
Contribution of Blood Vessel Activation, Remodeling and Barrier Function to Inflammatory Bowel Diseases



Abstract
1. Introduction
2. The Intestinal Vasculature in Homeostasis
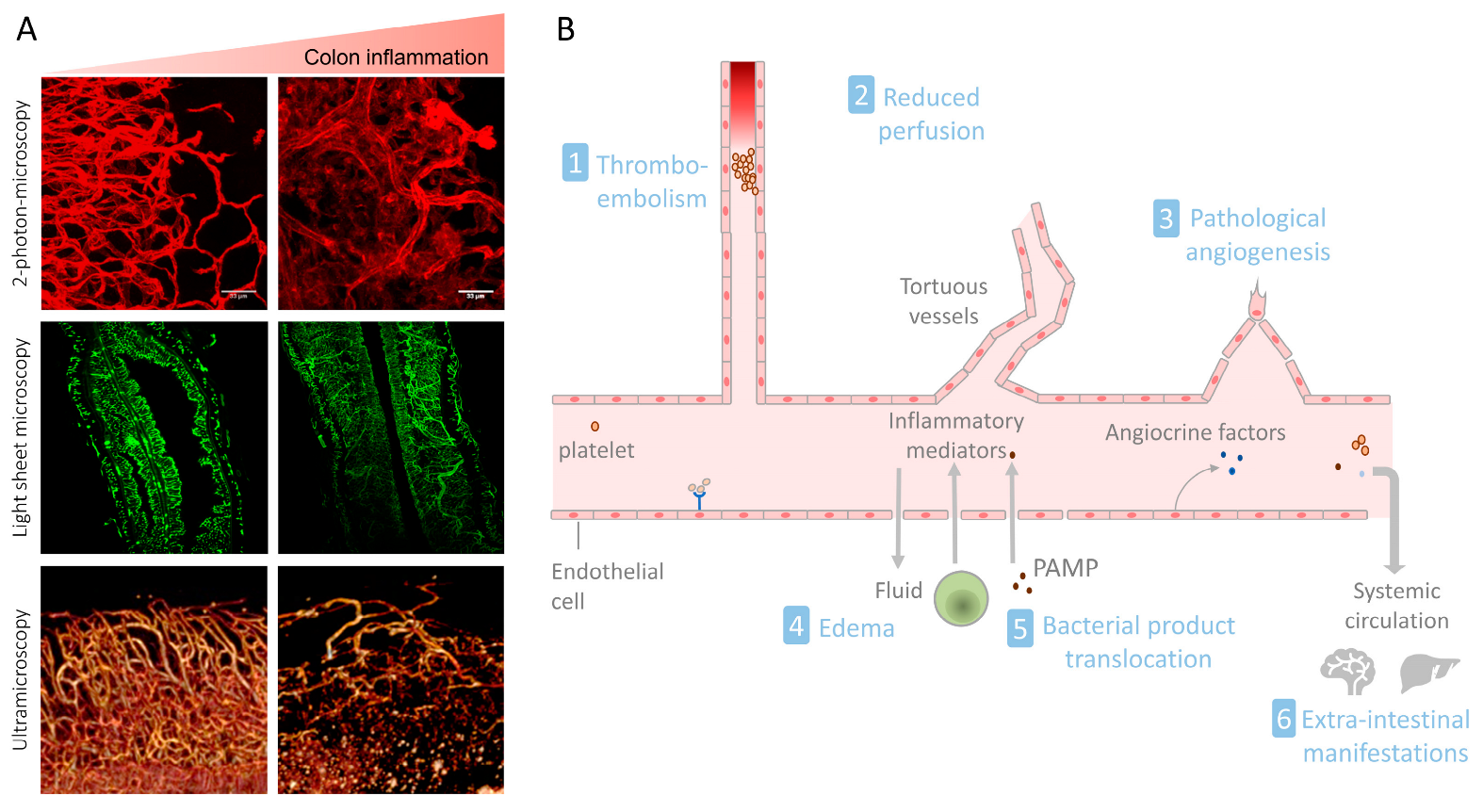
3. The Intestinal Vasculature in IBD
3.1. Endothelial Cell Activation and Leukocyte Recruitment
3.2. Pathological Angiogenesis in IBD
3.3. The Gut–Vascular Barrier in IBD
3.3.1. VE-Cadherin and Vascular Barrier Regulation in IBD
3.3.2. Vessel Coverage and Permeability
3.4. Microvascular Dysfunction in IBD
3.4.1. The Role of Nitric Oxide in Vascular Dysfunction during IBD
3.4.2. Coagulation
3.5. Regulatory Role of the Vasculature during Mucosal Inflammation
3.5.1. Vasculature and Innate Immunity
3.5.2. Paracrine Effects of the Inflamed Vasculature
3.6. Role of the Mesenteric Lymphatic Vasculature in IBD
3.7. Vascular Function and Extra-Intestinal Manifestations of IBD
3.7.1. Systemic Vascular Barrier Dysfunction in IBD
3.7.2. Endothelial Damage and Systemic Vascular Inflammation
3.7.3. Coagulation and Thrombosis in IBD
3.8. Targeting the Vasculature in IBD Therapy
4. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Zhang, Y.Z.; Li, Y.Y. Inflammatory bowel disease: Pathogenesis. World J. Gastroenterol. 2014, 20, 91–99. [Google Scholar] [CrossRef]
- Wallace, K.L.; Zheng, L.B.; Kanazawa, Y.; Shih, D.Q. Immunopathology of inflammatory bowel disease. World J. Gastroenterol. 2014, 20, 6–21. [Google Scholar] [CrossRef] [PubMed]
- Ni, J.; Wu, G.D.; Albenberg, L.; Tomov, V.T. Gut microbiota and IBD: Causation or correlation? Nat. Rev. Gastroenterol. Hepatol. 2017, 14, 573–584. [Google Scholar] [CrossRef]
- Thoo, L.; Noti, M.; Krebs, P. Keep calm: The intestinal barrier at the interface of peace and war. Cell Death Dis. 2019, 10, 849. [Google Scholar] [CrossRef]
- Cromer, W.E.; Mathis, J.M.; Granger, D.N.; Chaitanya, G.V.; Alexander, J.S. Role of the endothelium in inflammatory bowel diseases. World J. Gastroenterol. 2011, 17, 578–593. [Google Scholar] [CrossRef] [PubMed]
- Kvietys, P.R. Integrated Systems Physiology: From Molecule to Function. In The Gastrointestinal Circulation; Morgan & Claypool Life Sciences: San Rafael, CA, USA, 2010. [Google Scholar]
- Geboes, K.; Geboes, K.P.; Maleux, G. Vascular anatomy of the gastrointestinal tract. Best Pract. Res. Clin. Gastroenterol. 2001, 15, 1–14. [Google Scholar] [CrossRef]
- Arfors, K.E.; Rutili, G.; Svensjö, E. Microvascular transport of macromolecules in normal and inflammatory conditions. Acta Physiol. Scand. Suppl. 1979, 463, 93–103. [Google Scholar]
- Hasibeder, W. Gastrointestinal microcirculation: Still a mystery? Br. J. Anaesth. 2010, 105, 393–396. [Google Scholar] [CrossRef]
- Miller, M.J.; McDole, J.R.; Newberry, R.D. Microanatomy of the intestinal lymphatic system. Ann. N. Y. Acad. Sci. 2010, 1207 (Suppl. 1), E21–E28. [Google Scholar] [CrossRef]
- Cifarelli, V.; Eichmann, A. The Intestinal Lymphatic System: Functions and Metabolic Implications. Cell. Mol. Gastroenterol. Hepatol. 2019, 7, 503–513. [Google Scholar] [CrossRef] [PubMed]
- Brescia, P.; Rescigno, M. The gut vascular barrier: A new player in the gut-liver-brain axis. Trends Mol. Med. 2021, 27, 844–855. [Google Scholar] [CrossRef]
- Spadoni, I.; Zagato, E.; Bertocchi, A.; Paolinelli, R.; Hot, E.; Di Sabatino, A.; Caprioli, F.; Bottiglieri, L.; Oldani, A.; Viale, G.; et al. A gut-vascular barrier controls the systemic dissemination of bacteria. Science 2015, 350, 830–834. [Google Scholar] [CrossRef] [PubMed]
- Spadoni, I.; Fornasa, G.; Rescigno, M. Organ-specific protection mediated by cooperation between vascular and epithelial barriers. Nat. Rev. Immunol. 2017, 17, 761–773. [Google Scholar] [CrossRef] [PubMed]
- Bush, T.G.; Savidge, T.C.; Freeman, T.C.; Cox, H.J.; Campbell, E.A.; Mucke, L.; Johnson, M.H.; Sofroniew, M.V. Fulminant jejuno-ileitis following ablation of enteric glia in adult transgenic mice. Cell 1998, 93, 189–201. [Google Scholar] [CrossRef]
- Cornet, A.; Savidge, T.C.; Cabarrocas, J.; Deng, W.L.; Colombel, J.F.; Lassmann, H.; Desreumaux, P.; Liblau, R.S. Enterocolitis induced by autoimmune targeting of enteric glial cells: A possible mechanism in Crohn’s disease? Proc. Natl. Acad. Sci. USA 2001, 98, 13306–13311. [Google Scholar] [CrossRef]
- Kabouridis, P.S.; Lasrado, R.; McCallum, S.; Chng, S.H.; Snippert, H.J.; Clevers, H.; Pettersson, S.; Pachnis, V. Microbiota controls the homeostasis of glial cells in the gut lamina propria. Neuron 2015, 85, 289–295. [Google Scholar] [CrossRef] [PubMed]
- Reinhardt, C.; Bergentall, M.; Greiner, T.U.; Schaffner, F.; Ostergren-Lundén, G.; Petersen, L.C.; Ruf, W.; Bäckhed, F. Tissue factor and PAR1 promote microbiota-induced intestinal vascular remodelling. Nature 2012, 483, 627–631. [Google Scholar] [CrossRef]
- Schirbel, A.; Kessler, S.; Rieder, F.; West, G.; Rebert, N.; Asosingh, K.; McDonald, C.; Fiocchi, C. Pro-angiogenic activity of TLRs and NLRs: A novel link between gut microbiota and intestinal angiogenesis. Gastroenterology 2013, 144, 613–623.e9. [Google Scholar] [CrossRef]
- Stappenbeck, T.S.; Hooper, L.V.; Gordon, J.I. Developmental regulation of intestinal angiogenesis by indigenous microbes via Paneth cells. Proc. Natl. Acad. Sci. USA 2002, 99, 15451–15455. [Google Scholar] [CrossRef] [PubMed]
- Komarova, Y.A.; Kruse, K.; Mehta, D.; Malik, A.B. Protein Interactions at Endothelial Junctions and Signaling Mechanisms Regulating Endothelial Permeability. Circ. Res. 2017, 120, 179–206. [Google Scholar] [CrossRef]
- Scalise, A.A.; Kakogiannos, N.; Zanardi, F.; Iannelli, F.; Giannotta, M. The blood–brain and gut–vascular barriers: From the perspective of claudins. Tissue Barriers 2021, 9, 1926190. [Google Scholar] [CrossRef]
- Richards, M.; Nwadozi, E.; Pal, S.; Martinsson, P.; Kaakinen, M.; Gloger, M.; Sjöberg, E.; Koltowska, K.; Betsholtz, C.; Eklund, L.; et al. Claudin5 protects the peripheral endothelial barrier in an organ and vessel-type-specific manner. eLife 2022, 11, e78517. [Google Scholar] [CrossRef] [PubMed]
- Kalucka, J.; de Rooij, L.P.M.H.; Goveia, J.; Rohlenova, K.; Dumas, S.J.; Meta, E.; Conchinha, N.V.; Taverna, F.; Teuwen, L.-A.; Veys, K.; et al. Single—Cell Transcriptome Atlas of Murine Endothelial Cells. Cell 2020, 180, 764–779.e20. [Google Scholar] [CrossRef] [PubMed]
- Günzel, D.; Yu, A.S.L. Claudins and the modulation of tight junction permeability. Physiol. Rev. 2013, 93, 525–569. [Google Scholar] [CrossRef]
- Garcia-Hernandez, V.; Quiros, M.; Nusrat, A. Intestinal epithelial claudins: Expression and regulation in homeostasis and inflammation. Ann. N. Y. Acad. Sci. 2017, 1397, 66–79. [Google Scholar] [CrossRef]
- Haraldsen, G.; Kvale, D.; Lien, B.; Farstad, I.N.; Brandtzaeg, P. Cytokine-regulated expression of E-selectin, intercellular adhesion molecule-1 (ICAM-1), and vascular cell adhesion molecule-1 (VCAM-1) in human microvascular endothelial cells. J. Immunol. 1996, 156, 2558–2565. [Google Scholar] [CrossRef] [PubMed]
- Nilsen, E.M.; Johansen, F.E.; Jahnsen, F.L.; Lundin, K.E.A.; Scholz, T.; Brandtzaeg, P.; Haraldsen, G. Cytokine profiles of cultured microvascular endothelial cells from the human intestine. Gut 1998, 42, 635–642. [Google Scholar] [CrossRef]
- Binion, D.G.; Fu, S.; Ramanujam, K.S.; Chai, Y.C.; Dweik, R.A.; Drazba, J.A.; Wade, J.G.; Ziats, N.P.; Erzurum, S.C.; Wilson, K.T. iNOS expression in human intestinal microvascular endothelial cells inhibits leukocyte adhesion. Am. J. Physiol. 1998, 275, G592–G603. [Google Scholar] [CrossRef]
- Sessa, W.C. Molecular control of blood flow and angiogenesis: Role of nitric oxide. J. Thromb. Haemost. 2009, 7 (Suppl. 1), 35–37. [Google Scholar] [CrossRef]
- Pober, J.S.; Sessa, W.C. Evolving functions of endothelial cells in inflammation. Nat. Rev. Immunol. 2007, 7, 803–815. [Google Scholar] [CrossRef]
- Papa, A.; Scaldaferri, F.; Danese, S.; Guglielmo, S.; Roberto, I.; Bonizzi, M.; Mocci, G.; Felice, C.; Ricci, C.; Andrisani, G.; et al. Vascular involvement in inflammatory bowel disease: Pathogenesis and clinical aspects. Dig. Dis. 2008, 26, 149–155. [Google Scholar] [CrossRef]
- Haep, L.; Britzen-Laurent, N.; Weber, T.G.; Naschberger, E.; Schaefer, A.; Kremmer, E.; Foersch, S.; Vieth, M.; Scheuer, W.; Wirtz, S.; et al. Interferon Gamma Counteracts the Angiogenic Switch and Induces Vascular Permeability in Dextran Sulfate Sodium Colitis in Mice. Inflamm. Bowel Dis. 2015, 21, 2360–2371. [Google Scholar] [CrossRef]
- Langer, V.; Vivi, E.; Regensburger, D.; Winkler, T.H.; Waldner, M.J.; Rath, T.; Schmid, B.; Skottke, L.; Lee, S.; Jeon, N.L.; et al. IFN-γ drives inflammatory bowel disease pathogenesis through VE-cadherin-directed vascular barrier disruption. J. Clin. Investig. 2019, 129, 4691–4707. [Google Scholar] [CrossRef]
- Nourshargh, S.; Alon, R. Leukocyte Migration into Inflamed Tissues. Immunity 2014, 41, 694–707. [Google Scholar] [CrossRef]
- Binion, D.G.; West, G.A.; Volk, E.E.; Drazba, J.A.; Ziats, N.P.; Petras, R.E.; Fiocchi, C. Acquired increase in leucocyte binding by intestinal microvascular endothelium in inflammatory bowel disease. Lancet 1998, 352, 1742–1746. [Google Scholar] [CrossRef]
- Scaldaferri, F.; Sans, M.; Vetrano, S.; Correale, C.; Arena, V.; Pagano, N.; Rando, G.; Romeo, F.; Potenza, A.E.; Repici, A.; et al. The role of MAPK in governing lymphocyte adhesion to and migration across the microvasculature in inflammatory bowel disease. Eur. J. Immunol. 2009, 39, 290–300. [Google Scholar] [CrossRef] [PubMed]
- Fägerstam, J.P.; Whiss, P.A.; Ström, M.; Andersson, R.G. Expression of platelet P-selectin and detection of soluble P-selectin, NPY and RANTES in patients with inflammatory bowel disease. Inflamm. Res. 2000, 49, 466–472. [Google Scholar] [CrossRef] [PubMed]
- Fägerstam, J.P.; Whiss, P.A. Higher platelet P-selectin in male patients with inflammatory bowel disease compared to healthy males. World J. Gastroenterol. 2006, 12, 1270–1272. [Google Scholar] [CrossRef]
- Ostanin, D.V.; Bao, J.; Koboziev, I.; Gray, L.; Robinson-Jackson, S.A.; Kosloski-Davidson, M.; Price, V.H.; Grisham, M.B. T cell transfer model of chronic colitis: Concepts, considerations, and tricks of the trade. Am. J. Physiol. Gastrointest. Liver Physiol. 2009, 296, G135–G146. [Google Scholar] [CrossRef]
- Zittermann, S.I.; Issekutz, A.C. Endothelial growth factors VEGF and bFGF differentially enhance monocyte and neutrophil recruitment to inflammation. J. Leukoc. Biol. 2006, 80, 247–257. [Google Scholar] [CrossRef] [PubMed]
- Goebel, S.; Huang, M.; Davis, W.C.; Jennings, M.; Siahaan, T.J.; Alexander, J.S.; Kevil, C.G. VEGF-A stimulation of leukocyte adhesion to colonic microvascular endothelium: Implications for inflammatory bowel disease. Am. J. Physiol. Gastrointest. Liver Physiol. 2006, 290, G648–G654. [Google Scholar] [CrossRef]
- Sans, M.; Fuster, D.; Vázquez, A.; Setoain, F.J.; Piera, C.; Piqué, J.M.; Panés, J. 123Iodine-labelled anti-VCAM-1 antibody scintigraphy in the assessment of experimental colitis. Eur. J. Gastroenterol. Hepatol. 2001, 13, 31–38. [Google Scholar] [CrossRef]
- Soriano, A.; Salas, A.; Salas, A.; Sans, M.; Gironella, M.; Elena, M.; Anderson, D.C.; Piqué, J.M.; Panés, J. VCAM-1, but not ICAM-1 or MAdCAM-1, immunoblockade ameliorates DSS-induced colitis in mice. Lab. Investig. 2000, 80, 1541–1551. [Google Scholar] [CrossRef] [PubMed]
- Rijcken, E.; Mennigen, R.B.; Schaefer, S.D.; Laukoetter, M.G.; Anthoni, C.; Spiegel, H.U.; Bruewer, M.; Senninger, N.; Krieglstein, C.F. PECAM-1 (CD 31) mediates transendothelial leukocyte migration in experimental colitis. Am. J. Physiol. Gastrointest. Liver Physiol. 2007, 293, G446–G452. [Google Scholar] [CrossRef] [PubMed]
- Briskin, M.; Winsor-Hines, D.; Shyjan, A.; Cochran, N.; Bloom, S.; Wilson, J.; McEvoy, L.M.; Butcher, E.C.; Kassam, N.; Mackay, C.R.; et al. Human mucosal addressin cell adhesion molecule-1 is preferentially expressed in intestinal tract and associated lymphoid tissue. Am. J. Pathol. 1997, 151, 97–110. [Google Scholar]
- Ogawa, H.; Binion, D.G.; Heidemann, J.; Theriot, M.; Fisher, P.J.; Johnson, N.A.; Otterson, M.F.; Rafiee, P. Mechanisms of MAdCAM-1 gene expression in human intestinal microvascular endothelial cells. Am. J. Physiol. Cell Physiol. 2005, 288, C272–C281. [Google Scholar] [CrossRef] [PubMed]
- Ando, T.; Jordan, P.; Wang, Y.; Itoh, M.; Joh, T.; Sasaki, M.; Elrod, J.W.; Carpenter, A.; Jennings, M.H.; Minagar, A.; et al. MAdCAM-1 expression and regulation in murine colonic endothelial cells in vitro. Inflamm. Bowel Dis. 2005, 11, 258–264. [Google Scholar] [CrossRef]
- Tyler, C.J.; Guzman, M.; Lundborg, L.R.; Yeasmin, S.; Zgajnar, N.; Jedlicka, P.; Bamias, G.; Rivera-Nieves, J. Antibody secreting cells are critically dependent on integrin α4β7/MAdCAM-1 for intestinal recruitment and control of the microbiota during chronic colitis. Mucosal Immunol. 2022, 15, 109–119. [Google Scholar] [CrossRef]
- Jackson, J.R.; Seed, M.P.; Kircher, C.H.; Willoughby, D.A.; Winkler, J.D. The codependence of angiogenesis and chronic inflammation. FASEB J. 1997, 11, 457–465. [Google Scholar] [CrossRef]
- Danese, S.; Sans, M.; de la Motte, C.; Graziani, C.; West, G.; Phillips, M.H.; Pola, R.; Rutella, S.; Willis, J.; Gasbarrini, A.; et al. Angiogenesis as a novel component of inflammatory bowel disease pathogenesis. Gastroenterology 2006, 130, 2060–2073. [Google Scholar] [CrossRef]
- Chidlow, J.H., Jr.; Langston, W.; Greer, J.J.; Ostanin, D.; Abdelbaqi, M.; Houghton, J.; Senthilkumar, A.; Shukla, D.; Mazar, A.P.; Grisham, M.B.; et al. Differential angiogenic regulation of experimental colitis. Am. J. Pathol. 2006, 169, 2014–2030. [Google Scholar] [CrossRef] [PubMed]
- Alkim, C.; Alkim, H.; Koksal, A.R.; Boga, S.; Sen, I. Angiogenesis in Inflammatory Bowel Disease. Int. J. Inflamm. 2015, 2015, 970890. [Google Scholar] [CrossRef]
- Danese, S.; Sans, M.; Spencer, D.M.; Beck, I.; Doñate, F.; Plunkett, M.L.; de la Motte, C.; Redline, R.; Shaw, D.E.; Levine, A.D.; et al. Angiogenesis blockade as a new therapeutic approach to experimental colitis. Gut 2007, 56, 855–862. [Google Scholar] [CrossRef] [PubMed]
- Danese, S. Inflammation and the mucosal microcirculation in inflammatory bowel disease: The ebb and flow. Curr. Opin. Gastroenterol. 2007, 23, 384–389. [Google Scholar] [CrossRef]
- Binion, D.G.; Rafiee, P. Is inflammatory bowel disease a vascular disease? Targeting angiogenesis improves chronic inflammation in inflammatory bowel disease. Gastroenterology 2009, 136, 400–403. [Google Scholar] [CrossRef] [PubMed]
- Danese, S. Role of the vascular and lymphatic endothelium in the pathogenesis of inflammatory bowel disease: ‘Brothers in arms’. Gut 2011, 60, 998–1008. [Google Scholar] [CrossRef] [PubMed]
- Dulic-Sills, A.; Blunden, M.J.; Mawdsley, J.; Bastin, A.J.; McAuley, D.; Griffiths, M.; Rampton, D.S.; Yaqoob, M.M.; Macey, M.G.; Agrawal, S.G. New flow cytometric technique for the evaluation of circulating endothelial progenitor cell levels in various disease groups. J. Immunol. Methods 2006, 316, 107–115. [Google Scholar] [CrossRef] [PubMed]
- Garolla, A.; D’Incà, R.; Checchin, D.; Biagioli, A.; De Toni, L.; Nicoletti, V.; Scarpa, M.; Bolzonello, E.; Sturniolo, G.C.; Foresta, C. Reduced endothelial progenitor cell number and function in inflammatory bowel disease: A possible link to the pathogenesis. Am. J. Gastroenterol. 2009, 104, 2500–2507. [Google Scholar] [CrossRef]
- Deng, X.; Szabo, S.; Chen, L.; Paunovic, B.; Khomenko, T.; Tolstanova, G.; Tarnawski, A.S.; Jones, M.K.; Sandor, Z. New cell therapy using bone marrow-derived stem cells/endothelial progenitor cells to accelerate neovascularization in healing of experimental ulcerative colitis. Curr. Pharm. Des. 2011, 17, 1643–1651. [Google Scholar] [CrossRef]
- Giatromanolaki, A.; Sivridis, E.; Maltezos, E.; Papazoglou, D.; Simopoulos, C.; Gatter, K.C.; Harris, A.L.; Koukourakis, M.I. Hypoxia inducible factor 1alpha and 2alpha overexpression in inflammatory bowel disease. J. Clin. Pathol. 2003, 56, 209–213. [Google Scholar] [CrossRef]
- Alkim, C.; Savas, B.; Ensari, A.; Alkim, H.; Dagli, U.; Parlak, E.; Ulker, A.; Sahin, B. Expression of p53, VEGF, microvessel density, and cyclin-D1 in noncancerous tissue of inflammatory bowel disease. Dig. Dis. Sci. 2009, 54, 1979–1984. [Google Scholar] [CrossRef] [PubMed]
- Alkim, C.; Sakiz, D.; Alkim, H.; Livaoglu, A.; Kendir, T.; Demirsoy, H.; Erdem, L.; Akbayir, N.; Sokmen, M. Thrombospondin-1 and VEGF in inflammatory bowel disease. Libyan J. Med. 2012, 7, 8942. [Google Scholar] [CrossRef] [PubMed]
- Scaldaferri, F.; Vetrano, S.; Sans, M.; Arena, V.; Straface, G.; Stigliano, E.; Repici, A.; Sturm, A.; Malesci, A.; Panes, J.; et al. VEGF-A links angiogenesis and inflammation in inflammatory bowel disease pathogenesis. Gastroenterology 2009, 136, 585–595.e5. [Google Scholar] [CrossRef]
- Griga, T.; Gutzeit, A.; Sommerkamp, C.; May, B. Increased production of vascular endothelial growth factor by peripheral blood mononuclear cells in patients with inflammatory bowel disease. Eur. J. Gastroenterol. Hepatol. 1999, 11, 175–179. [Google Scholar] [CrossRef] [PubMed]
- Bousvaros, A.; Leichtner, A.; Zurakowski, D.; Kwon, J.; Law, T.; Keough, K.; Fishman, S. Elevated serum vascular endothelial growth factor in children and young adults with Crohn’s disease. Dig. Dis. Sci. 1999, 44, 424–430. [Google Scholar] [CrossRef] [PubMed]
- Pousa, I.D.; Maté, J.; Gisbert, J.P. Angiogenesis in inflammatory bowel disease. Eur. J. Clin. Investig. 2008, 38, 73–81. [Google Scholar] [CrossRef] [PubMed]
- Krzystek-Korpacka, M.; Neubauer, K.; Matusiewicz, M. Platelet-derived growth factor-BB reflects clinical, inflammatory and angiogenic disease activity and oxidative stress in inflammatory bowel disease. Clin. Biochem. 2009, 42, 1602–1609. [Google Scholar] [CrossRef] [PubMed]
- Macé, V.; Ahluwalia, A.; Coron, E.; Le Rhun, M.; Boureille, A.; Bossard, C.; Mosnier, J.F.; Matysiak-Budnik, T.; Tarnawski, A.S. Confocal laser endomicroscopy: A new gold standard for the assessment of mucosal healing in ulcerative colitis. J. Gastroenterol. Hepatol. 2015, 30 (Suppl. 1), 85–92. [Google Scholar] [CrossRef]
- Ippolito, C.; Colucci, R.; Segnani, C.; Errede, M.; Girolamo, F.; Virgintino, D.; Dolfi, A.; Tirotta, E.; Buccianti, P.; Di Candio, G.; et al. Fibrotic and Vascular Remodelling of Colonic Wall in Patients with Active Ulcerative Colitis. J. Crohn’s Colitis 2016, 10, 1194–1204. [Google Scholar] [CrossRef]
- Kapsoritakis, A.; Sfiridaki, A.; Maltezos, E.; Simopoulos, K.; Giatromanolaki, A.; Sivridis, E.; Koukourakis, M.I. Vascular endothelial growth factor in inflammatory bowel disease. Int. J. Color. Dis. 2003, 18, 418–422. [Google Scholar] [CrossRef]
- Konno, S.; Iizuka, M.; Yukawa, M.; Sasaki, K.; Sato, A.; Horie, Y.; Nanjo, H.; Fukushima, T.; Watanabe, S. Altered expression of angiogenic factors in the VEGF-Ets-1 cascades in inflammatory bowel disease. J. Gastroenterol. 2004, 39, 931–939. [Google Scholar] [CrossRef] [PubMed]
- Magro, F.; Araujo, F.; Pereira, P.; Meireles, E.; Diniz-Ribeiro, M.; Velosom, F.T. Soluble selectins, sICAM, sVCAM, and angiogenic proteins in different activity groups of patients with inflammatory bowel disease. Dig. Dis. Sci. 2004, 49, 1265–1274. [Google Scholar] [CrossRef] [PubMed]
- Tolstanova, G.; Khomenko, T.; Deng, X.; Chen, L.; Tarnawski, A.; Ahluwalia, A.; Szabo, S.; Sandor, Z. Neutralizing anti-vascular endothelial growth factor (VEGF) antibody reduces severity of experimental ulcerative colitis in rats: Direct evidence for the pathogenic role of VEGF. J. Pharmacol. Exp. Ther. 2009, 328, 749–757. [Google Scholar] [CrossRef] [PubMed]
- Cromer, W.E.; Ganta, C.V.; Patel, M.; Traylor, J.; Kevil, C.G.; Alexander, J.S.; Mathis, J.M. VEGF-A isoform modulation in an preclinical TNBS model of ulcerative colitis: Protective effects of a VEGF164b therapy. J. Transl. Med. 2013, 11, 207. [Google Scholar] [CrossRef] [PubMed]
- Ozsoy, Z.; Ozsoy, S.; Gevrek, F.; Demir, E.; Benli, I.; Daldal, E.; Yenidogan, E. Effect of bevacizumab on acetic acid–induced ulcerative colitis in rats. J. Surg. Res. 2017, 216, 191–200. [Google Scholar] [CrossRef]
- Hindryckx, P.; Waeytens, A.; Laukens, D.; Peeters, H.; Van Huysse, J.; Ferdinande, L.; Carmeliet, P.; De Vos, M. Absence of placental growth factor blocks dextran sodium sulfate-induced colonic mucosal angiogenesis, increases mucosal hypoxia and aggravates acute colonic injury. Lab. Investig. 2010, 90, 566–576. [Google Scholar] [CrossRef]
- Hapani, S.; Chu, D.; Wu, S. Risk of gastrointestinal perforation in patients with cancer treated with bevacizumab: A meta-analysis. Lancet Oncol. 2009, 10, 559–568. [Google Scholar] [CrossRef]
- Coriat, R.; Mir, O.; Leblanc, S.; Ropert, S.; Brezault, C.; Chaussade, S.; Goldwasser, F. Feasibility of anti-VEGF agent bevacizumab in patients with Crohn’s disease. Inflamm. Bowel Dis. 2010, 17, 1632. [Google Scholar] [CrossRef]
- Tanaka, M.; Ishii, H.; Azuma, K.; Saisho, C.; Matsuo, N.; Imamura, Y.; Tokito, T.; Kinoshita, T.; Yamada, K.; Takedatsu, H.; et al. Ulcerative colitis in a patient with non-small-cell lung cancer receiving bevacizumab. Investig. New Drugs 2015, 33, 1133–1135. [Google Scholar] [CrossRef]
- Herrera-Gómez, R.G.; Grecea, M.; Gallois, C.; Boige, V.; Pautier, P.; Pistilli, B.; Planchard, D.; Malka, D.; Ducreux, M.; Mir, O. Safety and Efficacy of Bevacizumab in Cancer Patients with Inflammatory Bowel Disease. Cancers 2022, 14, 2914. [Google Scholar] [CrossRef]
- Loriot, Y.; Boudou-Rouquette, P.; Billemont, B.; Ropert, S.; Goldwasser, F. Acute exacerbation of hemorrhagic rectocolitis during antiangiogenic therapy with sunitinib and sorafenib. Ann. Oncol. 2008, 19, 1975. [Google Scholar] [CrossRef] [PubMed]
- Fukunaga, S.; Mori, A.; Ohuchi, A.; Yoshioka, S.; Akiba, J.; Mistuyama, K.; Tsuruta, O.; Torimura, T. Gastrointestinal: Abdominal pain, diarrhea, and bloody stools in a patient treated for renal cell carcinoma with sunitinib. J. Gastroenterol. Hepatol. 2020, 35, 10. [Google Scholar] [CrossRef] [PubMed]
- Gündoğdu, Y.; Deniz, O.C.; Saka, D.; Şişman, G.; Erdamar, S.; Köksal, İ. Sunitinib induced colitis manifesting as invasive diarrhea in a patient with renal cell carcinoma. J. Oncol. Pharm. Pract. 2022, 28, 516–518. [Google Scholar] [CrossRef] [PubMed]
- Guenzi, E.; Töpolt, K.; Cornali, E.; Lubeseder-Martellato, C.; Jörg, A.; Matzen, K.; Zietz, C.; Kremmer, E.; Nappi, F.; Schwemmle, M.; et al. The helical domain of GBP-1 mediates the inhibition of endothelial cell proliferation by inflammatory cytokines. EMBO J. 2001, 20, 5568–5577. [Google Scholar] [CrossRef] [PubMed]
- Guenzi, E.; Töpolt, K.; Lubeseder-Martellato, C.; Jörg, A.; Naschberger, E.; Benelli, R.; Albini, A.; Stürzl, M. The guanylate binding protein-1 GTPase controls the invasive and angiogenic capability of endothelial cells through inhibition of MMP-1 expression. EMBO J. 2003, 22, 3772–3782. [Google Scholar] [CrossRef]
- Lubeseder-Martellato, C.; Guenzi, E.; Jörg, A.; Töpolt, K.; Naschberger, E.; Kremmer, E.; Zietz, C.; Tschachler, E.; Hutzler, P.; Schwemmle, M.; et al. Guanylate-binding protein-1 expression is selectively induced by inflammatory cytokines and is an activation marker of endothelial cells during inflammatory diseases. Am. J. Pathol. 2002, 161, 1749–1759. [Google Scholar] [CrossRef]
- Cornelius, L.A.; Nehring, L.C.; Harding, E.; Bolanowski, M.; Welgus, H.G.; Kobayashi, D.K.; Pierce, R.A.; Shapiro, S.D. Matrix metalloproteinases generate angiostatin: Effects on neovascularization. J. Immunol. 1998, 161, 6845–6852. [Google Scholar] [CrossRef]
- Danese, S. Negative regulators of angiogenesis in inflammatory bowel disease: Thrombospondin in the spotlight. Pathobiology 2008, 75, 22–24. [Google Scholar] [CrossRef]
- Punekar, S.; Zak, S.; Kalter, V.G.; Dobransky, L.; Punekar, I.; Lawler, J.W.; Gutierrez, L.S. Thrombospondin 1 and its mimetic peptide ABT-510 decrease angiogenesis and inflammation in a murine model of inflammatory bowel disease. Pathobiology 2008, 75, 9–21. [Google Scholar] [CrossRef]
- Sandor, Z.; Deng, X.M.; Khomenko, T.; Tarnawski, A.S.; Szabo, S. Altered angiogenic balance in ulcerative colitis: A key to impaired healing? Biochem. Biophys. Res. Commun. 2006, 350, 147–150. [Google Scholar] [CrossRef]
- Singh, U.P.; Singh, N.P.; Murphy, E.A.; Price, R.L.; Fayad, R.; Nagarkatti, M.; Nagarkatti, P.S. Chemokine and cytokine levels in inflammatory bowel disease patients. Cytokine 2016, 77, 44–49. [Google Scholar] [CrossRef] [PubMed]
- Ozawa, C.R.; Banfi, A.; Glazer, N.L.; Thurston, G.; Springer, M.L.; Kraft, P.E.; McDonald, D.M.; Blau, H.M. Microenvironmental VEGF concentration, not total dose, determines a threshold between normal and aberrant angiogenesis. J. Clin. Investig. 2004, 113, 516–527. [Google Scholar] [CrossRef] [PubMed]
- Xing, S.; Luo, Y.; Liu, Z.; Bu, P.; Duan, H.; Liu, D.; Wang, P.; Yang, J.; Song, L.; Feng, J.; et al. Targeting endothelial CD146 attenuates colitis and prevents colitis-associated carcinogenesis. Am. J. Pathol. 2014, 184, 1604–1616. [Google Scholar] [CrossRef] [PubMed]
- Yan, H.; Zhang, C.; Wang, Z.; Tu, T.; Duan, H.; Luo, Y.; Feng, J.; Liu, F.; Yan, X. CD146 is required for VEGF-C-induced lymphatic sprouting during lymphangiogenesis. Sci. Rep. 2017, 7, 7442. [Google Scholar] [CrossRef] [PubMed]
- Tsiolakidou, G.; Koutroubakis, I.E.; Tzardi, M.; Kouroumalis, E.A. Increased expression of VEGF and CD146 in patients with inflammatory bowel disease. Dig. Liver Dis. 2008, 40, 673–679. [Google Scholar] [CrossRef]
- Danese, S.; de la Motte, C.; Sturm, A.; Vogel, J.D.; West, G.A.; Strong, S.A.; Katz, J.A.; Fiocchi, C. Platelets trigger a CD40-dependent inflammatory response in the microvasculature of inflammatory bowel disease patients. Gastroenterology 2003, 124, 1249–1264. [Google Scholar] [CrossRef]
- Vogel, J.D.; West, G.A.; Danese, S.; De La Motte, C.; Phillips, M.H.; Strong, S.A.; Willis, J.; Fiocchi, C. CD40-mediated immune-nonimmune cell interactions induce mucosal fibroblast chemokines leading to T-cell transmigration. Gastroenterology 2004, 126, 63–80. [Google Scholar] [CrossRef]
- Danese, S.; Scaldaferri, F.; Vetrano, S.; Stefanelli, T.; Graziani, C.; Repici, A.; Ricci, R.; Straface, G.; Sgambato, A.; Malesci, A.; et al. Critical role of the CD40 CD40-ligand pathway in regulating mucosal inflammation-driven angiogenesis in inflammatory bowel disease. Gut 2007, 56, 1248–1256. [Google Scholar] [CrossRef]
- Konerding, M.A.; Turhan, A.; Ravnic, D.J.; Lin, M.; Fuchs, C.; Secomb, T.W.; Tsuda, A.; Mentzer, S.J. Inflammation-induced intussusceptive angiogenesis in murine colitis. Anat. Rec. 2010, 293, 849–857. [Google Scholar] [CrossRef]
- Djonov, V.; Baum, O.; Burri, P.H. Vascular remodeling by intussusceptive angiogenesis. Cell Tissue Res. 2003, 314, 107–117. [Google Scholar] [CrossRef]
- Burri, P.H.; Hlushchuk, R.; Djonov, V. Intussusceptive angiogenesis: Its emergence, its characteristics, and its significance. Dev. Dyn. 2004, 231, 474–488. [Google Scholar] [CrossRef]
- Styp-Rekowska, B.; Hlushchuk, R.; Pries, A.R.; Djonov, V. Intussusceptive angiogenesis: Pillars against the blood flow. Acta Physiol. 2011, 202, 213–223. [Google Scholar] [CrossRef] [PubMed]
- Hlushchuk, R.; Styp-Rekowska, B.; Dzambazi, J.; Wnuk, M.; Huynh-Do, U.; Makanya, A.; Djonov, V. Endoglin inhibition leads to intussusceptive angiogenesis via activation of factors related to COUP-TFII signaling pathway. PLoS ONE 2017, 12, e0182813. [Google Scholar] [CrossRef] [PubMed]
- Groppa, E.; Brkic, S.; Uccelli, A.; Wirth, G.; Korpisalo-Pirinen, P.; Filippova, M.; Dasen, B.; Sacchi, V.; Muraro, M.G.; Trani, M.; et al. EphrinB2/EphB4 signaling regulates non-sprouting angiogenesis by VEGF. EMBO Rep. 2018, 19, e45054. [Google Scholar] [CrossRef] [PubMed]
- Ackermann, M.; Tsuda, A.; Secomb, T.W.; Mentzer, S.J.; Konerding, M.A. Intussusceptive remodeling of vascular branch angles in chemically-induced murine colitis. Microvasc. Res. 2013, 87, 75–82. [Google Scholar] [CrossRef]
- Esteban, S.; Clemente, C.; Koziol, A.; Gonzalo, P.; Rius, C.; Martínez, F.; Linares, P.M.; Chaparro, M.; Urzainqui, A.; Andrés, V.; et al. Endothelial MT1-MMP targeting limits intussusceptive angiogenesis and colitis via TSP1/nitric oxide axis. EMBO Mol. Med. 2020, 12, e10862. [Google Scholar] [CrossRef]
- Betto, T.; Amano, H.; Ito, Y.; Eshima, K.; Yoshida, T.; Matsui, Y.; Yamane, S.; Inoue, T.; Otaka, F.; Kobayashi, K.; et al. Vascular endothelial growth factor receptor 1 tyrosine kinase signaling facilitates healing of DSS-induced colitis by accumulation of Tregs in ulcer area. Biomed. Pharm. 2019, 111, 131–141. [Google Scholar] [CrossRef]
- Yoshimi, K.; Tanaka, T.; Serikawa, T.; Kuramoto, T. Tumor suppressor APC protein is essential in mucosal repair from colonic inflammation through angiogenesis. Am. J. Pathol. 2013, 182, 1263–1274. [Google Scholar] [CrossRef]
- Jerkic, M.; Peter, M.; Ardelean, D.; Fine, M.; Konerding, M.A.; Letarte, M. Dextran sulfate sodium leads to chronic colitis and pathological angiogenesis in endoglin heterozygous mice. Inflamm. Bowel Dis. 2010, 16, 1859–1870. [Google Scholar] [CrossRef]
- Ardelean, D.S.; Yin, M.; Jerkic, M.; Peter, M.; Ngan, B.; Kerbel, R.S.; Foster, F.S.; Letarte, M. Anti-VEGF therapy reduces intestinal inflammation in Endoglin heterozygous mice subjected to experimental colitis. Angiogenesis 2014, 17, 641–659. [Google Scholar] [CrossRef]
- Kobayashi, M.; Fukuda, M.; Nakayama, J. Role of sulfated O-glycans expressed by high endothelial venule-like vessels in pathogenesis of chronic inflammatory gastrointestinal diseases. Biol. Pharm. Bull. 2009, 32, 774–779. [Google Scholar] [CrossRef] [PubMed]
- Horjus Talabur Horje, C.S.; Smids, C.; Meijer, J.W.; Groenen, M.J.; Rijnders, M.K.; van Lochem, E.G.; Wahab, P.J. High endothelial venules associated with T cell subsets in the inflamed gut of newly diagnosed inflammatory bowel disease patients. Clin. Exp. Immunol. 2017, 188, 163–173. [Google Scholar] [CrossRef] [PubMed]
- Roosenboom, B.; Lochem, E.G.V.; Meijer, J.; Smids, C.; Nierkens, S.; Brand, E.C.; Erp, L.W.V.; Kemperman, L.; Groenen, M.J.M.; Horje, C.; et al. Development of Mucosal PNAd+ and MAdCAM-1+ Venules during Disease Course in Ulcerative Colitis. Cells 2020, 9, 891. [Google Scholar] [CrossRef] [PubMed]
- Kobayashi, M.; Hoshino, H.; Masumoto, J.; Fukushima, M.; Suzawa, K.; Kageyama, S.; Suzuki, M.; Ohtani, H.; Fukuda, M.; Nakayama, J. GlcNAc6ST-1-mediated decoration of MAdCAM-1 protein with L-selectin ligand carbohydrates directs disease activity of ulcerative colitis. Inflamm. Bowel Dis. 2009, 15, 697–706. [Google Scholar] [CrossRef]
- Nakai, D.; Miyake, M. The change of the electrophysiological parameters using human intestinal tissues from ulcerative colitis and Crohn’s disease. J. Pharmacol. Sci. 2022, 150, 90–93. [Google Scholar] [CrossRef]
- Jergens, A.E.; Parvinroo, S.; Kopper, J.; Wannemuehler, M.J. Rules of Engagement: Epithelial—Microbe Interactions and Inflammatory Bowel Disease. Front. Med. 2021, 8, 669913. [Google Scholar] [CrossRef]
- Neurath, M.F. Cytokines in inflammatory bowel disease. Nat. Rev. Immunol. 2014, 14, 329–342. [Google Scholar] [CrossRef]
- Buda, A.; Hatem, G.; Neumann, H.; D’Incà, R.; Mescoli, C.; Piselli, P.; Jackson, J.; Bruno, M.; Sturniolo, G.C. Confocal laser endomicroscopy for prediction of disease relapse in ulcerative colitis: A pilot study. J. Crohn’s Colitis 2014, 8, 304–311. [Google Scholar] [CrossRef]
- Taniguchi, T.; Inoue, A.; Okahisa, T.; Kimura, T.; Gohji, T.; Niki, M.; Kitamura, S.; Takeuchi, H.; Okamoto, K.; Kaji, M.; et al. Increased Angiogenesis and Vascular Permeability in Patient with Ulcerative Colitis. Gastrointest. Endosc. 2009, 69, AB365. [Google Scholar] [CrossRef]
- Oshima, T.; Laroux, F.S.; Coe, L.L.; Morise, Z.; Kawachi, S.; Bauer, P.; Grisham, M.B.; Specian, R.D.; Carter, P.; Jennings, S.; et al. Interferon-gamma and interleukin-10 reciprocally regulate endothelial junction integrity and barrier function. Microvasc. Res. 2001, 61, 130–143. [Google Scholar] [CrossRef]
- Bardin, N.; Reumaux, D.; Geboes, K.; Colombel, J.F.; Blot-Chabaud, M.; Sampol, J.; Duthilleul, P.; Dignat-George, F. Increased expression of CD146, a new marker of the endothelial junction in active inflammatory bowel disease. Inflamm. Bowel Dis. 2006, 12, 16–21. [Google Scholar] [CrossRef] [PubMed]
- Tolstanova, G.; Deng, X.; French, S.W.; Lungo, W.; Paunovic, B.; Khomenko, T.; Ahluwalia, A.; Kaplan, T.; Dacosta-Iyer, M.; Tarnawski, A.; et al. Early endothelial damage and increased colonic vascular permeability in the development of experimental ulcerative colitis in rats and mice. Lab. Investig. 2012, 92, 9–21. [Google Scholar] [CrossRef] [PubMed]
- Laroux, F.S.; Grisham, M.B. Immunological basis of inflammatory bowel disease: Role of the microcirculation. Microcirculation 2001, 8, 283–301. [Google Scholar] [CrossRef] [PubMed]
- Vestweber, D.; Wessel, F.; Nottebaum, A.F. Similarities and differences in the regulation of leukocyte extravasation and vascular permeability. Semin. Immunopathol. 2014, 36, 177–192. [Google Scholar] [CrossRef] [PubMed]
- Wautier, J.L.; Wautier, M.P. Vascular Permeability in Diseases. Int. J. Mol. Sci. 2022, 23, 3645. [Google Scholar] [CrossRef] [PubMed]
- Tolstanova, G.; Khomenko, T.; Deng, X.; Szabo, S.; Sandor, Z. New molecular mechanisms of the unexpectedly complex role of VEGF in ulcerative colitis. Biochem. Biophys. Res. Commun. 2010, 399, 613–616. [Google Scholar] [CrossRef] [PubMed]
- Weis, S.M.; Cheresh, D.A. Pathophysiological consequences of VEGF-induced vascular permeability. Nature 2005, 437, 497–504. [Google Scholar] [CrossRef]
- Gavard, J.; Gutkind, J.S. VEGF controls endothelial-cell permeability by promoting the beta-arrestin-dependent endocytosis of VE-cadherin. Nat. Cell Biol. 2006, 8, 1223–1234. [Google Scholar] [CrossRef]
- Zhang, C.; Chen, H.; He, Q.; Luo, Y.; He, A.; Tao, A.; Yan, J. Fibrinogen/AKT/Microfilament Axis Promotes Colitis by Enhancing Vascular Permeability. Cell. Mol. Gastroenterol. Hepatol. 2021, 11, 683–696. [Google Scholar] [CrossRef]
- Matsumoto, K.; Yamaba, R.; Inoue, K.; Utsumi, D.; Tsukahara, T.; Amagase, K.; Tominaga, M.; Kato, S. Transient receptor potential vanilloid 4 channel regulates vascular endothelial permeability during colonic inflammation in dextran sulphate sodium-induced murine colitis. Br. J. Pharmacol. 2018, 175, 84–99. [Google Scholar] [CrossRef]
- Dejana, E.; Orsenigo, F.; Lampugnani, M.G. The role of adherens junctions and VE-cadherin in the control of vascular permeability. J. Cell Sci. 2008, 121, 2115–2122. [Google Scholar] [CrossRef]
- Vestweber, D. VE-cadherin: The major endothelial adhesion molecule controlling cellular junctions and blood vessel formation. Arter. Thromb. Vasc. Biol. 2008, 28, 223–232. [Google Scholar] [CrossRef] [PubMed]
- Giannotta, M.; Trani, M.; Dejana, E. VE-cadherin and endothelial adherens junctions: Active guardians of vascular integrity. Dev. Cell 2013, 26, 441–454. [Google Scholar] [CrossRef] [PubMed]
- Xiao, K.; Allison, D.F.; Kottke, M.D.; Summers, S.; Sorescu, G.P.; Faundez, V.; Kowalczyk, A.P. Mechanisms of VE-cadherin processing and degradation in microvascular endothelial cells. J. Biol. Chem. 2003, 278, 19199–19208. [Google Scholar] [CrossRef]
- Schulz, B.; Pruessmeyer, J.; Maretzky, T.; Ludwig, A.; Blobel, C.P.; Saftig, P.; Reiss, K. ADAM10 regulates endothelial permeability and T-Cell transmigration by proteolysis of vascular endothelial cadherin. Circ. Res. 2008, 102, 1192–1201. [Google Scholar] [CrossRef]
- Arihiro, S.; Ohtani, H.; Hiwatashi, N.; Torii, A.; Sorsa, T.; Nagura, H. Vascular smooth muscle cells and pericytes express MMP-1, MMP-9, TIMP-1 and type I procollagen in inflammatory bowel disease. Histopathology 2001, 39, 50–59. [Google Scholar] [CrossRef] [PubMed]
- Angelini, D.J.; Hyun, S.W.; Grigoryev, D.N.; Garg, P.; Gong, P.; Singh, I.S.; Passaniti, A.; Hasday, J.D.; Goldblum, S.E. TNF-alpha increases tyrosine phosphorylation of vascular endothelial cadherin and opens the paracellular pathway through fyn activation in human lung endothelia. Am. J. Physiol. Lung Cell. Mol. Physiol. 2006, 291, L1232–L1245. [Google Scholar] [CrossRef]
- Baugh, M.D.; Perry, M.J.; Hollander, A.P.; Davies, D.R.; Cross, S.S.; Lobo, A.J.; Taylor, C.J.; Evans, G.S. Matrix metalloproteinase levels are elevated in inflammatory bowel disease. Gastroenterology 1999, 117, 814–822. [Google Scholar] [CrossRef]
- Matusiewicz, M.; Neubauer, K.; Mierzchala-Pasierb, M.; Gamian, A.; Krzystek-Korpacka, M. Matrix metalloproteinase-9: Its interplay with angiogenic factors in inflammatory bowel diseases. Dis. Mark. 2014, 2014, 643645. [Google Scholar] [CrossRef]
- Meijer, M.J.; Mieremet-Ooms, M.A.; van der Zon, A.M.; van Duijn, W.; van Hogezand, R.A.; Sier, C.F.; Hommes, D.W.; Lamers, C.B.; Verspaget, H.W. Increased mucosal matrix metalloproteinase-1, -2, -3 and -9 activity in patients with inflammatory bowel disease and the relation with Crohn’s disease phenotype. Dig. Liver Dis. 2007, 39, 733–739. [Google Scholar] [CrossRef] [PubMed]
- Santana, A.; Medina, C.; Paz-Cabrera, M.C.; Díaz-Gonzalez, F.; Farré, E.; Salas, A.; Radomski, M.W.; Quintero, E. Attenuation of dextran sodium sulphate induced colitis in matrix metalloproteinase-9 deficient mice. World J. Gastroenterol. 2006, 12, 6464–6472. [Google Scholar] [CrossRef] [PubMed]
- Medina, C.; Santana, A.; Paz, M.C.; Díaz-Gonzalez, F.; Farre, E.; Salas, A.; Radomski, M.W.; Quintero, E. Matrix metalloproteinase-9 modulates intestinal injury in rats with transmural colitis. J. Leukoc. Biol. 2006, 79, 954–962. [Google Scholar] [CrossRef] [PubMed]
- Nighot, P.; Al-Sadi, R.; Rawat, M.; Guo, S.; Watterson, D.M.; Ma, T. Matrix metalloproteinase 9-induced increase in intestinal epithelial tight junction permeability contributes to the severity of experimental DSS colitis. Am. J. Physiol. Gastrointest. Liver Physiol. 2015, 309, G988–G997. [Google Scholar] [CrossRef]
- Sidibé, A.; Mannic, T.; Arboleas, M.; Subileau, M.; Gulino-Debrac, D.; Bouillet, L.; Jan, M.; Vandhuick, T.; Le Loet, X.; Vittecoq, O.; et al. Soluble VE-cadherin in rheumatoid arthritis patients correlates with disease activity: Evidence for tumor necrosis factor alpha-induced VE-cadherin cleavage. Arthritis Rheum. 2012, 64, 77–87. [Google Scholar] [CrossRef] [PubMed]
- Flemming, S.; Burkard, N.; Renschler, M.; Vielmuth, F.; Meir, M.; Schick, M.A.; Wunder, C.; Germer, C.T.; Spindler, V.; Waschke, J.; et al. Soluble VE-cadherin is involved in endothelial barrier breakdown in systemic inflammation and sepsis. Cardiovasc. Res. 2015, 107, 32–44. [Google Scholar] [CrossRef] [PubMed]
- Polena, H.; Creuzet, J.; Dufies, M.; Sidibé, A.; Khalil-Mgharbel, A.; Salomon, A.; Deroux, A.; Quesada, J.L.; Roelants, C.; Filhol, O.; et al. The tyrosine-kinase inhibitor sunitinib targets vascular endothelial (VE)-cadherin: A marker of response to antitumoural treatment in metastatic renal cell carcinoma. Br. J. Cancer 2018, 118, 1179–1188. [Google Scholar] [CrossRef] [PubMed]
- Wallez, Y.; Cand, F.; Cruzalegui, F.; Wernstedt, C.; Souchelnytskyi, S.; Vilgrain, I.; Huber, P. Src kinase phosphorylates vascular endothelial-cadherin in response to vascular endothelial growth factor: Identification of tyrosine 685 as the unique target site. Oncogene 2007, 26, 1067–1077. [Google Scholar] [CrossRef]
- Nottebaum, A.F.; Cagna, G.; Winderlich, M.; Gamp, A.C.; Linnepe, R.; Polaschegg, C.; Filippova, K.; Lyck, R.; Engelhardt, B.; Kamenyeva, O.; et al. VE-PTP maintains the endothelial barrier via plakoglobin and becomes dissociated from VE-cadherin by leukocytes and by VEGF. J. Exp. Med. 2008, 205, 2929–2945. [Google Scholar] [CrossRef]
- Gioelli, N.; Neilson, L.J.; Wei, N.; Villari, G.; Chen, W.; Kuhle, B.; Ehling, M.; Maione, F.; Willox, S.; Brundu, S.; et al. Neuropilin 1 and its inhibitory ligand mini-tryptophanyl-tRNA synthetase inversely regulate VE-cadherin turnover and vascular permeability. Nat. Commun. 2022, 13, 4188. [Google Scholar] [CrossRef]
- Wong, R.K.; Baldwin, A.L.; Heimark, R.L. Cadherin-5 redistribution at sites of TNF-alpha and IFN-gamma-induced permeability in mesenteric venules. Am. J. Physiol. 1999, 276, H736–H748. [Google Scholar]
- Gavard, J. Endothelial permeability and VE-cadherin: A wacky comradeship. Cell Adhes. Migr. 2013, 7, 455–461. [Google Scholar] [CrossRef] [PubMed]
- Abu Taha, A.; Schnittler, H.J. Dynamics between actin and the VE-cadherin/catenin complex: Novel aspects of the ARP2/3 complex in regulation of endothelial junctions. Cell Adhes. Migr. 2014, 8, 125–135. [Google Scholar] [CrossRef] [PubMed]
- Brandt, M.; Gerke, V.; Betz, T. Human endothelial cells display a rapid tensional stress increase in response to tumor necrosis factor-α. PLoS ONE 2022, 17, e0270197. [Google Scholar] [CrossRef] [PubMed]
- Morales-Ruiz, M.; Fulton, D.; Sowa, G.; Languino, L.R.; Fujio, Y.; Walsh, K.; Sessa, W.C. Vascular endothelial growth factor-stimulated actin reorganization and migration of endothelial cells is regulated via the serine/threonine kinase Akt. Circ. Res. 2000, 86, 892–896. [Google Scholar] [CrossRef] [PubMed]
- Ostler, N.; Britzen-Laurent, N.; Liebl, A.; Naschberger, E.; Lochnit, G.; Ostler, M.; Forster, F.; Kunzelmann, P.; Ince, S.; Supper, V.; et al. Gamma interferon-induced guanylate binding protein 1 is a novel actin cytoskeleton remodeling factor. Mol. Cell. Biol. 2014, 34, 196–209. [Google Scholar] [CrossRef]
- Liu, P.; Bian, Y.; Fan, Y.; Zhong, J.; Liu, Z. Protective Effect of Naringin on In Vitro Gut-Vascular Barrier Disruption of Intestinal Microvascular Endothelial Cells Induced by TNF-α. J. Agric. Food Chem. 2020, 68, 168–175. [Google Scholar] [CrossRef]
- Carloni, S.; Bertocchi, A.; Mancinelli, S.; Bellini, M.; Erreni, M.; Borreca, A.; Braga, D.; Giugliano, S.; Mozzarelli, A.M.; Manganaro, D.; et al. Identification of a choroid plexus vascular barrier closing during intestinal inflammation. Science 2021, 374, 439–448. [Google Scholar] [CrossRef]
- Armulik, A.; Abramsson, A.; Betsholtz, C. Endothelial/pericyte interactions. Circ. Res. 2005, 97, 512–523. [Google Scholar] [CrossRef]
- Ganta, V.C.; Cromer, W.; Mills, G.L.; Traylor, J.; Jennings, M.; Daley, S.; Clark, B.; Mathis, J.M.; Bernas, M.; Boktor, M.; et al. Angiopoietin-2 in experimental colitis. Inflamm. Bowel Dis. 2010, 16, 1029–1039. [Google Scholar] [CrossRef]
- Sweeney, M.; Foldes, G. It Takes Two: Endothelial-Perivascular Cell Cross-Talk in Vascular Development and Disease. Front. Cardiovasc. Med. 2018, 5, 154. [Google Scholar] [CrossRef]
- Armulik, A.; Genove, G.; Mae, M.; Nisancioglu, M.H.; Wallgard, E.; Niaudet, C.; He, L.; Norlin, J.; Lindblom, P.; Strittmatter, K.; et al. Pericytes regulate the blood-brain barrier. Nature 2010, 468, 557–561. [Google Scholar] [CrossRef]
- Alexander, J.S.; Chaitanya, G.V.; Grisham, M.B.; Boktor, M. Emerging roles of lymphatics in inflammatory bowel disease. Ann. N. Y. Acad. Sci. 2010, 1207 (Suppl. 1), E75–E85. [Google Scholar] [CrossRef] [PubMed]
- Regensburger, D.; Tenkerian, C.; Pürzer, V.; Schmid, B.; Wohlfahrt, T.; Stolzer, I.; López-Posadas, R.; Günther, C.; Waldner, M.J.; Becker, C.; et al. Matricellular Protein SPARCL1 Regulates Blood Vessel Integrity and Antagonizes Inflammatory Bowel Disease. Inflamm. Bowel Dis. 2021, 27, 1491–1502. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, H.; Shibano, K.; Okane, M.; Kono, I.; Matsui, Y.; Yamane, K.; Kashiwagi, H. Interferon-gamma modulates messenger RNA levels of c-sis (PDGF-B chain), PDGF-A chain, and IL-1 beta genes in human vascular endothelial cells. Am. J. Pathol. 1989, 134, 35–43. [Google Scholar] [PubMed]
- Darden, J.; Payne, L.B.; Zhao, H.; Chappell, J.C. Excess vascular endothelial growth factor-A disrupts pericyte recruitment during blood vessel formation. Angiogenesis 2019, 22, 167–183. [Google Scholar] [CrossRef] [PubMed]
- Sprague, A.H.; Khalil, R.A. Inflammatory cytokines in vascular dysfunction and vascular disease. Biochem. Pharmacol. 2009, 78, 539–552. [Google Scholar] [CrossRef]
- Hatoum, O.A.; Binion, D.G.; Gutterman, D.D. Paradox of simultaneous intestinal ischaemia and hyperaemia in inflammatory bowel disease. Eur. J. Clin. Investig. 2005, 35, 599–609. [Google Scholar] [CrossRef]
- Binion, D.G.; Rafiee, P.; Ramanujam, K.S.; Fu, S.; Fisher, P.J.; Rivera, M.T.; Johnson, C.P.; Otterson, M.F.; Telford, G.L.; Wilson, K.T. Deficient iNOS in inflammatory bowel disease intestinal microvascular endothelial cells results in increased leukocyte adhesion. Free Radic. Biol. Med. 2000, 29, 881–888. [Google Scholar] [CrossRef]
- Horowitz, S.; Binion, D.G.; Nelson, V.M.; Kanaa, Y.; Javadi, P.; Lazarova, Z.; Andrekopoulos, C.; Kalyanaraman, B.; Otterson, M.F.; Rafiee, P. Increased arginase activity and endothelial dysfunction in human inflammatory bowel disease. Am. J. Physiol. Gastrointest. Liver Physiol. 2007, 292, G1323–G1336. [Google Scholar] [CrossRef]
- Hatoum, O.A.; Binion, D.G.; Otterson, M.F.; Gutterman, D.D. Acquired microvascular dysfunction in inflammatory bowel disease: Loss of nitric oxide-mediated vasodilation. Gastroenterology 2003, 125, 58–69. [Google Scholar] [CrossRef]
- Hatoum, O.A.; Gauthier, K.M.; Binion, D.G.; Miura, H.; Telford, G.; Otterson, M.F.; Campbell, W.B.; Gutterman, D.D. Novel mechanism of vasodilation in inflammatory bowel disease. Arter. Thromb. Vasc. Biol. 2005, 25, 2355–2361. [Google Scholar] [CrossRef] [PubMed]
- Thibeault, S.; Rautureau, Y.; Oubaha, M.; Faubert, D.; Wilkes, B.C.; Delisle, C.; Gratton, J.P. S-nitrosylation of beta-catenin by eNOS-derived NO promotes VEGF-induced endothelial cell permeability. Mol. Cell 2010, 39, 468–476. [Google Scholar] [CrossRef] [PubMed]
- Di Lorenzo, A.; Lin, M.I.; Murata, T.; Landskroner-Eiger, S.; Schleicher, M.; Kothiya, M.; Iwakiri, Y.; Yu, J.; Huang, P.L.; Sessa, W.C. eNOS-derived nitric oxide regulates endothelial barrier function through VE-cadherin and Rho GTPases. J. Cell Sci. 2013, 126, 5541–5552. [Google Scholar] [CrossRef] [PubMed]
- Seerapu, H.; Subramaniam, G.P.; Majumder, S.; Sinha, S.; Bisana, S.; Mahajan, S.; Kolluru, G.K.; Muley, A.; Siamwala, J.H.; Illavazagan, G.; et al. Inhibition of dynamin-2 confers endothelial barrier dysfunctions by attenuating nitric oxide production. Cell Biol. Int. 2010, 34, 755–761. [Google Scholar] [CrossRef] [PubMed]
- Beck, P.L.; Xavier, R.; Wong, J.; Ezedi, I.; Mashimo, H.; Mizoguchi, A.; Mizoguchi, E.; Bhan, A.K.; Podolsky, D.K. Paradoxical roles of different nitric oxide synthase isoforms in colonic injury. Am. J. Physiol. Gastrointest. Liver Physiol. 2004, 286, G137–G147. [Google Scholar] [CrossRef] [PubMed]
- Hokari, R.; Kato, S.; Matsuzaki, K.; Kuroki, M.; Iwai, A.; Kawaguchi, A.; Nagao, S.; Miyahara, T.; Itoh, K.; Sekizuka, E.; et al. Reduced sensitivity of inducible nitric oxide synthase-deficient mice to chronic colitis. Free Radic. Biol. Med. 2001, 31, 153–163. [Google Scholar] [CrossRef]
- Sasaki, M.; Bharwani, S.; Jordan, P.; Elrod, J.W.; Grisham, M.B.; Jackson, T.H.; Lefer, D.J.; Alexander, J.S. Increased disease activity in eNOS-deficient mice in experimental colitis. Free Radic. Biol. Med. 2003, 35, 1679–1687. [Google Scholar] [CrossRef]
- Vallance, B.A.; Dijkstra, G.; Qiu, B.; van der Waaij, L.A.; van Goor, H.; Jansen, P.L.; Mashimo, H.; Collins, S.M. Relative contributions of NOS isoforms during experimental colitis: Endothelial-derived NOS maintains mucosal integrity. Am. J. Physiol. Gastrointest. Liver Physiol. 2004, 287, G865–G874. [Google Scholar] [CrossRef]
- Collins, C.E.; Rampton, D.S.; Rogers, J.; Williams, N.S. Platelet aggregation and neutrophil sequestration in the mesenteric circulation in inflammatory bowel disease. Eur. J. Gastroenterol. Hepatol. 1997, 9, 1213–1217. [Google Scholar]
- Tekelioglu, Y.; Uzun, H.; Sisman, G. Activated platelets in patients suffering from inflammatory bowel disease. Bratisl. Lek. Listy 2014, 115, 83–85. [Google Scholar] [CrossRef]
- Koutroubakis, I.E.; Theodoropoulou, A.; Xidakis, C.; Sfiridaki, A.; Notas, G.; Kolios, G.; Kouroumalis, E.A. Association between enhanced soluble CD40 ligand and prothrombotic state in inflammatory bowel disease. Eur. J. Gastroenterol. Hepatol. 2004, 16, 1147–1152. [Google Scholar] [CrossRef] [PubMed]
- Danese, S.; Katz, J.A.; Saibeni, S.; Papa, A.; Gasbarrini, A.; Vecchi, M.; Fiocchi, C. Activated platelets are the source of elevated levels of soluble CD40 ligand in the circulation of inflammatory bowel disease patients. Gut 2003, 52, 1435–1441. [Google Scholar] [CrossRef] [PubMed]
- Yoshida, H.; Russell, J.; Stokes, K.Y.; Yilmaz, C.E.; Esmon, C.T.; Granger, D.N. Role of the protein C pathway in the extraintestinal thrombosis associated with murine colitis. Gastroenterology 2008, 135, 882–888. [Google Scholar] [CrossRef] [PubMed]
- Scaldaferri, F.; Sans, M.; Vetrano, S.; Graziani, C.; De Cristofaro, R.; Gerlitz, B.; Repici, A.; Arena, V.; Malesci, A.; Panes, J.; et al. Crucial role of the protein C pathway in governing microvascular inflammation in inflammatory bowel disease. J. Clin. Investig. 2007, 117, 1951–1960. [Google Scholar] [CrossRef]
- Faioni, E.M.; Ferrero, S.; Fontana, G.; Gianelli, U.; Ciulla, M.M.; Vecchi, M.; Saibeni, S.; Biguzzi, E.; Cordani, N.; Franchi, F.; et al. Expression of endothelial protein C receptor and thrombomodulin in the intestinal tissue of patients with inflammatory bowel disease. Crit. Care Med. 2004, 32, S266–S270. [Google Scholar] [CrossRef]
- Vetrano, S.; Ploplis, V.A.; Sala, E.; Sandoval-Cooper, M.; Donahue, D.L.; Correale, C.; Arena, V.; Spinelli, A.; Repici, A.; Malesci, A.; et al. Unexpected role of anticoagulant protein C in controlling epithelial barrier integrity and intestinal inflammation. Proc. Natl. Acad. Sci. USA 2011, 108, 19830–19835. [Google Scholar] [CrossRef]
- Ogawa, H.; Rafiee, P.; Heidemann, J.; Fisher, P.J.; Johnson, N.A.; Otterson, M.F.; Kalyanaraman, B.; Pritchard, K.A., Jr.; Binion, D.G. Mechanisms of endotoxin tolerance in human intestinal microvascular endothelial cells. J. Immunol. 2003, 170, 5956–5964. [Google Scholar] [CrossRef]
- Heidemann, J.; Domschke, W.; Kucharzik, T.; Maaser, C. Intestinal microvascular endothelium and innate immunity in inflammatory bowel disease: A second line of defense? Infect. Immun. 2006, 74, 5425–5432. [Google Scholar] [CrossRef]
- Maaser, C.; Heidemann, J.; von Eiff, C.; Lugering, A.; Spahn, T.W.; Binion, D.G.; Domschke, W.; Lugering, N.; Kucharzik, T. Human intestinal microvascular endothelial cells express Toll-like receptor 5: A binding partner for bacterial flagellin. J. Immunol. 2004, 172, 5056–5062. [Google Scholar] [CrossRef]
- Vijay-Kumar, M.; Aitken, J.D.; Gewirtz, A.T. Toll like receptor-5: Protecting the gut from enteric microbes. Semin. Immunopathol. 2008, 30, 11–21. [Google Scholar] [CrossRef]
- Heidemann, J.; Rüther, C.; Kebschull, M.; Domschke, W.; Brüwer, M.; Koch, S.; Kucharzik, T.; Maaser, C. Expression of IL-12-related molecules in human intestinal microvascular endothelial cells is regulated by TLR3. Am. J. Physiol. Gastrointest. Liver Physiol. 2007, 293, G1315–G1324. [Google Scholar] [CrossRef] [PubMed]
- Lodes, M.J.; Cong, Y.; Elson, C.O.; Mohamath, R.; Landers, C.J.; Targan, S.R.; Fort, M.; Hershberg, R.M. Bacterial flagellin is a dominant antigen in Crohn disease. J. Clin. Investig. 2004, 113, 1296–1306. [Google Scholar] [CrossRef] [PubMed]
- Sitaraman, S.V.; Klapproth, J.M.; Moore, D.A., 3rd; Landers, C.; Targan, S.; Williams, I.R.; Gewirtz, A.T. Elevated flagellin-specific immunoglobulins in Crohn’s disease. Am. J. Physiol. Gastrointest. Liver Physiol. 2005, 288, G403–G406. [Google Scholar] [CrossRef] [PubMed]
- Targan, S.R.; Landers, C.J.; Yang, H.; Lodes, M.J.; Cong, Y.; Papadakis, K.A.; Vasiliauskas, E.; Elson, C.O.; Hershberg, R.M. Antibodies to CBir1 flagellin define a unique response that is associated independently with complicated Crohn’s disease. Gastroenterology 2005, 128, 2020–2028. [Google Scholar] [CrossRef]
- Vijay-Kumar, M.; Sanders, C.J.; Taylor, R.T.; Kumar, A.; Aitken, J.D.; Sitaraman, S.V.; Neish, A.S.; Uematsu, S.; Akira, S.; Williams, I.R.; et al. Deletion of TLR5 results in spontaneous colitis in mice. J. Clin. Investig. 2007, 117, 3909–3921. [Google Scholar] [CrossRef] [PubMed]
- Vijay-Kumar, M.; Wu, H.; Aitken, J.; Kolachala, V.L.; Neish, A.S.; Sitaraman, S.V.; Gewirtz, A.T. Activation of toll-like receptor 3 protects against DSS-induced acute colitis. Inflamm. Bowel Dis. 2007, 13, 856–864. [Google Scholar] [CrossRef]
- Hu, G.; Xue, J.; Duan, H.; Yang, Z.; Gao, L.; Luo, H.; Mu, X.; Cui, S. IFN-γ induces IFN-α and IFN-β expressions in cultured rat intestinal mucosa microvascular endothelial cells. Immunopharmacol. Immunotoxicol. 2010, 32, 656–662. [Google Scholar] [CrossRef]
- Stürzl, M.; Kunz, M.; Krug, S.M.; Naschberger, E. Angiocrine Regulation of Epithelial Barrier Integrity in Inflammatory Bowel Disease. Front. Med. 2021, 8, 643607. [Google Scholar] [CrossRef]
- Imaizumi, T.; Yoshida, H.; Satoh, K. Regulation of CX3CL1/fractalkine expression in endothelial cells. J. Atheroscler. Thromb. 2004, 11, 15–21. [Google Scholar] [CrossRef]
- Nishimura, M.; Kuboi, Y.; Muramoto, K.; Kawano, T.; Imai, T. Chemokines as novel therapeutic targets for inflammatory bowel disease. Ann. N. Y. Acad. Sci. 2009, 1173, 350–356. [Google Scholar] [CrossRef]
- Sans, M.; Danese, S.; de la Motte, C.; de Souza, H.S.; Rivera-Reyes, B.M.; West, G.A.; Phillips, M.; Katz, J.A.; Fiocchi, C. Enhanced recruitment of CX3CR1+ T cells by mucosal endothelial cell-derived fractalkine in inflammatory bowel disease. Gastroenterology 2007, 132, 139–153. [Google Scholar] [CrossRef] [PubMed]
- Wu, X.; Sun, M.; Yang, Z.; Lu, C.; Wang, Q.; Wang, H.; Deng, C.; Liu, Y.; Yang, Y. The Roles of CCR9/CCL25 in Inflammation and Inflammation-Associated Diseases. Front. Cell Dev. Biol. 2021, 9, 686548. [Google Scholar] [CrossRef] [PubMed]
- Bernier-Latmani, J.; Petrova, T.V. Intestinal lymphatic vasculature: Structure, mechanisms and functions. Nat. Rev. Gastroenterol. Hepatol. 2017, 14, 510–526. [Google Scholar] [CrossRef]
- Kim, H.; Kataru, R.P.; Koh, G.Y. Inflammation-associated lymphangiogenesis: A double-edged sword? J. Clin. Investig. 2014, 124, 936–942. [Google Scholar] [CrossRef]
- Geleff, S.; Schoppmann, S.F.; Oberhuber, G. Increase in podoplanin-expressing intestinal lymphatic vessels in inflammatory bowel disease. Virchows Arch. 2003, 442, 231–237. [Google Scholar] [CrossRef] [PubMed]
- Fogt, F.; Pascha, T.L.; Zhang, P.J.; Gausas, R.E.; Rahemtulla, A.; Zimmerman, R.L. Proliferation of D2-40-expressing intestinal lymphatic vessels in the lamina propria in inflammatory bowel disease. Int. J. Mol. Med. 2004, 13, 211–214. [Google Scholar] [CrossRef]
- Rahier, J.F.; De Beauce, S.; Dubuquoy, L.; Erdual, E.; Colombel, J.F.; Jouret-Mourin, A.; Geboes, K.; Desreumaux, P. Increased lymphatic vessel density and lymphangiogenesis in inflammatory bowel disease. Aliment. Pharmacol. Ther. 2011, 34, 533–543. [Google Scholar] [CrossRef] [PubMed]
- Wu, T.F.; MacNaughton, W.K.; von der Weid, P.Y. Lymphatic vessel contractile activity and intestinal inflammation. Mem. Inst. Oswaldo Cruz 2005, 100 (Suppl. 1), 107–110. [Google Scholar] [CrossRef]
- Von Der Weid, P.Y.; Rehal, S. Lymphatic pump function in the inflamed gut. Ann. N. Y. Acad. Sci. 2010, 1207 (Suppl. 1), E69–E74. [Google Scholar] [CrossRef]
- Nikolakis, D.; de Voogd, F.A.E.; Pruijt, M.J.; Grootjans, J.; van de Sande, M.G.; D’Haens, G.R. The Role of the Lymphatic System in the Pathogenesis and Treatment of Inflammatory Bowel Disease. Int. J. Mol. Sci. 2022, 23, 1854. [Google Scholar] [CrossRef]
- Zhang, L.; Ocansey, D.K.W.; Liu, L.; Olovo, C.V.; Zhang, X.; Qian, H.; Xu, W.; Mao, F. Implications of lymphatic alterations in the pathogenesis and treatment of inflammatory bowel disease. Biomed Pharm. 2021, 140, 111752. [Google Scholar] [CrossRef]
- von der Weid, P.Y.; Rainey, K.J. Review article: Lymphatic system and associated adipose tissue in the development of inflammatory bowel disease. Aliment. Pharmacol. Ther. 2010, 32, 697–711. [Google Scholar] [CrossRef] [PubMed]
- Agollah, G.D.; Wu, G.; Peng, H.L.; Kwon, S. Dextran sulfate sodium-induced acute colitis impairs dermal lymphatic function in mice. World J. Gastroenterol. 2015, 21, 12767–12777. [Google Scholar] [CrossRef] [PubMed]
- Ott, C.; Schölmerich, J. Extraintestinal manifestations and complications in IBD. Nat. Rev. Gastroenterol. Hepatol. 2013, 10, 585–595. [Google Scholar] [CrossRef] [PubMed]
- Navabi, S.; Gorrepati, V.S.; Yadav, S.; Chintanaboina, J.; Maher, S.; Demuth, P.; Stern, B.; Stuart, A.; Tinsley, A.; Clarke, K.; et al. Influences and Impact of Anxiety and Depression in the Setting of Inflammatory Bowel Disease. Inflamm. Bowel Dis. 2018, 24, 2303–2308. [Google Scholar] [CrossRef] [PubMed]
- Gupta, G.; Gelfand, J.M.; Lewis, J.D. Increased Risk for Demyelinating Diseases in Patients With Inflammatory Bowel Disease. Gastroenterology 2005, 129, 819–826. [Google Scholar] [CrossRef]
- Ferro, J.M.; Oliveira, S.N.; Correia, L. Neurologic manifestations of inflammatory bowel diseases. Handb. Clin. Neurol. 2014, 120, 595–605. [Google Scholar] [CrossRef]
- Mouries, J.; Brescia, P.; Silvestri, A.; Spadoni, I.; Sorribas, M.; Wiest, R.; Mileti, E.; Galbiati, M.; Invernizzi, P.; Adorini, L.; et al. Microbiota-driven gut vascular barrier disruption is a prerequisite for non-alcoholic steatohepatitis development. J. Hepatol. 2019, 71, 1216–1228. [Google Scholar] [CrossRef]
- Bertocchi, A.; Carloni, S.; Ravenda, P.S.; Bertalot, G.; Spadoni, I.; Lo Cascio, A.; Gandini, S.; Lizier, M.; Braga, D.; Asnicar, F.; et al. Gut vascular barrier impairment leads to intestinal bacteria dissemination and colorectal cancer metastasis to liver. Cancer Cell 2021, 39, 708–724.e11. [Google Scholar] [CrossRef]
- Golan, D.; Gross, B.; Miller, A.; Klil-Drori, S.; Lavi, I.; Shiller, M.; Honigman, S.; Almog, R.; Segol, O. Cognitive Function of Patients with Crohn’s Disease is Associated with Intestinal Disease Activity. Inflamm. Bowel Dis. 2016, 22, 364–371. [Google Scholar] [CrossRef]
- Byrne, G.; Rosenfeld, G.; Leung, Y.; Qian, H.; Raudzus, J.; Nunez, C.; Bressler, B. Prevalence of Anxiety and Depression in Patients with Inflammatory Bowel Disease. Can. J. Gastroenterol. Hepatol. 2017, 2017, 6496727. [Google Scholar] [CrossRef]
- Tadin Hadjina, I.; Zivkovic, P.M.; Matetic, A.; Rusic, D.; Vilovic, M.; Bajo, D.; Puljiz, Z.; Tonkic, A.; Bozic, J. Impaired neurocognitive and psychomotor performance in patients with inflammatory bowel disease. Sci. Rep. 2019, 9, 13740. [Google Scholar] [CrossRef] [PubMed]
- Cluny, N.L.; Nyuyki, K.D.; Almishri, W.; Griffin, L.; Lee, B.H.; Hirota, S.A.; Pittman, Q.J.; Swain, M.G.; Sharkey, K.A. Recruitment of α4β7 monocytes and neutrophils to the brain in experimental colitis is associated with elevated cytokines and anxiety-like behavior. J. Neuroinflammation 2022, 19, 73. [Google Scholar] [CrossRef] [PubMed]
- Do, J.; Woo, J. From Gut to Brain: Alteration in Inflammation Markers in the Brain of Dextran Sodium Sulfate-induced Colitis Model Mice. Clin. Psychopharmacol. Neurosci. 2018, 16, 422–433. [Google Scholar] [CrossRef] [PubMed]
- Nyuyki, K.D.; Cluny, N.L.; Swain, M.G.; Sharkey, K.A.; Pittman, Q.J. Altered Brain Excitability and Increased Anxiety in Mice With Experimental Colitis: Consideration of Hyperalgesia and Sex Differences. Front. Behav. Neurosci. 2018, 12, 58. [Google Scholar] [CrossRef] [PubMed]
- Dempsey, E.; Abautret-Daly, Á.; Docherty, N.G.; Medina, C.; Harkin, A. Persistent central inflammation and region specific cellular activation accompany depression- and anxiety-like behaviours during the resolution phase of experimental colitis. Brain Behav. Immun. 2019, 80, 616–632. [Google Scholar] [CrossRef]
- Natah, S.S.; Mouihate, A.; Pittman, Q.J.; Sharkey, K.A. Disruption of the blood-brain barrier during TNBS colitis. Neurogastroenterol. Motil. 2005, 17, 433–446. [Google Scholar] [CrossRef]
- Bernstein, C.N.; Nugent, Z.; Targownik, L.E.; Singh, H.; Lix, L.M. Predictors and risks for death in a population-based study of persons with IBD in Manitoba. Gut 2015, 64, 1403–1411. [Google Scholar] [CrossRef]
- Ludvigsson, J.F.; Holmgren, J.; Grip, O.; Halfvarson, J.; Askling, J.; Sachs, M.C.; Olén, O. Adult-onset inflammatory bowel disease and rate of serious infections compared to the general population: A nationwide register-based cohort study 2002-2017. Scand. J. Gastroenterol. 2021, 56, 1152–1162. [Google Scholar] [CrossRef]
- Ocón, B.; Aranda, C.J.; Gámez-Belmonte, R.; Suárez, M.D.; Zarzuelo, A.; Martínez-Augustin, O.; Sánchez de Medina, F. The glucocorticoid budesonide has protective and deleterious effects in experimental colitis in mice. Biochem. Pharm. 2016, 116, 73–88. [Google Scholar] [CrossRef]
- Gutiérrez, A.; Francés, R.; Amorós, A.; Zapater, P.; Garmendia, M.; Ndongo, M.; Cano, R.; Jover, R.; Such, J.; Perez-Mateo, M. Cytokine association with bacterial DNA in serum of patients with inflammatory bowel disease. Inflamm. Bowel Dis. 2009, 15, 508–514. [Google Scholar] [CrossRef] [PubMed]
- Gutiérrez, A.; Zapater, P.; Juanola, O.; Sempere, L.; Garcia, M.; Laveda, R.; Martinez, A.; Scharl, M.; Gonzalez-Navajas, J.M.; Such, J.; et al. Gut Bacterial DNA Translocation is an Independent Risk Factor of Flare at Short Term in Patients With Crohn’s Disease. Am. J. Gastroenterol. 2016, 111, 529–540. [Google Scholar] [CrossRef] [PubMed]
- Pasternak, B.A.; D’Mello, S.; Jurickova, I.I.; Han, X.; Willson, T.; Flick, L.; Petiniot, L.; Uozumi, N.; Divanovic, S.; Traurnicht, A.; et al. Lipopolysaccharide exposure is linked to activation of the acute phase response and growth failure in pediatric Crohn’s disease and murine colitis. Inflamm. Bowel Dis. 2010, 16, 856–869. [Google Scholar] [CrossRef] [PubMed]
- Rojo, Ó.P.; Román, A.L.S.; Arbizu, E.A.; de la Hera Martínez, A.; Sevillano, E.R.; Martínez, A.A. Serum lipopolysaccharide-binding protein in endotoxemic patients with inflammatory bowel disease. Inflamm. Bowel Dis. 2006, 13, 269–277. [Google Scholar] [CrossRef] [PubMed]
- Luchetti, M.M.; Ciccia, F.; Avellini, C.; Benfaremo, D.; Rizzo, A.; Spadoni, T.; Svegliati, S.; Marzioni, D.; Santinelli, A.; Costantini, A.; et al. Gut epithelial impairment, microbial translocation and immune system activation in inflammatory bowel disease-associated spondyloarthritis. Rheumatology 2021, 60, 92–102. [Google Scholar] [CrossRef]
- Pastorelli, L.; Dozio, E.; Pisani, L.F.; Boscolo-Anzoletti, M.; Vianello, E.; Munizio, N.; Spina, L.; Tontini, G.E.; Peyvandi, F.; Corsi Romanelli, M.M.; et al. Procoagulatory state in inflammatory bowel diseases is promoted by impaired intestinal barrier function. Gastroenterol. Res. Pract. 2015, 2015, 189341. [Google Scholar] [CrossRef]
- Tulkens, J.; Vergauwen, G.; Van Deun, J.; Geeurickx, E.; Dhondt, B.; Lippens, L.; De Scheerder, M.A.; Miinalainen, I.; Rappu, P.; De Geest, B.G.; et al. Increased levels of systemic LPS-positive bacterial extracellular vesicles in patients with intestinal barrier dysfunction. Gut 2020, 69, 191–193. [Google Scholar] [CrossRef]
- Marques, F.; Sousa, J.C.; Coppola, G.; Falcao, A.M.; Rodrigues, A.J.; Geschwind, D.H.; Sousa, N.; Correia-Neves, M.; Palha, J.A. Kinetic profile of the transcriptome changes induced in the choroid plexus by peripheral inflammation. J. Cereb. Blood Flow Metab. 2009, 29, 921–932. [Google Scholar] [CrossRef]
- Marques, F.; Sousa, J.C.; Coppola, G.; Geschwind, D.H.; Sousa, N.; Palha, J.A.; Correia-Neves, M. The choroid plexus response to a repeated peripheral inflammatory stimulus. BMC Neurosci. 2009, 10, 135. [Google Scholar] [CrossRef]
- Neuendorf, R.; Harding, A.; Stello, N.; Hanes, D.; Wahbeh, H. Depression and anxiety in patients with Inflammatory Bowel Disease: A systematic review. J. Psychosom. Res. 2016, 87, 70–80. [Google Scholar] [CrossRef]
- Aloi, M.; Tromba, L.; Di Nardo, G.; Dilillo, A.; Del Giudice, E.; Marocchi, E.; Viola, F.; Civitelli, F.; Berni, A.; Cucchiara, S. Premature subclinical atherosclerosis in pediatric inflammatory bowel disease. J. Pediatr. 2012, 161, 589–594.e1. [Google Scholar] [CrossRef] [PubMed]
- Aloi, M.; Tromba, L.; Rizzo, V.; D’Arcangelo, G.; Dilillo, A.; Blasi, S.; Civitelli, F.; Kiltzanidi, D.; Redler, A.; Viola, F. Aortic Intima-Media Thickness as an Early Marker of Atherosclerosis in Children With Inflammatory Bowel Disease. J. Pediatr. Gastroenterol. Nutr. 2015, 61, 41–46. [Google Scholar] [CrossRef] [PubMed]
- Caliskan, Z.; Gokturk, H.S.; Caliskan, M.; Gullu, H.; Ciftci, O.; Ozgur, G.T.; Guven, A.; Selcuk, H. Impaired coronary microvascular and left ventricular diastolic function in patients with inflammatory bowel disease. Microvasc. Res. 2015, 97, 25–30. [Google Scholar] [CrossRef]
- Cappello, M.; Licata, A.; Calvaruso, V.; Bravatà, I.; Aiello, A.; Torres, D.; Della Corte, V.; Tuttolomondo, A.; Perticone, M.; Licata, G.; et al. Increased expression of markers of early atherosclerosis in patients with inflammatory bowel disease. Eur. J. Intern. Med. 2017, 37, 83–89. [Google Scholar] [CrossRef] [PubMed]
- Erolu, E.; Polat, E. Cardiac functions and aortic elasticity in children with inflammatory bowel disease: Effect of age at disease onset. Cardiol. Young 2020, 30, 313–317. [Google Scholar] [CrossRef]
- Triantafyllou, C.; Nikolaou, M.; Ikonomidis, I.; Bamias, G.; Papaconstantinou, I. Endothelial and Cardiac Dysfunction in Inflammatory Bowel Diseases: Does Treatment Modify the Inflammatory Load on Arterial and Cardiac Structure and Function? Curr. Vasc. Pharmacol. 2020, 18, 27–37. [Google Scholar] [CrossRef]
- Kakuta, K.; Dohi, K.; Yamamoto, T.; Fujimoto, N.; Shimoyama, T.; Umegae, S.; Ito, M. Coronary Microvascular Dysfunction Restored After Surgery in Inflammatory Bowel Disease: A Prospective Observational Study. J. Am. Heart Assoc. 2021, 10, e019125. [Google Scholar] [CrossRef]
- Sy, A.; Khalidi, N.; Dehghan, N.; Barra, L.; Carette, S.; Cuthbertson, D.; Hoffman, G.S.; Koening, C.L.; Langford, C.A.; McAlear, C.; et al. Vasculitis in patients with inflammatory bowel diseases: A study of 32 patients and systematic review of the literature. Semin. Arthritis Rheum. 2016, 45, 475–482. [Google Scholar] [CrossRef]
- Ho, T.; Orenstein, L.A.V.; Boos, M.D.; White, K.P.; Fett, N. Cutaneous Small-Vessel Vasculitis in Two Children with Inflammatory Bowel Disease: Case Series and Review of the Literature. Pediatr. Dermatol. 2017, 34, e235–e240. [Google Scholar] [CrossRef]
- Romas, E.; Paspaliaris, B.; d’Apice, A.J.; Elliott, P.R. Autoantibodies to neutrophil cytoplasmic (ANCA) and endothelial cell surface antigens (AECA) in chronic inflammatory bowel disease. Aust. N. Z. J. Med. 1992, 22, 652–659. [Google Scholar] [CrossRef]
- Stevens, T.R.; Harley, S.L.; Groom, J.S.; Cambridge, G.; Leaker, B.; Blake, D.R.; Rampton, D.S. Anti-endothelial cell antibodies in inflammatory bowel disease. Dig. Dis. Sci. 1993, 38, 426–432. [Google Scholar] [CrossRef] [PubMed]
- Aldebert, D.; Notteghem, B.; Reumaux, D.; Lassalle, P.; Lion, G.; Desreumaux, P.; Duthilleul, P.; Colombel, J.F. Anti-endothelial cell antibodies in sera from patients with inflammatory bowel disease. Gastroenterol. Clin. Biol. 1995, 19, 867–870. [Google Scholar] [PubMed]
- Agnoletti, D.; Piani, F.; Cicero, A.F.G.; Borghi, C. The Gut Microbiota and Vascular Aging: A State-of-the-Art and Systematic Review of the Literature. J. Clin. Med. 2022, 11, 3557. [Google Scholar] [CrossRef] [PubMed]
- Yoshida, H.; Russell, J.; Granger, D.N. Thrombin mediates the extraintestinal thrombosis associated with experimental colitis. Am. J. Physiol. Gastrointest. Liver Physiol. 2008, 295, G904–G908. [Google Scholar] [CrossRef]
- Papay, P.; Miehsler, W.; Tilg, H.; Petritsch, W.; Reinisch, W.; Mayer, A.; Haas, T.; Kaser, A.; Feichtenschlager, T.; Fuchssteiner, H.; et al. Clinical presentation of venous thromboembolism in inflammatory bowel disease. J. Crohn’s Colitis 2013, 7, 723–729. [Google Scholar] [CrossRef]
- Di Fabio, F.; Obrand, D.; Satin, R.; Gordon, P.H. Intra-abdominal venous and arterial thromboembolism in inflammatory bowel disease. Dis. Colon Rectum 2009, 52, 336–342. [Google Scholar] [CrossRef]
- Cognat, E.; Crassard, I.; Denier, C.; Vahedi, K.; Bousser, M.G. Cerebral venous thrombosis in inflammatory bowel diseases: Eight cases and literature review. Int. J. Stroke 2011, 6, 487–492. [Google Scholar] [CrossRef]
- Awab, A.; Elahmadi, B.; Elmoussaoui, R.; Elhijri, A.; Alilou, M.; Azzouzi, A. Cerebral venous thrombosis and inflammatory bowel disease: Reflections on pathogenesis. Colorectal. Dis. 2012, 14, 1153–1154. [Google Scholar] [CrossRef]
- Tan, V.P.; Chung, A.; Yan, B.P.; Gibson, P.R. Venous and arterial disease in inflammatory bowel disease. J. Gastroenterol. Hepatol. 2013, 28, 1095–1113. [Google Scholar] [CrossRef]
- Principi, M.; Mastrolonardo, M.; Scicchitano, P.; Gesualdo, M.; Sassara, M.; Guida, P.; Bucci, A.; Zito, A.; Caputo, P.; Albano, F.; et al. Endothelial function and cardiovascular risk in active inflammatory bowel diseases. J. Crohn’s Colitis 2013, 7, e427–e433. [Google Scholar] [CrossRef]
- Orfei, M.; Gasparetto, M.; Torrente, F. Headache and inflammatory bowel disease: Think cerebral vein! BMJ Case Rep. 2019, 12, e227228. [Google Scholar] [CrossRef] [PubMed]
- Rohani, P.; Taraghikhah, N.; Nasehi, M.M.; Alimadadi, H.; Assadzadeh Aghdaei, H. Cerebrovascular Events in Pediatric Inflammatory Bowel Disease: A Review of Published Cases. Pediatr. Gastroenterol. Hepatol. Nutr. 2022, 25, 180–193. [Google Scholar] [CrossRef] [PubMed]
- Carty, E.; MacEy, M.; Rampton, D.S. Inhibition of platelet activation by 5-aminosalicylic acid in inflammatory bowel disease. Aliment. Pharmacol. Ther. 2000, 14, 1169–1179. [Google Scholar] [CrossRef] [PubMed]
- Hommes, D.W.; van Dullemen, H.M.; Levi, M.; van der Ende, A.; Woody, J.; Tytgat, G.N.; van Deventer, S.J. Beneficial effect of treatment with a monoclonal anti-tumor necrosis factor-alpha antibody on markers of coagulation and fibrinolysis in patients with active Crohn’s disease. Haemostasis 1997, 27, 269–277. [Google Scholar] [CrossRef]
- Schinzari, F.; Armuzzi, A.; De Pascalis, B.; Mores, N.; Tesauro, M.; Melina, D.; Cardillo, C. Tumor necrosis factor-alpha antagonism improves endothelial dysfunction in patients with Crohn’s disease. Clin. Pharmacol. Ther. 2008, 83, 70–76. [Google Scholar] [CrossRef]
- Danese, S.; Sans, M.; Scaldaferri, F.; Sgambato, A.; Rutella, S.; Cittadini, A.; Piqué, J.M.; Panes, J.; Katz, J.A.; Gasbarrini, A.; et al. TNF-alpha blockade down-regulates the CD40/CD40L pathway in the mucosal microcirculation: A novel anti-inflammatory mechanism of infliximab in Crohn’s disease. J. Immunol. 2006, 176, 2617–2624. [Google Scholar] [CrossRef]
- Löwenberg, M.; D’Haens, G. Next-Generation Therapeutics for IBD. Curr. Gastroenterol. Rep. 2015, 17, 21. [Google Scholar] [CrossRef]
- Neurath, M.F. Current and emerging therapeutic targets for IBD. Nat. Rev. Gastroenterol. Hepatol. 2017, 14, 269–278. [Google Scholar] [CrossRef]
- Park, S.C.; Jeen, Y.T. Anti-integrin therapy for inflammatory bowel disease. World J. Gastroenterol. 2018, 24, 1868–1880. [Google Scholar] [CrossRef]
- Feagan, B.G.; Rutgeerts, P.; Sands, B.E.; Hanauer, S.; Colombel, J.F.; Sandborn, W.J.; Van Assche, G.; Axler, J.; Kim, H.J.; Danese, S.; et al. Vedolizumab as induction and maintenance therapy for ulcerative colitis. N. Engl. J. Med. 2013, 369, 699–710. [Google Scholar] [CrossRef]
- Sandborn, W.J.; Feagan, B.G.; Rutgeerts, P.; Hanauer, S.; Colombel, J.F.; Sands, B.E.; Lukas, M.; Fedorak, R.N.; Lee, S.; Bressler, B.; et al. Vedolizumab as induction and maintenance therapy for Crohn’s disease. N. Engl. J. Med. 2013, 369, 711–721. [Google Scholar] [CrossRef] [PubMed]
- Zundler, S.; Schillinger, D.; Fischer, A.; Atreya, R.; Lopez-Posadas, R.; Watson, A.; Neufert, C.; Atreya, I.; Neurath, M.F. Blockade of alphaEbeta7 integrin suppresses accumulation of CD8+ and Th9 lymphocytes from patients with IBD in the inflamed gut in vivo. Gut 2017, 66, 1936–1948. [Google Scholar] [CrossRef] [PubMed]
- Picardo, S.; Panaccione, R. Anti-MADCAM therapy for ulcerative colitis. Expert Opin. Biol. Ther. 2020, 20, 437–442. [Google Scholar] [CrossRef] [PubMed]
- Reinisch, W.; Sandborn, W.J.; Danese, S.; Hébuterne, X.; Kłopocka, M.; Tarabar, D.; Vaňásek, T.; Greguš, M.; Hellstern, P.A.; Kim, J.S.; et al. Long-term Safety and Efficacy of the Anti-MAdCAM-1 Monoclonal Antibody Ontamalimab [SHP647] for the Treatment of Ulcerative Colitis: The Open-label Study TURANDOT II. J. Crohn’s Colitis 2021, 15, 938–949. [Google Scholar] [CrossRef]
- Lightner, A.L.; Sklow, B.; Click, B.; Regueiro, M.; McMichael, J.J.; Jia, X.; Vaidya, P.; Delaney, C.P.; Cohen, B.; Wexner, S.D.; et al. Venous Thromboembolism in Admitted Patients with Inflammatory Bowel Disease: An Enterprise—Wide Experience of 86,000 Hospital Encounters. Dis. Colon Rectum 2022, 66, 410–418. [Google Scholar] [CrossRef]
- Chande, N.; Wang, Y.; McDonald, J.W.; MacDonald, J.K. Unfractionated or low-molecular weight heparin for induction of remission in ulcerative colitis. Cochrane Database Syst. Rev. 2015, 8, Cd006774. [Google Scholar] [CrossRef]
- Hong, L.; Chen, G.; Cai, Z.; Liu, H.; Zhang, C.; Wang, F.; Xiao, Z.; Zhong, J.; Wang, L.; Wang, Z.; et al. Balancing Microthrombosis and Inflammation via Injectable Protein Hydrogel for Inflammatory Bowel Disease. Adv. Sci. 2022, 9, e2200281. [Google Scholar] [CrossRef]
- Prajapati, D.N.; Newcomer, J.R.; Emmons, J.; Abu-Hajir, M.; Binion, D.G. Successful treatment of an acute flare of steroid-resistant Crohn’s colitis during pregnancy with unfractionated heparin. Inflamm. Bowel Dis. 2002, 8, 192–195. [Google Scholar] [CrossRef]
- Chande, N.; MacDonald, J.K.; Wang, J.J.; McDonald, J.W. Unfractionated or low molecular weight heparin for induction of remission in ulcerative colitis: A Cochrane inflammatory bowel disease and functional bowel disorders systematic review of randomized trials. Inflamm. Bowel Dis. 2011, 17, 1979–1986. [Google Scholar] [CrossRef]
- Day, R.; Forbes, A. Heparin, cell adhesion, and pathogenesis of inflammatory bowel disease. Lancet 1999, 354, 62–65. [Google Scholar] [CrossRef]
- Papa, A.; Danese, S.; Gasbarrini, A.; Gasbarrini, G. Review article: Potential therapeutic applications and mechanisms of action of heparin in inflammatory bowel disease. Aliment. Pharmacol. Ther. 2000, 14, 1403–1409. [Google Scholar] [CrossRef] [PubMed]
- Yazeji, T.; Moulari, B.; Beduneau, A.; Stein, V.; Dietrich, D.; Pellequer, Y.; Lamprecht, A. Nanoparticle-based delivery enhances anti-inflammatory effect of low molecular weight heparin in experimental ulcerative colitis. Drug Deliv. 2017, 24, 811–817. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S.; Cho, W.J.; Jin, A.T.; Kok, L.Y.; Shi, Y.; Heller, D.E.; Lee, Y.L.; Zhou, Y.; Xie, X.; Korzenik, J.R.; et al. Heparin-Coated Albumin Nanoparticles for Drug Combination in Targeting Inflamed Intestine. Adv. Healthc. Mater. 2020, 9, e2000536. [Google Scholar] [CrossRef]
- Wang, S.Y.; Tao, P.; Hu, H.Y.; Yuan, J.Y.; Zhao, L.; Sun, B.Y.; Zhang, W.J.; Lin, J. Effects of initiating time and dosage of Panax notoginseng on mucosal microvascular injury in experimental colitis. World J. Gastroenterol. 2017, 23, 8308–8320. [Google Scholar] [CrossRef]
- Zhang, X.; Song, L.; Li, L.; Zhu, B.; Huo, L.; Hu, Z.; Wang, X.; Wang, J.; Gao, M.; Zhang, J.; et al. Phosphatidylserine externalized on the colonic capillaries as a novel pharmacological target for IBD therapy. Signal Transduct. Target. Ther. 2021, 6, 235. [Google Scholar] [CrossRef] [PubMed]
- Adzemovic, M.V.; Zeitelhofer, M.; Eriksson, U.; Olsson, T.; Nilsson, I. Imatinib ameliorates neuroinflammation in a rat model of multiple sclerosis by enhancing blood-brain barrier integrity and by modulating the peripheral immune response. PLoS ONE 2013, 8, e56586. [Google Scholar] [CrossRef]
- Aman, J.; van Bezu, J.; Damanafshan, A.; Huveneers, S.; Eringa, E.C.; Vogel, S.M.; Groeneveld, A.B.; Vonk Noordegraaf, A.; van Hinsbergh, V.W.; van Nieuw Amerongen, G.P. Effective treatment of edema and endothelial barrier dysfunction with imatinib. Circulation 2012, 126, 2728–2738. [Google Scholar] [CrossRef] [PubMed]
- Hosaka, K.; Yang, Y.; Seki, T.; Nakamura, M.; Andersson, P.; Rouhi, P.; Yang, X.; Jensen, L.; Lim, S.; Feng, N.; et al. Tumour PDGF-BB expression levels determine dual effects of anti-PDGF drugs on vascular remodelling and metastasis. Nat. Commun. 2013, 4, 2129. [Google Scholar] [CrossRef] [PubMed]
- Magro, F.; Costa, C. Long-standing remission of Crohn’s disease under imatinib therapy in a patient with Crohn’s disease. Inflamm. Bowel Dis. 2006, 12, 1087–1089. [Google Scholar] [CrossRef]
- Gonzalez-Cabrera, P.J.; Brown, S.; Studer, S.M.; Rosen, H. S1P signaling: New therapies and opportunities. F1000Prime Rep. 2014, 6, 109. [Google Scholar] [CrossRef]
- Verstockt, B.; Vetrano, S.; Salas, A.; Nayeri, S.; Duijvestein, M.; Vande Casteele, N. Sphingosine 1-phosphate modulation and immune cell trafficking in inflammatory bowel disease. Nat. Rev. Gastroenterol. Hepatol. 2022, 19, 351–366. [Google Scholar] [CrossRef] [PubMed]
- Obinata, H.; Hla, T. Sphingosine 1-phosphate and inflammation. Int. Immunol. 2019, 31, 617–625. [Google Scholar] [CrossRef] [PubMed]
- Oo, M.L.; Chang, S.H.; Thangada, S.; Wu, M.T.; Rezaul, K.; Blaho, V.; Hwang, S.I.; Han, D.K.; Hla, T. Engagement of S1P₁-degradative mechanisms leads to vascular leak in mice. J. Clin. Investig. 2011, 121, 2290–2300. [Google Scholar] [CrossRef] [PubMed]
- Jeya Paul, J.; Weigel, C.; Müller, T.; Heller, R.; Spiegel, S.; Gräler, M.H. Inflammatory Conditions Disrupt Constitutive Endothelial Cell Barrier Stabilization by Alleviating Autonomous Secretion of Sphingosine 1-Phosphate. Cells 2020, 9, 928. [Google Scholar] [CrossRef]
- Ziegler, A.C.; Gräler, M.H. Barrier maintenance by S1P during inflammation and sepsis. Tissue Barriers 2021, 9, 1940069. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
| Cell Type | In Homeostasis | In IBD |
|---|---|---|
| HIMECs | Tolerance to bacterial products from the gut microbiota Constitutive iNOS expression | Leukocyte hyperadhesion Angiogenesis ↑ ανβ3 integrin expression ↑ vessel permeability ↓ iNOS and eNOS expression ↓ protein C system activation ↑ secretion of inflammatory mediators |
| HEVs | Recruitment and trafficking of lymphocytes from blood to lymph nodes and secondary lymphoid organs | ↑ density ↑ leukocyte binding Formation of extrafollicular HEVs |
| Lymphatic ECs | Absorption of fatty acids Immune regulation | ↑ density ↓ contractile activity Lymphangitis |
| Mural cells | Development and maintenance of the GVB | ↓ vessel coverage ↑ MMP expression |
| Enteric glial cells | Part of gut–vascular units Development and maintenance of the GVB | Sensing of bacterial translocation Closure of the PVB |
| Cytokines | Effect |
|---|---|
| IFN-γ | EC activation, ↑ CAM expression (notably MadCAM-1) ↑ vascular permeability Disassembly of VE-cadherin junctions, ↓ VE-cadherin expression ↓ EC proliferation and migration, ↓ angiogenesis ↓ vascular coverage, ↓ PDGF-B ↑ TLR3 expression ↑ CX3CL1 (fractalkine) |
| TNF-α | EC activation, ↑ CAM expression (notably MadCAM-1) ↑ vascular permeability, ↑ Phosphorylation of VE-cadherin, ↑ monolayer tension ↓ TJ protein expression in EC ↑ circulating levels, ↑ vascular dysfunction ↓ EC proliferation and migration, ↓ angiogenesis ↑ CX3CL1 (fractalkine) |
| IL-1β | EC activation, ↑ CAM expression (notably MadCAM-1) ↑ CX3CL1 (fractalkine) |
| Factor | Effect on the Intestinal Vasculature |
|---|---|
| Angiogenic growth factors | |
| VEGF | Expression increased in IBD ↑ sprouting angiogenesis ↑ EC proliferation and migration ↑ ICAM-1 and VCAM-1 expression ↑ recruitment of VEGFR- expressing immune cells ↑ vascular permeability, disassembly of VE-cadherin junctions ↓ vascular coverage ↑ wound healing |
| bFGF | Expression increased in IBD ↑ sprouting angiogenesis |
| PDGF | Expression increased in IBD ↑ sprouting angiogenesis ↑ vascular coverage Protective effect in UC |
| Nitric oxide (NO) | Decreased constitutive expression of eNOS and iNOS in intestinal EC during IBD ↓ CAM expression ↓ ROS production ↑ intestinal endothelial barrier function ↑ vasodilatation Potentiates VEGF-mediated effects |
| Coagulation factors | Increased platelet activation and thrombi in IBD ↓ thrombomodulin expression in IBD ↓ protein C receptor expression in IBD Impaired protein C activation in activated intestinal EC |
| Toll-like receptors (TLR) | Tolerance to endotoxin in intestinal EC Expression of TLR3 and TLR5 by intestinal EC, protective against colitis in mice ↑ endothelial barrier function |
| Angiocrine factors | ↑ CX3CL1 (fractalkine) → ↑ adhesion and activation of CX3CR1+ leukocytes ↑ CL25 → recruitment of CCR9+ immune cells → protective effect ↑ NO ↑ CXCL10 → epithelial cell survival |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Britzen-Laurent, N.; Weidinger, C.; Stürzl, M. Contribution of Blood Vessel Activation, Remodeling and Barrier Function to Inflammatory Bowel Diseases. Int. J. Mol. Sci. 2023, 24, 5517. https://doi.org/10.3390/ijms24065517
Britzen-Laurent N, Weidinger C, Stürzl M. Contribution of Blood Vessel Activation, Remodeling and Barrier Function to Inflammatory Bowel Diseases. International Journal of Molecular Sciences. 2023; 24(6):5517. https://doi.org/10.3390/ijms24065517
Chicago/Turabian StyleBritzen-Laurent, Nathalie, Carl Weidinger, and Michael Stürzl. 2023. "Contribution of Blood Vessel Activation, Remodeling and Barrier Function to Inflammatory Bowel Diseases" International Journal of Molecular Sciences 24, no. 6: 5517. https://doi.org/10.3390/ijms24065517
APA StyleBritzen-Laurent, N., Weidinger, C., & Stürzl, M. (2023). Contribution of Blood Vessel Activation, Remodeling and Barrier Function to Inflammatory Bowel Diseases. International Journal of Molecular Sciences, 24(6), 5517. https://doi.org/10.3390/ijms24065517