

Altering Calcium Sensitivity in Heart Failure: A Crossroads of Disease Etiology and Therapeutic Innovation
 , ,
, ,
Abstract
:1. Introduction
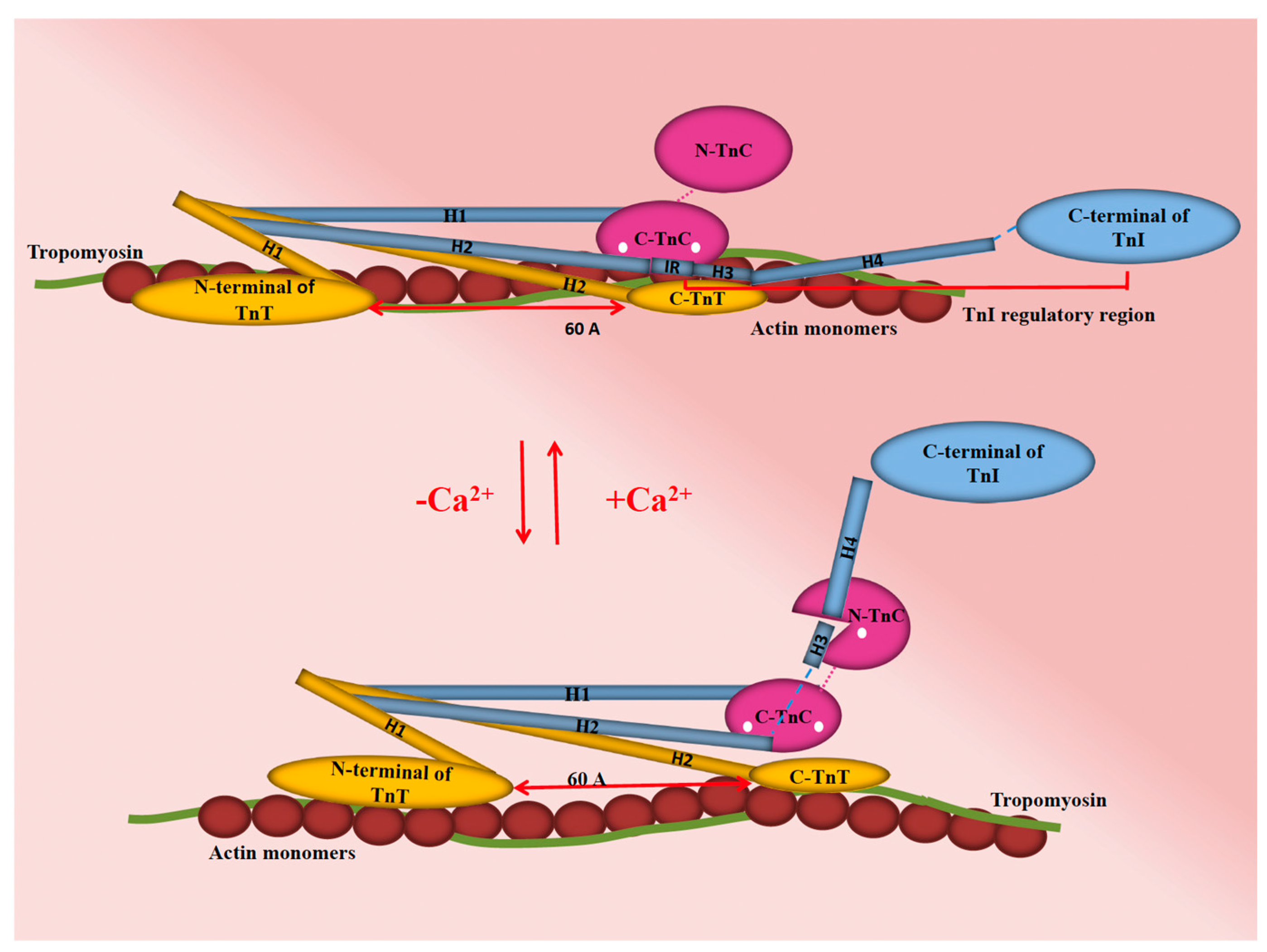
2. Overview of Ca2+ Cycling and Homeostasis
3. Alterations in Ca2+ Sensitivity in HF
4. Ca2+ Sensitivity Changes in HFpEF and HFrEF
5. Manipulating Ca2+ Sensitivity for Therapeutic Gain
6. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
References
- Virani, S.S.; Alonso, A.; Benjamin, E.J.; Bittencourt, M.S.; Callaway, C.W.; Carson, A.P.; Chamberlain, A.M.; Chang, A.R.; Cheng, S.; Delling, F.N. Heart disease and stroke statistics—2020 update: A report from the american heart association. Circulation 2020, 141, e139–e596. [Google Scholar] [CrossRef] [PubMed]
- Tsao, C.W.; Aday, A.W.; Almarzooq, Z.I.; Alonso, A.; Beaton, A.Z.; Bittencourt, M.S.; Boehme, A.K.; Buxton, A.E.; Carson, A.P.; Commodore-Mensah, Y. Heart disease and stroke statistics—2022 update: A report from the American Heart Association. Circulation 2022, 145, e153–e639. [Google Scholar] [CrossRef] [PubMed]
- Lindenfeld, J.; Albert, N.M.; Boehmer, J.P.; Collins, S.P.; Ezekowitz, J.A.; Givertz, M.M.; Katz, S.D.; Klapholz, M.; Moser, D.K.; Rogers, J.G. HFSA 2010 comprehensive heart failure practice guideline. J. Card. Fail. 2010, 16, e1–e194. [Google Scholar] [PubMed]
- Members, W.C.; Hunt, S.A.; Abraham, W.T.; Chin, M.H.; Feldman, A.M.; Francis, G.S.; Ganiats, T.G.; Jessup, M.; Konstam, M.A.; Mancini, D.M. 2009 focused update incorporated into the ACC/AHA 2005 guidelines for the diagnosis and management of heart failure in adults: A report of the american college of cardiology foundation/american heart association task force on practice guidelines: Developed in collaboration with the international society for heart and lung transplantation. Circulation 2009, 119, e391–e479. [Google Scholar]
- Dharmarajan, K.; Rich, M.W. Epidemiology, pathophysiology, and prognosis of heart failure in older adults. Heart Fail. Clin. 2017, 13, 417–426. [Google Scholar] [CrossRef] [PubMed]
- Gazewood, J.D.; Turner, P.L. Heart Failure with Preserved Ejection Fraction: Diagnosis and Management. Am. Fam. Physician 2017, 96, 582–588. [Google Scholar] [PubMed]
- Pieske, B.; Tschöpe, C.; De Boer, R.A.; Fraser, A.G.; Anker, S.D.; Donal, E.; Edelmann, F.; Fu, M.; Guazzi, M.; Lam, C.S. How to diagnose heart failure with preserved ejection fraction: The HFA–PEFF diagnostic algorithm: A consensus recommendation from the Heart Failure Association (HFA) of the European Society of Cardiology (ESC). Eur. Heart J. 2019, 40, 3297–3317. [Google Scholar] [CrossRef]
- Satpathy, C.; Mishra, T.K.; Satpathy, R.; Satpathy, H.K.; Barone, E. Diagnosis and management of diastolic dysfunction and heart failure. Am. Fam. Physician 2006, 73, 841–846. [Google Scholar]
- Shah, S.J.; Katz, D.H.; Selvaraj, S.; Burke, M.A.; Yancy, C.W.; Gheorghiade, M.; Bonow, R.O.; Huang, C.-C.; Deo, R.C. Phenomapping for novel classification of heart failure with preserved ejection fraction. Circulation 2015, 131, 269–279. [Google Scholar] [CrossRef]
- Ponikowski, P.; Voors, A.A.; Anker, S.D.; Bueno, H.; Cleland, J.G.; Coats, A.J.; Falk, V.; González-Juanatey, J.R.; Harjola, V.-P.; Jankowska, E.A. 2016 ESC Guidelines for the diagnosis and treatment of acute and chronic heart failure. Kardiol. Pol. 2016, 74, 1037–1147. [Google Scholar] [CrossRef]
- Yancy, C.W.; Jessup, M.; Bozkurt, B.; Butler, J.; Casey, D.E., Jr.; Drazner, M.H.; Fonarow, G.C.; Geraci, S.A.; Horwich, T.; Januzzi, J.L. 2013 ACCF/AHA guideline for the management of heart failure: Executive summary: A report of the American College of Cardiology Foundation/American Heart Association Task Force on practice guidelines. Circulation 2013, 128, 1810–1852. [Google Scholar] [CrossRef] [PubMed]
- Lompré, A.-M.; Hajjar, R.J.; Harding, S.E.; Kranias, E.G.; Lohse, M.J.; Marks, A.R. Ca2+ cycling and new therapeutic approaches for heart failure. Circulation 2010, 121, 822–830. [Google Scholar] [CrossRef] [PubMed]
- Bers, D.M. Altered cardiac myocyte Ca regulation in heart failure. Physiology 2006, 21, 380–387. [Google Scholar] [CrossRef] [PubMed]
- Chung, J.-H.; Biesiadecki, B.J.; Ziolo, M.T.; Davis, J.P.; Janssen, P.M. Myofilament calcium sensitivity: Role in regulation of in vivo cardiac contraction and relaxation. Front. Physiol. 2016, 7, 562. [Google Scholar] [CrossRef] [PubMed]
- Wen, Y.; Pinto, J.R.; Gomes, A.V.; Xu, Y.; Wang, Y.; Wang, Y.; Potter, J.D.; Kerrick, W.G.L. Functional consequences of the human cardiac troponin I hypertrophic cardiomyopathy mutation R145G in transgenic mice. J. Biol. Chem. 2008, 283, 20484–20494. [Google Scholar] [CrossRef]
- Sommese, R.F.; Nag, S.; Sutton, S.; Miller, S.M.; Spudich, J.A.; Ruppel, K.M. Effects of troponin T cardiomyopathy mutations on the calcium sensitivity of the regulated thin filament and the actomyosin cross-bridge kinetics of human β-cardiac myosin. PLoS ONE 2013, 8, e83403. [Google Scholar] [CrossRef]
- Du, C.-K.; Morimoto, S.; Nishii, K.; Minakami, R.; Ohta, M.; Tadano, N.; Lu, Q.-W.; Wang, Y.-Y.; Zhan, D.-Y.; Mochizuki, M. Knock-in mouse model of dilated cardiomyopathy caused by troponin mutation. Circ. Res. 2007, 101, 185–194. [Google Scholar] [CrossRef]
- Blanchard, E.; Seidman, C.; Seidman, J.; LeWinter, M.; Maughan, D. Altered crossbridge kinetics in the αMHC403/+ mouse model of familial hypertrophic cardiomyopathy. Circ. Res. 1999, 84, 475–483. [Google Scholar] [CrossRef]
- Palmer, B.M.; Wang, Y.; Teekakirikul, P.; Hinson, J.T.; Fatkin, D.; Strouse, S.; VanBuren, P.; Seidman, C.E.; Seidman, J.G.; Maughan, D.W. Myofilament mechanical performance is enhanced by R403Q myosin in mouse myocardium independent of sex. Am. J. Physiol. Heart Circ. Physiol. 2008, 294, H1939–H1947. [Google Scholar] [CrossRef]
- Chuan, P.; Sivaramakrishnan, S.; Ashley, E.A.; Spudich, J.A. Cell-intrinsic functional effects of the α-cardiac myosin Arg-403-Gln mutation in familial hypertrophic cardiomyopathy. Biophys. J. 2012, 102, 2782–2790. [Google Scholar] [CrossRef]
- Kim, S.-J.; Iizuka, K.; Kelly, R.A.; Geng, Y.-J.; Bishop, S.P.; Yang, G.; Kudej, A.; McConnell, B.K.; Seidman, C.E.; Seidman, J.G. An α-cardiac myosin heavy chain gene mutation impairs contraction and relaxation function of cardiac myocytes. Am. J. Physiol. Heart Circ. Physiol. 1999, 276, H1780–H1787. [Google Scholar] [CrossRef] [PubMed]
- Szczesna-Cordary, D.; Guzman, G.; Zhao, J.; Hernandez, O.; Wei, J.; Diaz-Perez, Z. The E22K mutation of myosin RLC that causes familial hypertrophic cardiomyopathy increases calcium sensitivity of force and ATPase in transgenic mice. J. Cell Sci. 2005, 118, 3675–3683. [Google Scholar] [CrossRef] [PubMed]
- Szczesna-Cordary, D.; Jones, M.; Moore, J.R.; Watt, J.; Kerrick, W.G.L.; Xu, Y.; Wang, Y.; Wagg, C.; Lopaschuk, G.D. Myosin regulatory light chain E22K mutation results in decreased cardiac intracellular calcium and force transients. FASEB J. 2007, 21, 3974–3985. [Google Scholar] [CrossRef] [PubMed]
- Berridge, M.J.; Bootman, M.D.; Roderick, H.L. Calcium signalling: Dynamics, homeostasis and remodelling. Nat. Rev. Mol. Cell Biol. 2003, 4, 517–529. [Google Scholar] [CrossRef] [PubMed]
- Huxley, A.F. Muscle structures and theories of contraction. Progr. Biophys. Chem. 1957, 7, 255–318. [Google Scholar] [CrossRef]
- Hibberd, M.; Jewell, B. Calcium-and length-dependent force production in rat ventricular muscle. J. Physiol. 1982, 329, 527–540. [Google Scholar] [CrossRef] [PubMed]
- Kentish, J.C.; Ter Keurs, H.; Ricciardi, L.; Bucx, J.; Noble, M. Comparison between the sarcomere length-force relations of intact and skinned trabeculae from rat right ventricle. Influence of calcium concentrations on these relations. Circ. Res. 1986, 58, 755–768. [Google Scholar] [CrossRef]
- Patterson, S.W.; Piper, H.; Starling, E. The regulation of the heart beat. J. Physiol. 1914, 48, 465. [Google Scholar] [CrossRef]
- Solaro, R.J. Modulation of cardiac myofilament activity by protein phosphorylation. Compr. Physiol. 2011, 264–300. [Google Scholar] [CrossRef]
- Mope, L.; McClellan, G.B.; Winegrad, S. Calcium sensitivity of the contractile system and phosphorylation of troponin in hyperpermeable cardiac cells. J. Gen. Physiol. 1980, 75, 271–282. [Google Scholar] [CrossRef]
- Zhang, R.; Zhao, J.; Mandveno, A.; Potter, J.D. Cardiac troponin I phosphorylation increases the rate of cardiac muscle relaxation. Circ. Res. 1995, 76, 1028–1035. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Desantiago, J.; Chu, G.; Kranias, E.G.; Bers, D.M. Phosphorylation of phospholamban and troponin I in β-adrenergic-induced acceleration of cardiac relaxation. Am. J. Physiol. Heart Circ. Physiol. 2000, 278, H769–H779. [Google Scholar] [CrossRef] [PubMed]
- Haworth, R.S.; Cuello, F.; Herron, T.J.; Franzen, G.; Kentish, J.C.; Gautel, M.; Avkiran, M. Protein kinase D is a novel mediator of cardiac troponin I phosphorylation and regulates myofilament function. Circ. Res. 2004, 95, 1091–1099. [Google Scholar] [CrossRef] [PubMed]
- Lim, C.C.; Yang, H.; Yang, M.; Wang, C.-K.; Shi, J.; Berg, E.A.; Pimentel, D.R.; Gwathmey, J.K.; Hajjar, R.J.; Helmes, M. A novel mutant cardiac troponin C disrupts molecular motions critical for calcium binding affinity and cardiomyocyte contractility. Biophys. J. 2008, 94, 3577–3589. [Google Scholar] [CrossRef]
- Saad, N.S.; Elnakish, M.T.; Brundage, E.A.; Biesiadecki, B.J.; Kilic, A.; Ahmed, A.A.; Mohler, P.J.; Janssen, P.M. Assessment of PKA and PKC inhibitors on force and kinetics of non-failing and failing human myocardium. Life Sci. 2018, 215, 119–127. [Google Scholar] [CrossRef]
- Anderson, P.; Malouf, N.; Oakeley, A.; Pagani, E.; Allen, P. Troponin T isoform expression in humans. A comparison among normal and failing adult heart, fetal heart, and adult and fetal skeletal muscle. Circ. Res. 1991, 69, 1226–1233. [Google Scholar] [CrossRef]
- Solaro, R.J.; Powers, F.M.; Gao, L.; Gwathmey, J.K. Adaptive and Maladaptive Processes: Control of Myofilament Activation in Heart Failure. Circulation 1993, 87, 38. [Google Scholar]
- Mesnard-Rouiller, L.; Mercadier, J.-J.; Butler-Browne, G.; Heimburger, M.; Logeart, D.; Allen, P.D.; Samson, F. Troponin T mRNA and protein isoforms in the human left ventricle: Pattern of expression in failing and control hearts. J. Mol. Cell. Cardiol. 1997, 29, 3043–3055. [Google Scholar] [CrossRef]
- Van der Velden, J.; Klein, L.; Van Der Bijl, M.; Huybregts, M.; Stooker, W.; Witkop, J.; Eijsman, L.; Visser, C.; Visser, F.; Stienen, G. Isometric tension development and its calcium sensitivity in skinned myocyte-sized preparations from different regions of the human heart. Cardiovasc. Res. 1999, 42, 706–719. [Google Scholar] [CrossRef]
- Morano, I.; Hädicke, K.; Haase, H.; Böhm, M.; Erdmann, E.; Schaub, M.C. Changes in essential myosin light chain isoform expression provide a molecular basis for isometric force regulation in the failing human heart. J. Mol. Cell. Cardiol. 1997, 29, 1177–1187. [Google Scholar] [CrossRef]
- Miyata, S.; Minobe, W.; Bristow, M.R.; Leinwand, L.A. Myosin heavy chain isoform expression in the failing and nonfailing human heart. Circ. Res. 2000, 86, 386–390. [Google Scholar] [CrossRef] [PubMed]
- Margossian, S.S.; Anderson, P.A.; Chantler, P.D.; Deziel, M.; Umeda, P.K.; Stafford, W.F.; Norton, P.; Malhotra, A.; Yang, F.; Caulfield, J.B. Calcium regulation in the human myocardium affected by dilated cardiomyopathy: A structural basis for impaired Ca2+-sensitivity. Mol. Cell. Biochem. 1999, 194, 301–313. [Google Scholar] [CrossRef] [PubMed]
- Harding, S.E.; Brown, L.A.; Wynne, D.G.; Davies, C.H.; Poole-Wilson, P.A. Mechanisms of β adrenoceptor desensitisation in the failing human heart. Cardiovasc. Res. 1994, 28, 1451–1460. [Google Scholar] [CrossRef] [PubMed]
- Pieske, B.; Beyermann, B.; Breu, V.; Löffler, B.M.; Schlotthauer, K.; Maier, L.S.; Schmidt-Schweda, S.; Just, H.R.; Hasenfuss, G. Functional effects of endothelin and regulation of endothelin receptors in isolated human nonfailing and failing myocardium. Circulation 1999, 99, 1802–1809. [Google Scholar] [CrossRef]
- Asano, K.; Dutcher, D.L.; Port, J.D.; Minobe, W.A.; Tremmel, K.D.; Roden, R.L.; Bohlmeyer, T.J.; Bush, E.W.; Jenkin, M.J.; Abraham, W.T. Selective downregulation of the angiotensin II AT1-receptor subtype in failing human ventricular myocardium. Circulation 1997, 95, 1193–1200. [Google Scholar] [CrossRef]
- Takeishi, Y.; Bhagwat, A.; Ball, N.A.; Kirkpatrick, D.L.; Periasamy, M.; Walsh, R.A. Effect of angiotensin-converting enzyme inhibition on protein kinase C and SR proteins in heart failure. Am. J. Physiol. Heart Circ. Physiol. 1999, 276, H53–H62. [Google Scholar] [CrossRef]
- Bowling, N.; Walsh, R.A.; Song, G.; Estridge, T.; Sandusky, G.E.; Fouts, R.L.; Mintze, K.; Pickard, T.; Roden, R.; Bristow, M.R. Increased protein kinase C activity and expression of Ca2+-sensitive isoforms in the failing human heart. Circulation 1999, 99, 384–391. [Google Scholar] [CrossRef]
- Neumann, J.; Eschenhagen, T.; Jones, L.R.; Linck, B.; Schmitz, W.; Scholz, H.; Zimmermann, N. Increased expression of cardiac phosphatases in patients with end-stage heart failure. J. Mol. Cell. Cardiol. 1997, 29, 265–272. [Google Scholar] [CrossRef]
- Willott, R.H.; Gomes, A.V.; Chang, A.N.; Parvatiyar, M.S.; Pinto, J.R.; Potter, J.D. Mutations in Troponin that cause HCM, DCM AND RCM: What can we learn about thin filament function? J. Mol. Cell. Cardiol. 2010, 48, 882–892. [Google Scholar] [CrossRef]
- Lang, R.; Gomes, A.V.; Zhao, J.; Miller, T.; Potter, J.D.; Housmans, P.R. Functional analysis of a troponin I (R145G) mutation associated with familial hypertrophic cardiomyopathy. J. Biol. Chem. 2002, 277, 11670–11678. [Google Scholar] [CrossRef]
- Elliott, K.; Watkins, H.; Redwood, C.S. Altered regulatory properties of human cardiac troponin I mutants that cause hypertrophic cardiomyopathy. J. Biol. Chem. 2000, 275, 22069–22074. [Google Scholar] [CrossRef] [PubMed]
- Blanchard, E.M.; Solaro, R.J. Inhibition of the activation and troponin calcium binding of dog cardiac myofibrils by acidic pH. Circ. Res. 1984, 55, 382–391. [Google Scholar] [CrossRef] [PubMed]
- Fabiato, A.; Fabiato, F. Effects of pH on the myofilaments and the sarcoplasmic reticulum of skinned cells from cardiace and skeletal muscles. J. Physiol. 1978, 276, 233–255. [Google Scholar] [CrossRef] [PubMed]
- Kentish, J.C. The effects of inorganic phosphate and creatine phosphate on force production in skinned muscles from rat ventricle. J. Physiol. 1986, 370, 585–604. [Google Scholar] [CrossRef] [PubMed]
- Gao, W.D.; Atar, D.; Liu, Y.; Perez, N.G.; Murphy, A.M.; Marban, E. Role of troponin I proteolysis in the pathogenesis of stunned myocardium. Circ. Res. 1997, 80, 393–399. [Google Scholar] [CrossRef] [PubMed]
- Van Eyk, J.E.; Powers, F.; Law, W.; Larue, C.; Hodges, R.S.; Solaro, R.J. Breakdown and release of myofilament proteins during ischemia and ischemia/reperfusion in rat hearts: Identification of degradation products and effects on the pCa-force relation. Circ. Res. 1998, 82, 261–271. [Google Scholar] [CrossRef]
- van Der Velden, J.; Klein, L.; Zaremba, R.; Boontje, N.; Huybregts, M.; Stooker, W.; Eijsman, L.; De Jong, J.; Visser, C.; Visser, F. Effects of calcium, inorganic phosphate, and pH on isometric force in single skinned cardiomyocytes from donor and failing human hearts. Circulation 2001, 104, 1140–1146. [Google Scholar] [CrossRef]
- Wolff, M.R.; Buck, S.H.; Stoker, S.W.; Greaser, M.L.; Mentzer, R.M. Myofibrillar calcium sensitivity of isometric tension is increased in human dilated cardiomyopathies: Role of altered beta-adrenergically mediated protein phosphorylation. J. Clin. Investig. 1996, 98, 167–176. [Google Scholar] [CrossRef]
- Van der Velden, J.; Boontje, N.; Papp, Z.; Klein, L.; Visser, F.; De Jong, J.; Owen, V.; Burton, P.; Stienen, G. Calcium sensitivity of force in human ventricular cardiomyocytes from donor and failing hearts. Basic Res. Cardiol. 2002, 97, I118–I126. [Google Scholar] [CrossRef]
- Margossian, S.; White, H.; Caulfield, J.; Norton, P.; Taylor, S.; Slayter, H. Light chain 2 profile and activity of human ventricular myosin during dilated cardiomyopathy. Identification of a causal agent for impaired myocardial function. Circulation 1992, 85, 1720–1733. [Google Scholar] [CrossRef]
- Kobayashi, T.; Solaro, R.J. Calcium, thin filaments, and the integrative biology of cardiac contractility. Annu. Rev. Physiol. 2005, 67, 39–67. [Google Scholar] [CrossRef] [PubMed]
- Metzger, J.M.; Westfall, M.V. Covalent and noncovalent modification of thin filament action the essential role of troponin in cardiac muscle regulation. Circ. Res. 2004, 94, 146–158. [Google Scholar] [CrossRef] [PubMed]
- Bodor, G.S.; Oakeley, A.E.; Allen, P.D.; Crimmins, D.L.; Ladenson, J.H.; Anderson, P.A. Troponin I phosphorylation in the normal and failing adult human heart. Circulation 1997, 96, 1495–1500. [Google Scholar] [CrossRef] [PubMed]
- Zakhary, D.R.; Moravec, C.S.; Stewart, R.W.; Bond, M. Protein kinase A (PKA)-dependent troponin-I phosphorylation and PKA regulatory subunits are decreased in human dilated cardiomyopathy. Circulation 1999, 99, 505–510. [Google Scholar] [CrossRef]
- van der Velden, J.; Papp, Z.; Zaremba, R.; Boontje, N.; de Jong, J.W.; Owen, V.; Burton, P.; Goldmann, P.; Jaquet, K.; Stienen, G. Increased Ca2+-sensitivity of the contractile apparatus in end-stage human heart failure results from altered phosphorylation of contractile proteins. Cardiovasc. Res. 2003, 57, 37–47. [Google Scholar] [CrossRef] [PubMed]
- Van der Velden, J.; Merkus, D.; Klarenbeek, B.; James, A.; Boontje, N.; Dekkers, D.; Stienen, G.; Lamers, J.; Duncker, D. Alterations in myofilament function contribute to left ventricular dysfunction in pigs early after myocardial infarction. Circ. Res. 2004, 95, e85–e95. [Google Scholar] [CrossRef] [PubMed]
- Wolff, M.R.; Whitesell, L.F.; Moss, R.L. Calcium sensitivity of isometric tension is increased in canine experimental heart failure. Circ. Res. 1995, 76, 781–789. [Google Scholar] [CrossRef]
- Lamberts, R.R.; Hamdani, N.; Soekhoe, T.W.; Boontje, N.M.; Zaremba, R.; Walker, L.A.; De Tombe, P.P.; Van Der Velden, J.; Stienen, G.J. Frequency-dependent myofilament Ca2+ desensitization in failing rat myocardium. J. Physiol. 2007, 582, 695–709. [Google Scholar] [CrossRef]
- Saad, N.S.; Elnakish, M.T.; Ahmed, A.A.; Janssen, P.M. Protein kinase A as a promising target for heart failure drug development. Arch. Med. Res. 2018, 49, 530–537. [Google Scholar] [CrossRef]
- Calderone, A.; Bouvier, M.; Li, K.; Juneau, C.; de Champlain, J.; Rouleau, J.-L. Dysfunction of the beta-and alpha-adrenergic systems in a model of congestive heart failure. The pacing-overdrive dog. Circ. Res. 1991, 69, 332–343. [Google Scholar] [CrossRef]
- Marzo, K.P.; Frey, M.J.; Wilson, J.R.; Liang, B.T.; Manning, D.R.; Lanoce, V.; Molinoff, P.B. Beta-adrenergic receptor-G protein-adenylate cyclase complex in experimental canine congestive heart failure produced by rapid ventricular pacing. Circ. Res. 1991, 69, 1546–1556. [Google Scholar] [CrossRef] [PubMed]
- Kiuchi, K.; Shannon, R.P.; Komamura, K.; Cohen, D.J.; Bianchi, C.; Homcy, C.J.; Vatner, S.F.; Vatner, D.E. Myocardial beta-adrenergic receptor function during the development of pacing-induced heart failure. J. Clin. Investig. 1993, 91, 907–914. [Google Scholar] [CrossRef] [PubMed]
- Delehanty, J.M.; Himura, Y.; Elam, H.; Hood, W.B., Jr.; Liang, C.-S. Beta-adrenoceptor downregulation in pacing-induced heart failure is associated with increased interstitial NE content. Am. J. Physiol. Heart Circ. Physiol. 1994, 266, H930–H935. [Google Scholar] [CrossRef] [PubMed]
- El-Armouche, A.; Pohlmann, L.; Schlossarek, S.; Starbatty, J.; Yeh, Y.-H.; Nattel, S.; Dobrev, D.; Eschenhagen, T.; Carrier, L. Decreased phosphorylation levels of cardiac myosin-binding protein-C in human and experimental heart failure. J. Mol. Cell. Cardiol. 2007, 43, 223–229. [Google Scholar] [CrossRef]
- Kobayashi, T.; Jin, L.; de Tombe, P.P. Cardiac thin filament regulation. Pflügers Arch. 2008, 457, 37–46. [Google Scholar] [CrossRef]
- Pagani, E.D.; Alousi, A.A.; Grant, A.M.; Older, T.M.; Dziuban, S.W., Jr.; Allen, P. Changes in myofibrillar content and Mg-ATPase activity in ventricular tissues from patients with heart failure caused by coronary artery disease, cardiomyopathy, or mitral valve insufficiency. Circ. Res. 1988, 63, 380–385. [Google Scholar] [CrossRef]
- Hajjar, R.J.; Gwathmey, J.K.; Briggs, G.; Morgan, J.P. Differential effect of DPI 201-106 on the sensitivity of the myofilaments to Ca2+ in intact and skinned trabeculae from control and myopathic human hearts. J. Clin. Investig. 1988, 82, 1578–1584. [Google Scholar] [CrossRef]
- Blair, E.; Redwood, C.; Ashrafian, H.; Oliveira, M.; Broxholme, J.; Kerr, B.; Salmon, A.; Östman-Smith, I.; Watkins, H. Mutations in the γ2 subunit of AMP-activated protein kinase cause familial hypertrophic cardiomyopathy: Evidence for the central role of energy compromise in disease pathogenesis. Hum. Mol. Genet. 2001, 10, 1215–1220. [Google Scholar] [CrossRef]
- Parvatiyar, M.S.; Landstrom, A.P.; Figueiredo-Freitas, C.; Potter, J.D.; Ackerman, M.J.; Pinto, J.R. A mutation in TNNC1-encoded cardiac troponin C, TNNC1-A31S, predisposes to hypertrophic cardiomyopathy and ventricular fibrillation. J. Biol. Chem. 2012, 287, 31845–31855. [Google Scholar] [CrossRef]
- Hajjar, R.J.; Schwinger, R.H.; Schmidt, U.; Kim, C.S.; Lebeche, D.; Doye, A.A.; Gwathmey, J.K. Myofilament calcium regulation in human myocardium. Circulation 2000, 101, 1679–1685. [Google Scholar] [CrossRef]
- Gwathmey, J.K.; Hajjar, R.J. Effect of protein kinase C activation on sarcoplasmic reticulum function and apparent myofibrillar Ca2+ sensitivity in intact and skinned muscles from normal and diseased human myocardium. Circ. Res. 1990, 67, 744–752. [Google Scholar] [CrossRef] [PubMed]
- European Heart Rhythm Association; Heart Rhythm Society; Zipes, D.P.; Camm, A.J.; Borggrefe, M.; Buxton, A.E.; Chaitman, B.; Fromer, M.; Gregoratos, G.; Klein, G.; et al. ACC/AHA/ESC 2006 guidelines for management of patients with ventricular arrhythmias and the prevention of sudden cardiac death: A report of the American College of Cardiology/American Heart Association Task Force and the European Society of Cardiology Committee for Practice Guidelines (Writing Committee to Develop Guidelines for Management of Patients With Ventricular Arrhythmias and the Prevention of Sudden Cardiac Death). J. Am. Coll. Cardiol. 2006, 48, e247–e346. [Google Scholar] [PubMed]
- Ter Keurs, H.E.; Boyden, P.A. Calcium and arrhythmogenesis. Physiol. Rev. 2007, 87, 457–506. [Google Scholar] [CrossRef] [PubMed]
- Baudenbacher, F.; Schober, T.; Pinto, J.R.; Sidorov, V.Y.; Hilliard, F.; Solaro, R.J.; Potter, J.D.; Knollmann, B.C. Myofilament Ca 2+ sensitization causes susceptibility to cardiac arrhythmia in mice. J. Clin. Investig. 2008, 118, 3893–3903. [Google Scholar] [PubMed]
- Robinson, P.; Mirza, M.; Knott, A.; Abdulrazzak, H.; Willott, R.; Marston, S.; Watkins, H.; Redwood, C. Alterations in thin filament regulation induced by a human cardiac troponin T mutant that causes dilated cardiomyopathy are distinct from those induced by troponin T mutants that cause hypertrophic cardiomyopathy. J. Biol. Chem. 2002, 277, 40710–40716. [Google Scholar] [CrossRef] [PubMed]
- Gomes, A.V.; Potter, J.D. Molecular and cellular aspects of troponin cardiomyopathies. Ann. N. Y. Acad. Sci. 2004, 1015, 214–224. [Google Scholar] [CrossRef]
- Redwood, C.; Lohmann, K.; Bing, W.; Esposito, G.M.; Elliott, K.; Abdulrazzak, H.; Knott, A.; Purcell, I.; Marston, S.; Watkins, H. Investigation of a truncated cardiac troponin T that causes familial hypertrophic cardiomyopathy: Ca2+ regulatory properties of reconstituted thin filaments depend on the ratio of mutant to wild-type protein. Circ. Res. 2000, 86, 1146–1152. [Google Scholar] [CrossRef]
- Clark, K.A.; Velazquez, E.J. Heart failure with preserved ejection fraction: Time for a reset. JAMA 2020, 324, 1506–1508. [Google Scholar] [CrossRef]
- Paulus, W.J.; Tschöpe, C. A novel paradigm for heart failure with preserved ejection fraction: Comorbidities drive myocardial dysfunction and remodeling through coronary microvascular endothelial inflammation. J. Am. Coll. Cardiol. 2013, 62, 263–271. [Google Scholar] [CrossRef]
- Hamdani, N.; Kooij, V.; van Dijk, S.; Merkus, D.; Paulus, W.J.; Remedios, C.D.; Duncker, D.J.; Stienen, G.J.; van der Velden, J. Sarcomeric dysfunction in heart failure. Cardiovasc. Res. 2008, 77, 649–658. [Google Scholar] [CrossRef]
- Solaro, R.J.; Kobayashi, T. Protein phosphorylation and signal transduction in cardiac thin filaments. J. Biol. Chem. 2011, 286, 9935–9940. [Google Scholar] [CrossRef] [PubMed]
- Burkart, E.M.; Sumandea, M.P.; Kobayashi, T.; Nili, M.; Martin, A.F.; Homsher, E.; Solaro, R.J. Phosphorylation or glutamic acid substitution at protein kinase C sites on cardiac troponin I differentially depress myofilament tension and shortening velocity. J. Biol. Chem. 2003, 278, 11265–11272. [Google Scholar] [CrossRef] [PubMed]
- Belin, R.J.; Sumandea, M.P.; Kobayashi, T.; Walker, L.A.; Rundell, V.L.; Urboniene, D.; Yuzhakova, M.; Ruch, S.H.; Geenen, D.L.; Solaro, R.J. Left ventricular myofilament dysfunction in rat experimental hypertrophy and congestive heart failure. Am. J. Physiol. Heart Circ. Physiol. 2006, 291, H2344–H2353. [Google Scholar] [CrossRef] [PubMed]
- Marston, S.B.; de Tombe, P.P. Troponin phosphorylation and myofilament Ca2+-sensitivity in heart failure: Increased or decreased? J. Mol. Cell. Cardiol. 2008, 45, 603–607. [Google Scholar] [CrossRef] [PubMed]
- Hamdani, N.; de Waard, M.; Messer, A.E.; Boontje, N.M.; Kooij, V.; van Dijk, S.; Versteilen, A.; Lamberts, R.; Merkus, D.; Dos Remedios, C. Myofilament dysfunction in cardiac disease from mice to men. J. Muscle Res. Cell Motil. 2008, 29, 189–201. [Google Scholar] [CrossRef]
- Solaro, R.J.; van der Velden, J. Why does troponin I have so many phosphorylation sites? Fact and fancy. J. Mol. Cell. Cardiol. 2010, 48, 810–816. [Google Scholar] [CrossRef]
- Dong, X.; Sumandea, C.A.; Chen, Y.-C.; Garcia-Cazarin, M.L.; Zhang, J.; Balke, C.W.; Sumandea, M.P.; Ge, Y. Augmented phosphorylation of cardiac troponin I in hypertensive heart failure. J. Biol. Chem. 2012, 287, 848–857. [Google Scholar] [CrossRef]
- Zima, A.V.; Bovo, E.; Mazurek, S.R.; Rochira, J.A.; Li, W.; Terentyev, D. Ca handling during excitation–contraction coupling in heart failure. Pflügers Arch. 2014, 466, 1129–1137. [Google Scholar] [CrossRef]
- Louch, W.E.; Stokke, M.K.; Sjaastad, I.; Christensen, G.; Sejersted, O.M. No rest for the weary: Diastolic calcium homeostasis in the normal and failing myocardium. Physiology 2012, 27, 308–323. [Google Scholar] [CrossRef]
- Go, L.O.; Moschella, M.; Watras, J.; Handa, K.; Fyfe, B.; Marks, A. Differential regulation of two types of intracellular calcium release channels during end-stage heart failure. J. Clin. Investig. 1995, 95, 888–894. [Google Scholar] [CrossRef]
- Frisk, M.; Ruud, M.; Espe, E.K.; Aronsen, J.M.; Røe, Å.T.; Zhang, L.; Norseng, P.A.; Sejersted, O.M.; Christensen, G.A.; Sjaastad, I. Elevated ventricular wall stress disrupts cardiomyocyte t-tubule structure and calcium homeostasis. Cardiovasc. Res. 2016, 112, 443–451. [Google Scholar] [CrossRef] [PubMed]
- Louch, W.E.; Bito, V.; Heinzel, F.R.; Macianskiene, R.; Vanhaecke, J.; Flameng, W.; Mubagwa, K.; Sipido, K.R. Reduced synchrony of Ca2+ release with loss of T-tubules—A comparison to Ca2+ release in human failing cardiomyocytes. Cardiovasc. Res. 2004, 62, 63–73. [Google Scholar] [CrossRef] [PubMed]
- Song, L.-S.; Sobie, E.A.; McCulle, S.; Lederer, W.; Balke, C.W.; Cheng, H. Orphaned ryanodine receptors in the failing heart. Proc. Natl. Acad. Sci. USA 2006, 103, 4305–4310. [Google Scholar] [CrossRef] [PubMed]
- Domeier, T.L.; Roberts, C.J.; Gibson, A.K.; Hanft, L.M.; McDonald, K.S.; Segal, S.S. Dantrolene suppresses spontaneous Ca2+ release without altering excitation-contraction coupling in cardiomyocytes of aged mice. Am. J. Physiol. Heart Circ. Physiol. 2014, 307, H818–H829. [Google Scholar] [CrossRef] [PubMed]
- Cooper, L.L.; Li, W.; Lu, Y.; Centracchio, J.; Terentyeva, R.; Koren, G.; Terentyev, D. Redox modification of ryanodine receptors by mitochondria-derived reactive oxygen species contributes to aberrant Ca2+ handling in ageing rabbit hearts. J. Physiol. 2013, 591, 5895–5911. [Google Scholar] [CrossRef] [PubMed]
- Primessnig, U.; Schönleitner, P.; Höll, A.; Pfeiffer, S.; Bracic, T.; Rau, T.; Kapl, M.; Stojakovic, T.; Glasnov, T.; Leineweber, K. Novel pathomechanisms of cardiomyocyte dysfunction in a model of heart failure with preserved ejection fraction. Eur. J. Heart Fail. 2016, 18, 987–997. [Google Scholar] [CrossRef] [PubMed]
- Howarth, F.; Qureshi, M.; Hassan, Z.; Al Kury, L.; Isaev, D.; Parekh, K.; Yammahi, S.; Oz, M.; Adrian, T.; Adeghate, E. Changing pattern of gene expression is associated with ventricular myocyte dysfunction and altered mechanisms of Ca2+ signalling in young type 2 Zucker diabetic fatty rat heart. Exp. Physiol. 2011, 96, 325–337. [Google Scholar] [CrossRef]
- Dibb, K.; Rueckschloss, U.; Eisner, D.; Isenberg, G.; Trafford, A. Mechanisms underlying enhanced cardiac excitation contraction coupling observed in the senescent sheep myocardium. J. Mol. Cell. Cardiol. 2004, 37, 1171–1181. [Google Scholar] [CrossRef]
- Harzheim, D.; Movassagh, M.; Foo, R.S.-Y.; Ritter, O.; Tashfeen, A.; Conway, S.J.; Bootman, M.D.; Roderick, H.L. Increased InsP3Rs in the junctional sarcoplasmic reticulum augment Ca2+ transients and arrhythmias associated with cardiac hypertrophy. Proc. Natl. Acad. Sci. USA 2009, 106, 11406–11411. [Google Scholar] [CrossRef]
- Shorofsky, S.R.; Aggarwal, R.; Corretti, M.; Baffa, J.M.; Strum, J.M.; Al-Seikhan, B.A.; Kobayashi, Y.M.; Jones, L.R.; Wier, W.G.; Balke, C.W. Cellular mechanisms of altered contractility in the hypertrophied heart: Big hearts, big sparks. Circ. Res. 1999, 84, 424–434. [Google Scholar] [CrossRef]
- Ljubojevic, S.; Radulovic, S.; Leitinger, G.; Sedej, S.; Sacherer, M.; Holzer, M.; Winkler, C.; Pritz, E.; Mittler, T.; Schmidt, A. Early remodeling of perinuclear Ca2+ stores and nucleoplasmic Ca2+ signaling during the development of hypertrophy and heart failure. Circulation 2014, 130, 244–255. [Google Scholar] [CrossRef] [PubMed]
- Ohba, T.; Watanabe, H.; Murakami, M.; Takahashi, Y.; Iino, K.; Kuromitsu, S.; Mori, Y.; Ono, K.; Iijima, T.; Ito, H. Upregulation of TRPC1 in the development of cardiac hypertrophy. J. Mol. Cell. Cardiol. 2007, 42, 498–507. [Google Scholar] [CrossRef] [PubMed]
- Bush, E.W.; Hood, D.B.; Papst, P.J.; Chapo, J.A.; Minobe, W.; Bristow, M.R.; Olson, E.N.; McKinsey, T.A. Canonical transient receptor potential channels promote cardiomyocyte hypertrophy through activation of calcineurin signaling. J. Biol. Chem. 2006, 281, 33487–33496. [Google Scholar] [CrossRef] [PubMed]
- Kuwahara, K.; Wang, Y.; McAnally, J.; Richardson, J.A.; Bassel-Duby, R.; Hill, J.A.; Olson, E.N. TRPC6 fulfills a calcineurin signaling circuit during pathologic cardiac remodeling. J. Clin. Investig. 2006, 116, 3114–3126. [Google Scholar] [CrossRef] [PubMed]
- Triposkiadis, F.; Briasoulis, A.; Sarafidis, P.; Magouliotis, D.; Athanasiou, T.; Paraskevaidis, I.; Skoularigis, J.; Xanthopoulos, A. The Sympathetic Nervous System in Hypertensive Heart Failure with Preserved LVEF. J. Clin. Med. 2023, 12, 6486. [Google Scholar] [CrossRef] [PubMed]
- Hegemann, N.; Primessnig, U.; Bode, D.; Wakula, P.; Beindorff, N.; Klopfleisch, R.; Michalick, L.; Grune, J.; Hohendanner, F.; Messroghli, D. Right-ventricular dysfunction in HFpEF is linked to altered cardiomyocyte Ca2+ homeostasis and myofilament sensitivity. ESC Heart Fail. 2021, 8, 3130–3144. [Google Scholar] [CrossRef] [PubMed]
- Bencivenga, L.; Palaia, M.E.; Sepe, I.; Gambino, G.; Komici, K.; Cannavo, A.; Femminella, G.D.; Rengo, G. Why do we not assess sympathetic nervous system activity in heart failure management: Might GRK2 serve as a new biomarker? Cells 2021, 10, 457. [Google Scholar] [CrossRef]
- Mayor, F., Jr.; Murga, C. G Protein-Coupled Receptor Kinases Take Central Stage. Cells 2022, 12, 23. [Google Scholar] [CrossRef]
- Carrier, L.; Mearini, G.; Stathopoulou, K.; Cuello, F. Cardiac myosin-binding protein C (MYBPC3) in cardiac pathophysiology. Gene 2015, 573, 188–197. [Google Scholar] [CrossRef]
- Moss, R.L.; Fitzsimons, D.P.; Ralphe, J.C. Cardiac MyBP-C regulates the rate and force of contraction in mammalian myocardium. Circ. Res. 2015, 116, 183–192. [Google Scholar] [CrossRef]
- Hamdani, N.; Bishu, K.G.; von Frieling-Salewsky, M.; Redfield, M.M.; Linke, W.A. Deranged myofilament phosphorylation and function in experimental heart failure with preserved ejection fraction. Cardiovasc. Res. 2013, 97, 464–471. [Google Scholar] [CrossRef] [PubMed]
- Røe, Å.T.; Aronsen, J.M.; Skårdal, K.; Hamdani, N.; Linke, W.A.; Danielsen, H.E.; Sejersted, O.M.; Sjaastad, I.; Louch, W.E. Increased passive stiffness promotes diastolic dysfunction despite improved Ca2+ handling during left ventricular concentric hypertrophy. Cardiovasc. Res. 2017, 113, 1161–1172. [Google Scholar] [CrossRef] [PubMed]
- D’Assante, R.; Arcopinto, M.; Rengo, G.; Salzano, A.; Walser, M.; Gambino, G.; Monti, M.G.; Bencivenga, L.; Marra, A.M.; Åberg, D.N. Myocardial expression of somatotropic axis, adrenergic signalling, and calcium handling genes in heart failure with preserved ejection fraction and heart failure with reduced ejection fraction. ESC Heart Fail. 2021, 8, 1681–1686. [Google Scholar] [CrossRef] [PubMed]
- Robinson, P.; Griffiths, P.J.; Watkins, H.; Redwood, C.S. Dilated and hypertrophic cardiomyopathy mutations in troponin and α-tropomyosin have opposing effects on the calcium affinity of cardiac thin filaments. Circ. Res. 2007, 101, 1266–1273. [Google Scholar] [CrossRef] [PubMed]
- Braunwald, E.; Lambrew, C.T.; Rockoff, S.D.; Ross, J., Jr.; Morrow, A.G. Idiopathic hypertrophic subaortic stenosis: I. A description of the disease based upon an analysis of 64 patients. Circulation 1964, 29, IV-3–IV-119. [Google Scholar] [CrossRef]
- van Heerebeek, L.; Hamdani, N.; Falcão-Pires, I.; Leite-Moreira, A.F.; Begieneman, M.P.; Bronzwaer, J.G.; van der Velden, J.; Stienen, G.J.; Laarman, G.J.; Somsen, A. Low myocardial protein kinase G activity in heart failure with preserved ejection fraction. Circulation 2012, 126, 830–839. [Google Scholar] [CrossRef]
- Scotcher, J.; Prysyazhna, O.; Boguslavskyi, A.; Kistamas, K.; Hadgraft, N.; Martin, E.D.; Worthington, J.; Rudyk, O.; Rodriguez Cutillas, P.; Cuello, F. Disulfide-activated protein kinase G Iα regulates cardiac diastolic relaxation and fine-tunes the Frank–Starling response. Nat. Commun. 2016, 7, 13187. [Google Scholar] [CrossRef]
- Hamdani, N.; Krysiak, J.; Kreusser, M.M.; Neef, S.; Dos Remedios, C.G.; Maier, L.S.; Krüger, M.; Backs, J.; Linke, W.A. Crucial role for Ca2+/calmodulin-dependent protein kinase-II in regulating diastolic stress of normal and failing hearts via titin phosphorylation. Circ. Res. 2013, 112, 664–674. [Google Scholar] [CrossRef]
- Yan, C.; Miller, C.L.; Abe, J.-I. Regulation of phosphodiesterase 3 and inducible cAMP early repressor in the heart. Circ. Res. 2007, 100, 489–501. [Google Scholar] [CrossRef]
- Lymperopoulos, A.; Rengo, G.; Koch, W.J. Adrenergic nervous system in heart failure: Pathophysiology and therapy. Circ. Res. 2013, 113, 739–753. [Google Scholar] [CrossRef]
- Solaro, R.J.; Rüegg, J. Stimulation of Ca++ binding and ATPase activity of dog cardiac myofibrils by AR-L 115BS, a novel cardiotonic agent. Circ. Res. 1982, 51, 290–294. [Google Scholar] [CrossRef] [PubMed]
- Shettigar, V.; Zhang, B.; Little, S.C.; Salhi, H.E.; Hansen, B.J.; Li, N.; Zhang, J.; Roof, S.R.; Ho, H.-T.; Brunello, L. Rationally engineered Troponin C modulates in vivo cardiac function and performance in health and disease. Nat. Commun. 2016, 7, 10794. [Google Scholar] [CrossRef] [PubMed]
- Perrone, S.V.; Kaplinsky, E.J. Calcium sensitizer agents: A new class of inotropic agents in the treatment of decompensated heart failure. Int. J. Cardiol. 2005, 103, 248–255. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.A.; Allen, D.G. Calcium sensitisers. BMJ 1990, 300, 551. [Google Scholar] [CrossRef] [PubMed]
- Blinks, J.; Endoh, M. Modification of myofibrillar responsiveness to Ca++ as an inotropic mechanism. Circulation 1986, 73, III85–III98. [Google Scholar] [PubMed]
- U.S. Food and Drug Administration. Novel Drug Approvals for 2017; U.S. Food and Drug Administration: Silver Spring, MD, USA, 2017.
- Brixius, K.; Savvidou-Zaroti, P.; Mehlhorn, U.; Bloch, W.; Kranias, E.G.; Schwinger, R.H. Increased Ca2+-sensitivity of myofibrillar tension in heart failure and its functional implication. Basic Res. Cardiol. 2002, 97, I111–I117. [Google Scholar] [CrossRef] [PubMed]
- Dou, Y.; Arlock, P.; Arner, A. Blebbistatin specifically inhibits actin-myosin interaction in mouse cardiac muscle. Am. J. Physiol. Cell. Physiol. 2007, 293, C1148–C1153. [Google Scholar] [CrossRef]
- Fedorov, V.V.; Lozinsky, I.T.; Sosunov, E.A.; Anyukhovsky, E.P.; Rosen, M.R.; Balke, C.W.; Efimov, I.R. Application of blebbistatin as an excitation–contraction uncoupler for electrophysiologic study of rat and rabbit hearts. Heart Rhythm. 2007, 4, 619–626. [Google Scholar] [CrossRef]
- U.S. Food and Drug Administration. CAMZYOS (Mavacamten): Prescribinginformation; U.S. Food and Drug Administration: Silver Spring, MD, USA, 2022.
- Green, E.M.; Wakimoto, H.; Anderson, R.L.; Evanchik, M.J.; Gorham, J.M.; Harrison, B.C.; Henze, M.; Kawas, R.; Oslob, J.D.; Rodriguez, H.M. A small-molecule inhibitor of sarcomere contractility suppresses hypertrophic cardiomyopathy in mice. Science 2016, 351, 617–621. [Google Scholar] [CrossRef]
- Olivotto, I.; Oreziak, A.; Barriales-Villa, R.; Abraham, T.P.; Masri, A.; Garcia-Pavia, P.; Saberi, S.; Lakdawala, N.K.; Wheeler, M.T.; Owens, A. Mavacamten for treatment of symptomatic obstructive hypertrophic cardiomyopathy (EXPLORER-HCM): A randomised, double-blind, placebo-controlled, phase 3 trial. Lancet 2020, 396, 759–769. [Google Scholar] [CrossRef]
- Awinda, P.O.; Watanabe, M.; Bishaw, Y.; Huckabee, A.M.; Agonias, K.B.; Kazmierczak, K.; Szczesna-Cordary, D.; Tanner, B.C. Mavacamten decreases maximal force and Ca2+ sensitivity in the N47K-myosin regulatory light chain mouse model of hypertrophic cardiomyopathy. Am. J. Physiol. Heart Circ. Physiol. 2021, 320, H881–H890. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
{kind=link}
| Cardiac Disease | Model | Pathway/Target | Variations in Ca2+ Sensitivity | Reference |
|---|---|---|---|---|
| Ischemic and DCM | Human-skinned LV papillary muscle fibers | Expression of ALC-1 in the LV in addition to the essential VLC-1 | Increased | [40] |
| IDC | Human right and left ventricular tissues | Proteolytic break down of MLC-2 | Increased | [60] |
| DCM | Human left ventricular myocytes | Reduction of the β-adrenergically mediated phosphorylation of TnI via PKA | Increased | [58] |
| HF | Human mechanically isolated Triton-skinned single myocytes from LV | MLC-2 phosphorylation was significantly lower | Increased | [57] |
| End-stage HF | Human left ventricular myocytes | Increased percentage of dephosphorylated MLC-2 and TnI | Increased | [65] |
| Mitral or aortic valvular disease | Human left ventricular and atrial skinned myocytes | Re-expression of a fetal TnT | Increased | [39] |
| FHC | Human genetic screening | AMPK γ2 mutations | Increased | [78] |
| DCM | Canine left ventricular myocytes | Chronic reductions in β-adrenergic-mediated (PKA-dependent) phosphorylation of myofilament regulatory proteins such as TnI and/or C-protein. | Increased | [67] |
| HF | Rat cardiomyocytes | Low levels of TnI phosphorylation | Increased | [68] |
| MI | Pig left ventricular myocytes | Reduced TnI phosphorylation | Increased | [66] |
| HCM | Porcine left ventricular papillary muscle strips | Mutations in TnC | Increased | [79] |
| Ischemic and IDC | Human left ventricular skinned-fiber | Acidic pH, cGMP | Decreased | [80] |
| End stage-HF | Human trabeculae carneae | PKC activation | Decreased | [81] |
| Normal | Dog LV myofibrils | Acidic pH | Decreased | [52] |
| Hypoxia or Ischemia | Rat-skinned right ventricular trabeculae | Presence of inorganic phosphate | Decreased | [54] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Saad, N.S.; Mashali, M.A.; Repas, S.J.; Janssen, P.M.L. Altering Calcium Sensitivity in Heart Failure: A Crossroads of Disease Etiology and Therapeutic Innovation. Int. J. Mol. Sci. 2023, 24, 17577. https://doi.org/10.3390/ijms242417577
Saad NS, Mashali MA, Repas SJ, Janssen PML. Altering Calcium Sensitivity in Heart Failure: A Crossroads of Disease Etiology and Therapeutic Innovation. International Journal of Molecular Sciences. 2023; 24(24):17577. https://doi.org/10.3390/ijms242417577
Chicago/Turabian StyleSaad, Nancy S., Mohammed A. Mashali, Steven J. Repas, and Paul M. L. Janssen. 2023. "Altering Calcium Sensitivity in Heart Failure: A Crossroads of Disease Etiology and Therapeutic Innovation" International Journal of Molecular Sciences 24, no. 24: 17577. https://doi.org/10.3390/ijms242417577
APA StyleSaad, N. S., Mashali, M. A., Repas, S. J., & Janssen, P. M. L. (2023). Altering Calcium Sensitivity in Heart Failure: A Crossroads of Disease Etiology and Therapeutic Innovation. International Journal of Molecular Sciences, 24(24), 17577. https://doi.org/10.3390/ijms242417577