
Acute ACAT1/SOAT1 Blockade Increases MAM Cholesterol and Strengthens ER-Mitochondria Connectivity

 , ,
, , {kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
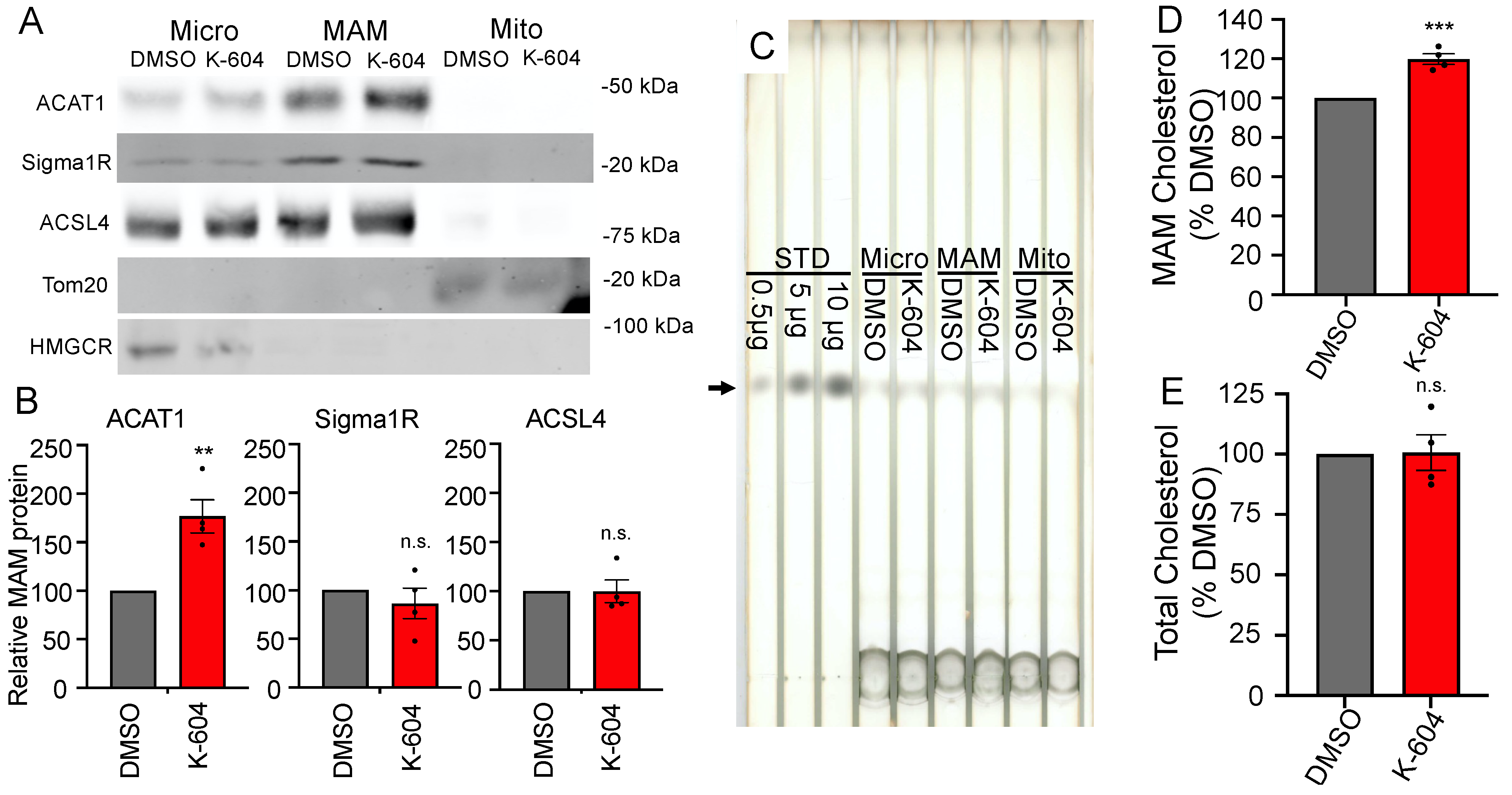
2.1. Initial Observation of the ACAT1/SOAT1 Blockade Cholesterol Pool
2.2. Direct Observation of Cholesterol Buildup around ACAT1/SOAT1
2.3. ACAT1/SOAT1 Inhibition Leads to Changes in ER-Mitochondria Connectivity
3. Discussion
4. Materials and Methods
4.1. Materials
4.2. Cell Culture
4.3. Western Blot
4.4. Measuring ACAT Activity by Mixed Micelle Assay
4.5. Measuring ACAT Activity by [3H] Oleate Pulse Assay
4.6. Optiprep Fractionation
4.7. MAM Fractionation
4.8. Proteomic Analysis of MAM Fractions
Both Buffers Adjusted to pH 10 with Ammonium Hydroxide for Offline Separation
4.9. Confocal Microscopy
4.10. Electron Microscopy
4.11. Data Analysis and Visualization
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Rowlands, L.J.; Marks, A.; Sanderson, J.M.; Law, R.V. 17O NMR spectroscopy as a tool to study hydrogen bonding of cholesterol in lipid bilayers. Chem. Commun. 2020, 56, 14499. [Google Scholar] [CrossRef]
- Slotte, J.P. The importance of hydrogen bonding in sphingomyelin’s membrane interactions with co-lipids. Biochim. Biophys. Acta 2016, 1858, 304–310. [Google Scholar] [CrossRef] [PubMed]
- Subczynski, W.K.; Pasenkiewicz-Gierula, M.; Widomska, J.; Mainali, L.; Raguz, M. High cholesterol/low cholesterol: Effects in biological membranes Review. Cell Biochem. Biophys. 2017, 75, 369–385. [Google Scholar] [CrossRef]
- Nezil, F.A.; Bloom, M. Combined influence of cholesterol and synthetic amphiphillic peptides upon bilayer thickness in model membranes. Biophys. J. 1992, 61, 1176–1183. [Google Scholar] [CrossRef]
- Fernández-Pérez, E.J.; Sepúlveda, F.J.; Peters, C.; Bascuñán, D.; Riffo-Lepe, N.O.; González-Sanmiguel, J.; Sánchez, S.A.; Peoples, R.W.; Vicente, B.; Aguayo, L.G. Effect of Cholesterol on Membrane Fluidity and Association of Aβ Oligomers and Subsequent Neuronal Damage: A Double-Edged Sword. Front. Aging Neurosci. 2018, 10, 226. [Google Scholar] [CrossRef] [PubMed]
- Simons, K.; Ikonen, E. Functional rafts in cell membranes. Nature 1997, 387, 569–572. [Google Scholar] [CrossRef]
- Yu, J.; Fischman, D.A.; Steck, T.L. Selective solubilization of proteins and phospholipids from red blood cell membranes by nonionic detergents. J. Supramol. Struct. 1973, 1, 233–248. [Google Scholar] [CrossRef]
- Baumgart, T.; Hammond, A.T.; Sengupta, P.; Hess, S.T.; Holowka, D.A.; Baird, B.A.; Webb, W.W. Large-scale fluid/fluid phase separation of proteins and lipids in giant plasma membrane vesicles. Proc. Natl. Acad. Sci. USA 2007, 104, 3165–3170. [Google Scholar] [CrossRef]
- Eggling, C.; Ringmaann, C.; Medda, R.; Schwrzmann, G.; Sandhoff, K.; Polyakova, S.; Belov, V.; von Middenorff, C.; Schonle, A.; Hell, S.W. Direct observation of the nanoscale dynamics of membrane lipids in a living cell. Nature 2009, 457, 1159–1163. [Google Scholar] [CrossRef] [PubMed]
- Sezgin, E.; Levental, I.; Mayor, S.; Eggeling, C. The mystery of membrane organization: Composition, regulation and roles of lipid rafts. Nat. Rev. 2017, 18, 361–374. [Google Scholar] [CrossRef]
- Brown, D.A. Lipid Rafts, Detergent-Resistant Membranes, and Raft Targeting Signals. Physiology 2006, 21, 430–439. [Google Scholar] [CrossRef]
- Stone, M.B.; Shelby, S.A.; Núñez, M.F.; Wisser, K.; Veatch, S.L. Protein sorting by lipid phase-like domains supports emergent signaling function in B lymphocyte plasma membranes. eLife 2017, 6, e19891. [Google Scholar] [CrossRef]
- Lorent, J.H.; Diaz-Rohrer, B.; Lin, X.; Spring, K.; Gorfe, A.A.; Levental, K.R.; Levental, I. Structural determinants and functional consequences of protein affinity for membrane rafts. Nat. Commun. 2017, 8, 1219. [Google Scholar] [CrossRef]
- Simons, K.; Toomre, D. Lipid rafts and signal transduction. Nat. Rev. Mol. Cell Biol. 2000, 1, 31–39. [Google Scholar] [CrossRef] [PubMed]
- King, C.; Sengupta, P.; Seo, A.Y.; Lippincott-Schwartz, J. ER membranes exhibit phase behavior at sites of organelle contact. Proc. Natl. Acad. Sci. USA 2020, 117, 7225–7235. [Google Scholar] [CrossRef]
- Copeland, D.E.; Dalton, A.J. An Association between Mitochondria and the Endoplasmic Reticulum in Cells of the Pseudobranch Gland of a Teleost. J. Biophys. Biochem. Cytol. 1959, 5, 393–396. [Google Scholar] [CrossRef] [PubMed]
- Vance, J.E. Phospholipid Synthesis in a Membrane Fraction Associated with Mitochondria. J. Biol. Chem. 1990, 265, 7248–7256. [Google Scholar] [CrossRef]
- Rowland, A.A.; Voeltz, G.K. Endoplasmic reticulum–mitochondria contacts: Function of the junction. Nat. Rev. Mol. Cell Biol. 2012, 13, 607–625. [Google Scholar] [CrossRef]
- Giacomello, M.; Pellegrini, L. The coming of age of the mitochondria–ER contact: A matter of thickness. Cell Death Differ. 2016, 23, 1417–1427. [Google Scholar] [CrossRef]
- Eisenberg-Bord, M.; Shai, N.; Schuldiner, M.; Bohnert, M. A Tether Is a Tether Is a Tether: Tethering at Membrane Contact Sites. Dev. Cell 2016, 39, 395–409. [Google Scholar] [CrossRef] [PubMed]
- Hayashi, T.; Fujimoto, M. Detergent-Resistant Microdomains Determine the Localization of σ-1 Receptors to the Endoplasmic Reticulum-Mitochondria Junction. Mol. Pharmacol. 2010, 77, 517–528. [Google Scholar] [CrossRef] [PubMed]
- Fujimoto, M.; Hayashi, T.; Su, T.-P. The role of cholesterol in the association of endoplasmic reticulum membranes with mitochondria. Biochem. Biophys. Res. Commun. 2012, 417, 635–639. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Hu, D.; Wang, R.; Sun, X.; Ropelewski, P.; Hubler, Z.; Lundberg, K.; Wang, Q.; Adams, D.J.; Xu, R.; et al. ATAD3A oligomerization promotes neuropathology and cognitive deficits in Alzheimer’s disease models. Nat. Commun. 2022, 13, 1121. [Google Scholar] [CrossRef]
- Nes, W.D. Biosynthesis of Cholesterol and Other Sterols. Chem. Rev. 2011, 111, 6423–6451. [Google Scholar] [CrossRef]
- Goldstein, J.L.; DeBose-Boyd, R.A.; Brown, M.S. Protein Sensors for Membrane Sterols. Cell 2006, 124, 35–46. [Google Scholar] [CrossRef]
- Oram, J.F.; Heinecke, J.W. ATP-Binding Cassette Transporter A1: A Cell Cholesterol Exporter That Protects Against Cardiovascular Disease. Physiol. Rev. 2006, 85, 1131–1417. [Google Scholar] [CrossRef] [PubMed]
- Venkateswaran, A.; Laffitte, B.A.; Joseph, S.B.; Mak, P.A.; Wilpitz, D.C.; Edwards, P.A.; Tontonoz, P. Control of cellular cholesterol efflux by the nuclear oxysterol receptor LXRα. Proc. Natl. Acad. Sci. USA 2000, 97, 12097–12102. [Google Scholar] [CrossRef]
- Chang, T.; Li, B.; Chang, C.C.Y.; Urano, Y. Acyl-coenzyme A: Cholesterol acyltransferases. Am. J. Physiol. Endocrinol. Metab. 2009, 297, 1–9. [Google Scholar] [CrossRef]
- Chang, C.C.Y.; Chen, J.; Thomas, M.A.; Cheng, D.; Del Priore Veronica, A.; Newton, R.S.; Pape, M.E.; Chang, T.-Y. Regulation and Immunolocalization of Acyl-Coenzyme A:Cholesterol Acyltransferase in Mammalian Cells as Studied with Specific Antibodies. J. Biol. Chem. 1995, 270, 29532–29540. [Google Scholar] [CrossRef]
- Cheng, D.; Chang, C.C.Y.; Qu, X.; Chang, T.-Y. Activation of Acyl-Coenzyme A:Cholesterol Acyltransferase by Cholesterol or by Oxysterol in a Cell-free System. J. Biol. Chem. 1995, 270, 685–695. [Google Scholar] [CrossRef]
- Yu, C.; Lin, S.; Liu, J.; Chang, C.C.Y.; Chang, T.-Y. Human Acyl-CoA:Cholesterol Acyltransferase-1 Is a Homotetrameric Enzyme in Intact Cells and in Vitro. J. Lipid Res. 1999, 274, 36139–36145. [Google Scholar] [CrossRef]
- Guo, Z.-Y.; Lin, S.; Heinen, J.A.; Chang, C.C.Y.; Chang, T.-Y. The Active Site His-460 of Human Acyl-coenzyme A:Cholesterol Acyltransferase 1 Resides in a Hitherto Undisclosed Transmembrane Domain. J. Biol. Chem. 2005, 280, 37814–37826. [Google Scholar] [CrossRef] [PubMed]
- Qian, H.; Zhao, X.; Yan, R.; Yao, X.; Gao, S.; Sun, X.; Du, X.; Yang, H.; Wong, C.C.L.; Yan, N. Structural basis for catalysis and substrate specificity of human ACAT1. Nature 2020, 581, 333–338. [Google Scholar] [CrossRef] [PubMed]
- Guan, C.; Niu, Y.; Chen, S.-C.; Kang, Y.; Wu, J.-X.; Nishi, K.; Chang, C.C.Y.; Chang, T.-Y.; Luo, T.; Chen, L. Structural insights into the inhibition mechanism of human sterol O-acyltransferase 1 by a competitive inhibitor. Nat. Commun. 2020, 11, 2478. [Google Scholar] [CrossRef]
- Long, T.; Sun, Y.; Hassan, A.; Qi, X.; Li, X. Structure of nevanimibe-bound tetrameric human ACAT1. Nature 2020, 581, 339–343. [Google Scholar] [CrossRef]
- Rusiñol, A.E.; Cui, Z.; Chen, M.H.; Vance, J.E. A unique mitochondria-associated membrane fraction from rat liver has a high capacity for lipid synthesis and contains pre-Golgi secretory proteins including nascent lipoproteins. J. Biol. Chem. 1994, 269, 27494–27502. [Google Scholar] [CrossRef]
- Area-Gomez, E.; del Carmen Lara Castillo, M.; Tambini, M.D.; Guardia-Laguarta, C.; de Groof, A.J.C.; Madra, M.; Ikenouchi, J.; Umeda, M.; Bird, T.D.; Sturley, S.L.; et al. Upregulated function of mitochondria-associated ER membranes in Alzheimer disease. EMBO J. 2012, 31, 4106–4123. [Google Scholar] [CrossRef]
- Alzheimer’s Association. 2022 Alzheimer’s Disease Facts and Figures; Alzheimers Dimentia 2022, 18, 700–789.
- Wenk, G.L. Neuropathologic Changes in Alzheimer’s Disease. J. Clin. Psychiatry 2003, 64, 7–10. [Google Scholar]
- Chan, R.B.; Oliveira, T.G.; Cortes, E.P.; Honig, L.S.; Duff, K.E.; Small, S.A.; Wenk, M.R.; Shui, G.; Di Paolo, G. Comparative Lipidomic Analysis of Mouse and Human Brain with Alzheimer Disease. J. Biol. Chem. 2012, 287, 2678–2688. [Google Scholar] [CrossRef]
- Tajima, Y.; Ishikawa, M.; Maekawa, K.; Murayama, M.; Senoo, Y.; Nishimaki-Mogami, T.; Nakanishi, H.; Ikeda, K.; Arita, M.; Taguchi, R.; et al. Lipidomic analysis of brain tissues and plasma in a mouse model expressing mutated human amyloid precursor protein/tau for Alzheimer’s disease. Lipids Health Dis. 2013, 12, 68. [Google Scholar] [CrossRef]
- Lim, W.L.F.; Lam, S.M.; Shui, G.; Mondal, A.; Ong, D.; Xinrui, D.; Creegan, R.; Martins, I.J.; Sharman, M.J.; Taddei, K.; et al. Effects of a high-fat, high-cholesterol diet on brain lipid profiles in apolipoprotein E e3 and e4 knock-in mice. Neurobiol. Aging 2013, 34, 2217–2224. [Google Scholar] [CrossRef] [PubMed]
- Strittmatter, W.J.; Saunders, A.M.; Schmechel, D.; Pericak-Vance, M.; Enghild, J.; Salvesen, G.S.; Roses, A.D. Apolipoprotein E: High-avidity binding to beta-amyloid and increased frequency of type 4 allele in late-onset familial Alzheimer disease. Proc. Natl. Acad. Sci. USA 1993, 90, 1977–1981. [Google Scholar] [CrossRef]
- Tanzi, R.E. The Genetics of Alzheimer Disease. Cold Spring Harb. Perspect. Med. 2012, 2, a006296. [Google Scholar] [CrossRef] [PubMed]
- Liu, C.-C.; Kanekiyo, T.; Xu, H.; Bu, G. Apolipoprotein E and Alzheimer disease: Risk, mechanisms, and therapy. Nat. Rev. Neurol. 2013, 9, 106–118. [Google Scholar] [CrossRef]
- Sienski, G.; Narayan, P.; Bonner, J.M.; Kory, N.; Boland, S.; Arczewska, A.A.; Ralvenius, W.T.; Akay, L.; Lockshin, E.; He, L.; et al. APOE4 disrupts intracellular lipid homeostasis in human iPSC-derived glia. Sci. Transl. Med. 2021, 13, eaaz4564. [Google Scholar] [CrossRef]
- Yamazaki, Y.; Zhao, N.; Caulfield, T.R.; Liu, C.-C.; Bu, G. Apolipoprotein E and Alzheimer disease: Pathobiology and targeting strategies. Nat. Rev. Neurol. 2019, 15, 501–518. [Google Scholar] [CrossRef] [PubMed]
- Qian, J.; Wolters, F.J.; Beiser, A.; Haan, M.; Ikram, M.A.; Karlawish, J.; Langbaum, J.B.; Neuhaus, J.M.; Reiman, E.M.; Roberts, J.S.; et al. APOE-related risk of mild cognitive impairment and dementia for prevention trials: An analysis of four cohorts. PLoS Med. 2017, 14, e1002254. [Google Scholar] [CrossRef]
- Simons, M.; Keller, P.; De Strooper, B.; Beyreuther, K.; Dotti, C.G.; Simons, K. Cholesterol depletion inhibits the generation of β-amyloid in hippocampal neurons. Proc. Natl. Acad. Sci. USA 1998, 95, 6460–6464. [Google Scholar] [CrossRef]
- Frears, E.R.; Stephens, D.J.; Walters, C.E.; Davies, H.; Austen, B.M. The role of cholesterol in the biosynthesis of β-amyloid. NeuroReport 1999, 10, 1699. [Google Scholar] [CrossRef]
- Refolo, L.M.R.; Pappolla, M.A.; LaFrancois, J.; Malester, B.; Schmidt, S.D.; Thomas-Bryant, T.; Tint, G.S.; Wang, R.; Mercken, M.; Petanceska, S.S.; et al. A Cholesterol-Lowering Drug Reduces β-Amyloid Pathology in a Transgenic Mouse Model of Alzheimer’s Disease. Neurobiol. Dis. 2001, 8, 890–899. [Google Scholar] [CrossRef]
- Fassbender, K.; Simons, M.; Bergmann, C.; Stroick, M.; Lütjohann, D.; Keller, P.; Runz, H.; Kühl, S.; Bertsch, T.; von Bergmann, K.; et al. Simvastatin strongly reduces levels of Alzheimer’s disease β-amyloid peptides Aβ42 and Aβ40 in vitro and in vivo. Proc. Natl. Acad. Sci. USA 2001, 98, 5856–5861. [Google Scholar] [CrossRef] [PubMed]
- Simons, M.; Schwärzler, F.; Lütjohann, D.; Von Bergmann, K.; Beyreuther, K.; Dichgans, J.; Wormstall, H.; Hartmann, T.; Schulz, J.B. Treatment with simvastatin in normocholesterolemic patients with Alzheimer’s disease: A 26-week randomized, placebo-controlled, double-blind trial. Ann. Neurol. 2002, 52, 346–350. [Google Scholar] [CrossRef]
- Sano, M.; Bell, K.L.; Galasko, D.; Galvin, J.E.; Thomas, R.G.; van Dyck, C.H.; Aisen, P.S. A randomized, double-blind, placebo-controlled trial of simvastatin to treat Alzheimer disease. Neurology 2011, 77, 556–563. [Google Scholar] [CrossRef]
- Puglielli, L.; Konopka, G.; Pack-Chung, E.; Ingano, L.A.M.; Berezovska, O.; Hyman, B.T.; Chang, T.-Y.; Tanzi, R.E.; Kovacs, D.M. Acyl-coenzyme A: Cholesterol acyltransferase modulates the generation of the amyloid β-peptide. Nat. Cell Biol. 2001, 3, 905–912. [Google Scholar] [CrossRef]
- Hutter-Paier, B.; Huttunen, H.J.; Puglielli, L.; Eckman, C.B.; Kim, D.Y.; Hofmeister, A.; Moir, R.D.; Domnitz, S.B.; Frosch, M.P.; Windisch, M.; et al. The ACAT Inhibitor CP-113,818 Markedly Reduces Amyloid Pathology in a Mouse Model of Alzheimer’s Disease. Neuron 2004, 44, 227–238. [Google Scholar] [CrossRef] [PubMed]
- Huttunen, H.J.; Havas, D.; Peach, C.; Barren, C.; Duller, S.; Xia, W.; Frosch, M.P.; Hutter-Paier, B.; Windisch, M.; Kovacs, D.M. The Acyl-Coenzyme A:Cholesterol Acyltransferase Inhibitor CI-1011 Reverses Diffuse Brain Amyloid Pathology in Aged Amyloid Precursor Protein Transgenic Mice. J. Neuropathol. Exp. Neurol. 2010, 69, 777–788. [Google Scholar] [CrossRef]
- Bryleva, E.Y.; Rogers, M.A.; Chang, C.C.Y.; Buen, F.; Harris, B.T.; Rousselet, E.; Seidah, N.G.; Oddo, S.; LaFerla, F.M.; Spencer, T.A.; et al. ACAT1 gene ablation increases 24(S)-hydroxycholesterol content in the brain and ameliorates amyloid pathology in mice with AD. Proc. Natl. Acad. Sci. USA 2010, 107, 3081–3086. [Google Scholar] [CrossRef]
- Murphy, S.R.; Chang, C.C.Y.; Dogbevia, G.; Bryleva, E.Y.; Bowen, Z.D.; Hasan, M.T.; Chang, T.-Y. Acat1 Knockdown Gene Therapy Decreases Amyloid-â in a Mouse Model of Alzheimer’s Disease. Am. Soc. Gene Cell Ther. 2013, 21, 1497–1506. [Google Scholar] [CrossRef]
- Shibuya, Y.; Chang, C.C.Y.; Huang, L.-H.; Bryleva, E.Y.; Chang, T.-Y. Inhibiting ACAT1/SOAT1 in Microglia Stimulates Autophagy-Mediated Lysosomal Proteolysis and Increases A 1-42 Clearance. J. Neurosci. 2014, 34, 14484–14501. [Google Scholar] [CrossRef] [PubMed]
- Shibuya, Y.; Niu, Z.; Bryleva, E.Y.; Harris, B.T.; Murphy, S.R.; Kheirollah, A.; Bowen, Z.D.; Chang, C.C.Y.; Chang, T.-Y. Acyl-CoA:cholesterol acyltransferase 1 blockage enhances autophagy in the neurons of triple transgenic Alzheimer’s disease mouse and reduces human P301L-tau content at the pre-symptomatic stage. Neurobiol. Aging 2015, 36, 2248–2259. [Google Scholar] [CrossRef] [PubMed]
- Nugent, A.A.; Lin, K.; van Lengerich, B.; Lianoglou, S.; Przybyla, L.; Davis, S.S.; Llapashtica, C.; Wang, J.; Kim, D.J.; Xia, D.; et al. TREM2 Regulates Microglial Cholesterol Metabolism upon Chronic Phagocytic Challenge. Neuron 2020, 105, 837–854.e9. [Google Scholar] [CrossRef] [PubMed]
- Langness, V.F.; van der Kant, R.; Das, U.; Wang, L.; dos Chaves, R.S.; Goldstein, L.S.B. Cholesterol-lowering drugs reduce APP processing to Aβ by inducing APP dimerization. Mol. Biol. Cell 2021, 32, 247–259. [Google Scholar] [CrossRef] [PubMed]
- Ikenoya, M.; Yoshinaka, Y.; Kobayashi, H.; Kawamine, K.; Shibuya, K.; Sato, F.; Sawanobori, K.; Watanabe, T.; Miyazaki, A. A selective ACAT-1 inhibitor, K-604, suppresses fatty streak lesions in fat-fed hamsters without affecting plasma cholesterol levels. Atherosclerosis 2007, 191, 290–297. [Google Scholar] [CrossRef] [PubMed]
- López-Farré, A.J.; Sacristán, D.; Zamorano-León, J.J.; San-Martín, N.; Macaya, C. Inhibition of acyl-CoA cholesterol acyltransferase by F12511 (eflucimibe): Could it be a new antiatherosclerotic therapeutic? Cardiovasc. Ther. 2008, 26, 65–74. [Google Scholar] [CrossRef]
- Chang, T.Y.; Chang, C.C.Y.; Harned, T.C.; De, A.L.; Torre, L.; Lee, J.; Huynh, T.N.; Gow, J.G. Blocking cholesterol storage to treat Alzheimer’s disease. Explor. Neuroprotective Ther. 2021, 1, 173–184. [Google Scholar] [CrossRef]
- Chang, C.C.Y.; Gregory Lee, C.-Y.; Chang, E.T.; Cruz, J.C.; Levesque, M.C.; Chang, T.-Y. Recombinant Acyl-CoA:cholesterol Acyltransferase-1 (ACAT-1) Purified to Essential Homogeneity Utilizes Cholesterol in Mixed Micelles or in Vesicles in a Highly Cooperative Manner. J. Biol. Chem. 1998, 273, 35132–35141. [Google Scholar] [CrossRef]
- Stansley, B.; Post, J.; Hensley, K. A comparative review of cell culture systems for the study of microglial biology in Alzheimer’s disease. J. Neuroinflammation 2012, 9, 115. [Google Scholar] [CrossRef]
- Chang, C.C.Y.; Doolittle, G.M.; Chang, T.Y. Cycloheximide sensitivity in regulation of acyl coenzyme A:cholesterol acyltransferase activity in Chinese hamster ovary cells. 1. Effect of exogenous sterols. Biochemistry 1986, 25, 1693–1699. [Google Scholar] [CrossRef]
- Goldstein, J.L.; Dana, S.E.; Brown, M.S. Esterification of Low Density Lipoprotein Cholesterol in Human Fibroblasts and Its Absence in Homozygous Familial Hypercholesterolemia. Proc. Natl. Acad. Sci. USA 1974, 71, 4288–4292. [Google Scholar] [CrossRef]
- De La Torre, A.L.; Smith, C.; Granger, J.; Anderson, F.L.; Harned, T.C.; Havrda, M.C.; Chang, C.C.Y.; Chang, T.-Y. Facile method to incorporate high-affinity ACAT/SOAT1 inhibitor F12511 into stealth liposome-based nanoparticle and demonstration of its efficacy in blocking cholesteryl ester biosynthesis without overt toxicity in neuronal cell culture. J. Neurosci. Methods 2022, 367, 109437. [Google Scholar] [CrossRef] [PubMed]
- Liscum, L. Pharmacological inhibition of the intracellular transport of low-density lipoprotein-derived cholesterol in Chinese hamster ovary cells. Biochem. Biophys. Acta 1990, 1045, 40–48. [Google Scholar] [CrossRef] [PubMed]
- Alberts, A.W.; Chen, J.; Kuron, G.; Hunt, V.; Huff, J.; Hoffman, C.; Rothrock, J.; Lopez, M.; Joshua, H.; Harris, E.; et al. Mevinolin: A highly potent competitive inhibitor of hydroxymethylglutaryl-coenzyme A reductase and a cholesterol-lowering agent. Proc. Natl. Acad. Sci. USA 1980, 77, 3957–3961. [Google Scholar] [CrossRef] [PubMed]
- Lange, Y.; Ye, J.; Rigney, M.; Steck, T. Cholesterol Movement in Niemann-Pick Type C Cells and in Cells Treated with Amphiphiles. J. Biol. Chem. 2000, 275, 17468–17475. [Google Scholar] [CrossRef]
- Lu, F.; Liang, Q.; Abi-Mosleh, L.; Das, A.; De Brabander, J.K.; Goldstein, J.L.; Brown, M.S. Identification of NPC1 as the target of U18666A, an inhibitor of lysosomal cholesterol export and Ebola infection. eLife 2015, 4, e12177. [Google Scholar] [CrossRef]
- Goldstein, J.L.; Brown, M.S. Regulation of the mevalonate pathway. Nature 1990, 343, 425–430. [Google Scholar] [CrossRef]
- Urano, Y.; Watanabe, H.; Murphy, S.R.; Shibuya, Y.; Geng, Y.; Peden, A.A.; Chang, C.C.Y.; Chang, T.Y.; Russell, D.W. Transport of LDL-derived cholesterol from the NPC1 compartment to the ER involves the trans-Golgi network and the SNARE protein complex. Proc. Natl. Acad. Sci. USA 2008, 105, 16513–16518. [Google Scholar] [CrossRef]
- Wieckowski, M.R.; Giorgi, C.; Lebiedzinska, M.; Duszynski, J.; Pinton, P. Isolation of mitochondria-associated membranes and mitochondria from animal tissues and cells. Nat. Protoc. 2009, 4, 1582–1590. [Google Scholar] [CrossRef]
- Tambini, M.D.; Pera, M.; Kanter, E.; Yang, H.; Guardia-Laguarta, C.; Holtzman, D.; Sulzer, D.; Area-Gomez, E.; Schon, E.A. ApoE4 upregulates the activity of mitochondria-associated ER membranes. EMBO Rep. 2016, 17, 27–36. [Google Scholar] [CrossRef]
- Area-Gomez, E.; Schon, E.A. Mitochondria-associated ER membranes and Alzheimer disease. Curr. Opin. Genet. Dev. 2016, 38, 90–96. [Google Scholar] [CrossRef]
- Lak, B.; Li, S.; Belevich, I.; Sree, S.; Butkovic, R.; Ikonen, E.; Jokitalo, E. Specific subdomain localization of ER resident proteins and membrane contact sites resolved by electron microscopy. Eur. J. Cell Biol. 2021, 100, 151180. [Google Scholar] [CrossRef] [PubMed]
- Chakrabarti, R.; Ji, W.-K.; Stan, R.V.; de Juan Sanz, J.; Ryan, T.A.; Higgs, H.N. INF2-mediated actin polymerization at the ER stimulates mitochondrial calcium uptake, inner membrane constriction, and division. J. Cell Biol. 2018, 217, 251–268. [Google Scholar] [CrossRef] [PubMed]
- McGuinness, B.; Craig, D.; Bullock, R.; Passmore, P. Statins for the prevention of dementia. Cochrane Database Syst. Rev. 2016, 2016, CD003160. [Google Scholar] [CrossRef] [PubMed]
- Bhattacharyya, R.; Barren, C.; Kovacs, D.M. Palmitoylation of Amyloid Precursor Protein Regulates Amyloidogenic Processing in Lipid Rafts—PMC. J. Neurosci. 2013, 33, 11169–11183. [Google Scholar] [CrossRef]
- Zhu, M.; Zhao, X.; Chen, J.; Xu, J.; Hu, G.; Guo, D.; Li, Q.; Zhang, X.; Chang, C.C.Y.; Song, B.; et al. ACAT1 regulates the dynamics of free cholesterols in plasma membrane which leads to the APP-α-processing alteration. Acta Biochim. Biophys. Sin. 2015, 47, 951–959. [Google Scholar] [CrossRef]
- Li, H.; Huynh, T.N.; Duong, M.T.; Gow, J.G.; Chang, C.C.Y.; Chang, T.-Y. ACAT1/SOAT1 Blockade Suppresses LPS-Mediated Neuroinflammation by Modulating the Fate of Toll-Like Receptor 4 in Microglia. bioRxiv 2022. [Google Scholar] [CrossRef]
- Rogers, M.A.; Chang, C.C.Y.; Maue, R.A.; Melton, E.M.; Peden, A.A.; Garver, W.S.; Lee, J.; Schroen, P.; Huang, M.; Chang, T.-Y. Acat1/Soat1 knockout extends the mutant Npc1 mouse lifespan and ameliorates functional deficiencies in multiple organelles of mutant cells. Proc. Natl. Acad. Sci. USA 2022, 119, e2201646119. [Google Scholar] [CrossRef]
- Frallicciardi, J.; Melcr, J.; Siginou, P.; Marrink, S.J.; Poolman, B. Membrane thickness, lipid phase and sterol type are determining factors in the permeability of membranes to small solutes. Nat. Commun. 2022, 13, 1605. [Google Scholar] [CrossRef]
- Ikonen, E. Cellular cholesterol trafficking and compartmentalization. Nat. Rev. Mol. Cell Biol. 2008, 9, 125–138. [Google Scholar] [CrossRef]
- Fantini, J.; Barrantes, F. How cholesterol interacts with membrane proteins: An exploration of cholesterol-binding sites including CRAC, CARC, and tilted domains. Front. Physiol. 2013, 4, 31. [Google Scholar] [CrossRef]
- Lee, A.G. Lipid-protein interactions. Biochem. Soc. Trans. 2011, 39, 761–766. [Google Scholar] [CrossRef] [PubMed]
- Johannsson, A.; Keightley, C.A.; Smith, G.A.; Richards, C.D.; Hesketh, T.R.; Metcalfe, J.C. The effect of bilayer thickness and n-alkanes on the activity of the (Ca2+ + Mg2+)-dependent ATPase of sarcoplasmic reticulum. J. Biol. Chem. 1981, 256, 1643–1650. [Google Scholar] [CrossRef]
- Lee, A.G. How lipids affect the activities of integral membrane proteins. Biochim. Biophys. Acta BBA-Biomembr. 2004, 1666, 62–87. [Google Scholar] [CrossRef] [PubMed]
- Milovanovic, D.; Honigmann, A.; Koike, S.; Göttfert, F.; Pähler, G.; Junius, M.; Müllar, S.; Diederichsen, U.; Janshoff, A.; Grubmüller, H.; et al. Hydrophobic mismatch sorts SNARE proteins into distinct membrane domains. Nat. Commun. 2015, 6, 5984. [Google Scholar] [CrossRef] [PubMed]
- Zhemkov, V.; Ditlev, J.A.; Lee, W.-R.; Wilson, M.; Liou, J.; Rosen, M.K.; Bezprozvanny, I. The role of sigma 1 receptor in organization of endoplasmic reticulum signaling microdomains. eLife 2021, 10, e65192. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Chang, C.C.Y.; Westover, E.J.; Covey, D.F.; Chang, T.-Y. Investigating the allosterism of acyl-CoA:cholesterol acyltransferase (ACAT) by using various sterols: In vitro and intact cell studies. Biochem. J. 2005, 391, 389–397. [Google Scholar] [CrossRef] [PubMed]
- Paillusson, S.; Stoica, R.; Gomez-Suaga, P.; Lau, D.H.W.; Mueller, S.; Miller, T.; Miller, C.C.J. There’s Something Wrong with my MAM; the ER-Mitochondria Axis and Neurodegenerative Diseases. Trends Neurosci. 2016, 39, 146–157. [Google Scholar] [CrossRef]
- Montesinos, J.; Pera, M.; Larrea, D.; Guardia-Laguarta, C.; Agrawal, R.R.; Velasco, K.R.; Yun, T.D.; Stavrovskaya, I.G.; Xu, Y.; Koo, S.Y.; et al. The Alzheimer’s disease-associated C99 fragment of APP regulates cellular cholesterol trafficking. EMBO J. 2020, 39, e103791. [Google Scholar] [CrossRef]
- Pera, M.; Larrea, D.; Guardia-Laguarta, C.; Montesinos, J.; Velasco, K.R.; Agrawal, R.R.; Xu, Y.; Chan, R.B.; Di Paolo, G.; Mehler, M.F.; et al. Increased localization of APP-C99 in mitochondria-associated ER membranes causes mitochondrial dysfunction in Alzheimer disease. EMBO J. 2017, 36, 3356–3371. [Google Scholar] [CrossRef]
- Zampese, E.; Fasolato, C.; Kipanyula, M.J.; Bortolozzi, M.; Pozzan, T.; Pizzo, P. Presenilin 2 modulates endoplasmic reticulum (ER)–mitochondria interactions and Ca2+ cross-talk. Proc. Natl. Acad. Sci. USA 2011, 108, 2777–2782. [Google Scholar] [CrossRef]
- Bishop, J.E.; Hajra, A.K. A method for the chemical synthesis of 14C-labeled fatty acyl coenzyme A’s of high specific activity. Anal. Biochem. 1980, 106, 344–350. [Google Scholar] [CrossRef] [PubMed]
- Friedman, J.R.; Lackner, L.L.; West, M.; DiBenedetto, J.R.; Nunnari, J.; Voeltz, G.K. ER Tubules Mark Sites of Mitochondrial Division. Science 2011, 334, 358–362. [Google Scholar] [CrossRef] [PubMed]
- Macala, L.J.; Yu, R.K.; Ando, S. Analysis of brain lipids by high performance thin-layer chromatography and densitometry Supplementary key words DEAE-Sephadex chromatography phos-pholipids plasmalogen acidic lipids. J. Lipid Res. 1983, 24, 1243–1250. [Google Scholar] [CrossRef] [PubMed]
- Nesvizhskii, A.I.; Keller, A.; Kolker, E.; Aebersold, R. A Statistical Model for Identifying Proteins by Tandem Mass Spectrometry. Anal. Chem. 2003, 75, 4646–4658. [Google Scholar] [CrossRef]
- Graw, S.; Tang, J.; Zafar, M.K.; Byrd, A.K.; Bolden, C.; Peterson, E.C.; Byrum, S.D. proteiNorm—A User-Friendly Tool for Normalization and Analysis of TMT and Label-Free Protein Quantification. ACS Omega 2020, 5, 25625–25633. [Google Scholar] [CrossRef]
- Ritchie, M.E.; Phipson, B.; Wu, D.; Hu, Y.; Law, C.W.; Shi, W.; Smyth, G.K. limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res. 2015, 43, e47. [Google Scholar] [CrossRef]
- Raudvere, U.; Kolberg, L.; Kuzmin, I.; Arak, T.; Adler, P.; Peterson, H.; Vilo, J. g:Profiler: A web server for functional enrichment analysis and conversions of gene lists (2019 update). Nucleic Acids Res. 2019, 47, W191–W198. [Google Scholar] [CrossRef]
- Kwak, C.; Shin, S.; Park, J.S.; Jung, M.; Nhung, T.T.M.; Kang, M.G.; Lee, C.; Kwon, T.K.; Park, S.K.; Mun, J.Y.; et al. Contact-ID, a tool for profiling organelle contact sites, reveals regulatory proteins of mitochondrial-associated membrane formation. Proc. Natl. Acad. Sci. USA 2020, 117, 12109–12120. [Google Scholar] [CrossRef]




Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Harned, T.C.; Stan, R.V.; Cao, Z.; Chakrabarti, R.; Higgs, H.N.; Chang, C.C.Y.; Chang, T.Y. Acute ACAT1/SOAT1 Blockade Increases MAM Cholesterol and Strengthens ER-Mitochondria Connectivity. Int. J. Mol. Sci. 2023, 24, 5525. https://doi.org/10.3390/ijms24065525
Harned TC, Stan RV, Cao Z, Chakrabarti R, Higgs HN, Chang CCY, Chang TY. Acute ACAT1/SOAT1 Blockade Increases MAM Cholesterol and Strengthens ER-Mitochondria Connectivity. International Journal of Molecular Sciences. 2023; 24(6):5525. https://doi.org/10.3390/ijms24065525
Chicago/Turabian StyleHarned, Taylor C., Radu V. Stan, Ze Cao, Rajarshi Chakrabarti, Henry N. Higgs, Catherine C. Y. Chang, and Ta Yuan Chang. 2023. "Acute ACAT1/SOAT1 Blockade Increases MAM Cholesterol and Strengthens ER-Mitochondria Connectivity" International Journal of Molecular Sciences 24, no. 6: 5525. https://doi.org/10.3390/ijms24065525
APA StyleHarned, T. C., Stan, R. V., Cao, Z., Chakrabarti, R., Higgs, H. N., Chang, C. C. Y., & Chang, T. Y. (2023). Acute ACAT1/SOAT1 Blockade Increases MAM Cholesterol and Strengthens ER-Mitochondria Connectivity. International Journal of Molecular Sciences, 24(6), 5525. https://doi.org/10.3390/ijms24065525