C. S., & Borovikov, Y. S. (2023). Molecular Mechanisms of Deregulation of Muscle Contractility Caused by the R168H Mutation in TPM3 and Its Attenuation by Therapeutic Agents. International Journal of Molecular Sciences, 24(6), 5829. https://doi.org/10.3390/ijms24065829