
ACAT1/SOAT1 Blockade Suppresses LPS-Mediated Neuroinflammation by Modulating the Fate of Toll-like Receptor 4 in Microglia

 , and
, and {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
2.1. ACAT1/SOAT1 Gene Expression Is Elevated in Neurodegenerative Diseases and in Acute Neuroinflammation
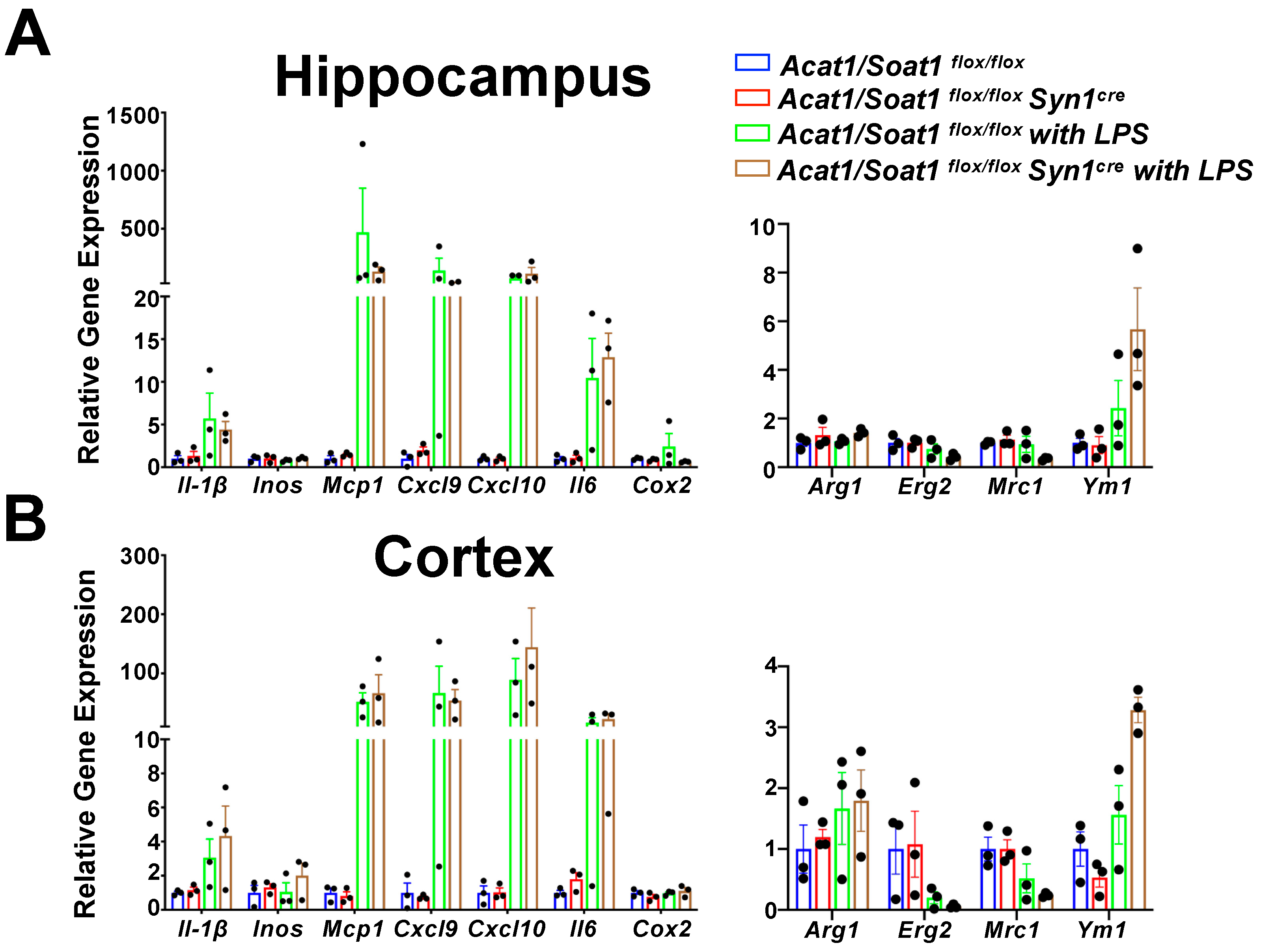
2.2. Blocking ACAT1/SOAT1 in Myeloid Attenuated LPS-Mediated Acute Neuroinflammation in the CNS
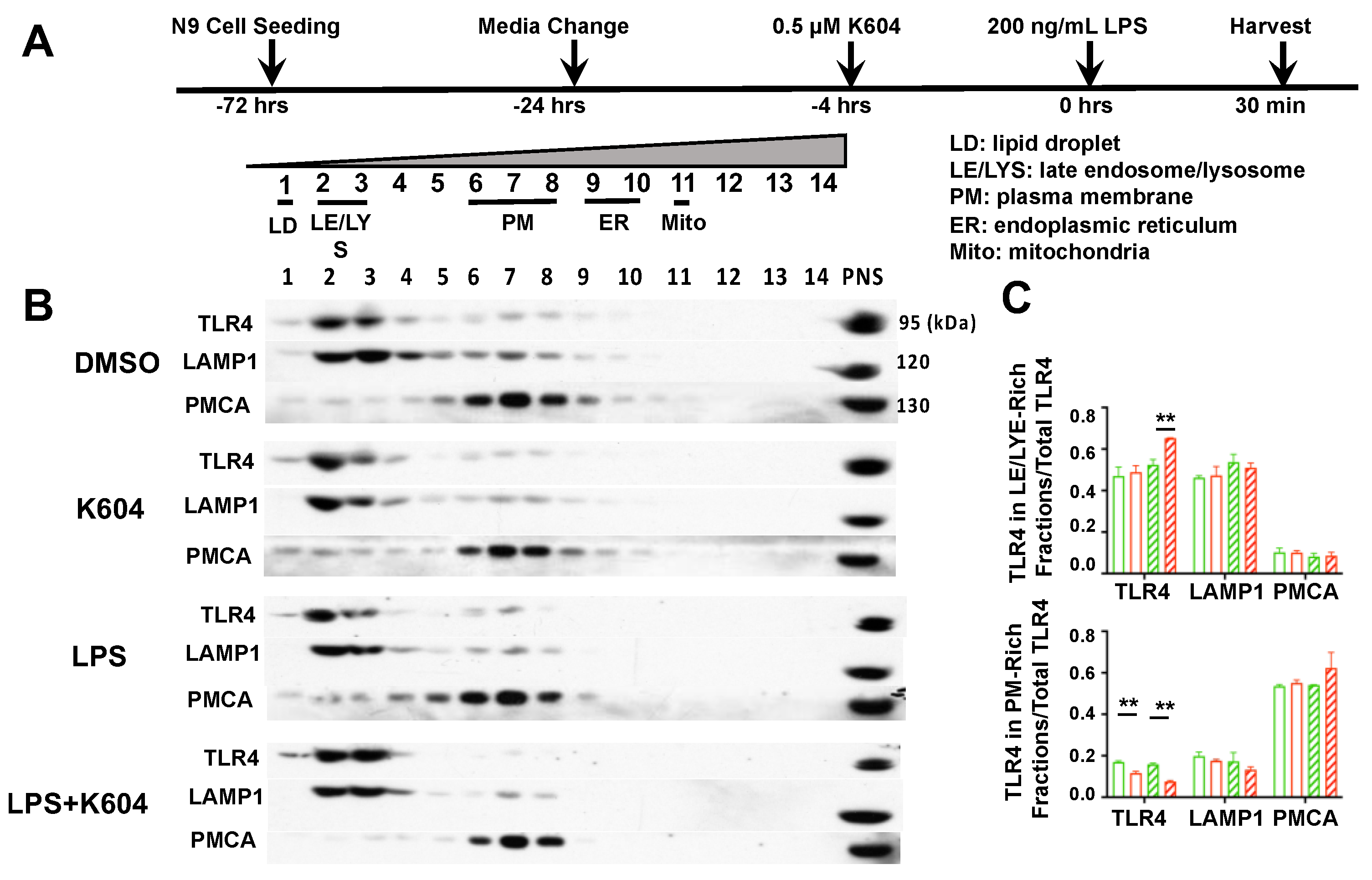
2.3. Blocking ACAT1/SOAT1 with ACAT1/SOAT1-Specific Small Molecule Inhibitor K-604 Attenuated the Pro-Inflammatory Responses of LPS-Treated N9 Microglial Cells through TLR4-Mediated Pathway
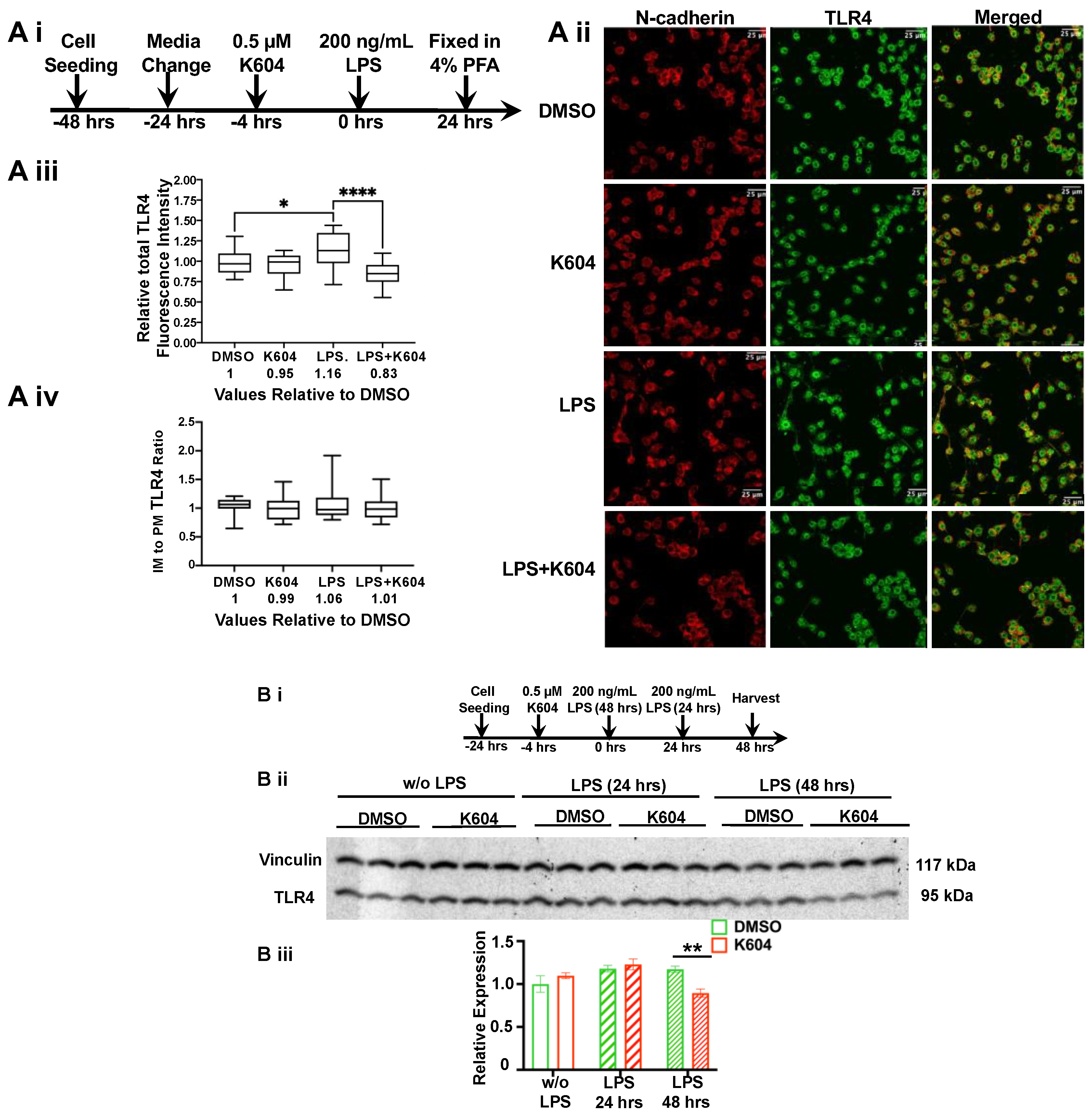
2.4. Blocking ACAT1/SOAT1 with ACAT1/SOAT1-Specific Small Molecule Inhibitor K-604 Alters the Fate of TLR4 in N9 Microglial Cells
2.5. A1B Decreases TLR4 Protein Content in Microglial N9 Cells Chronically Treated with LPS (for 24–48 h)
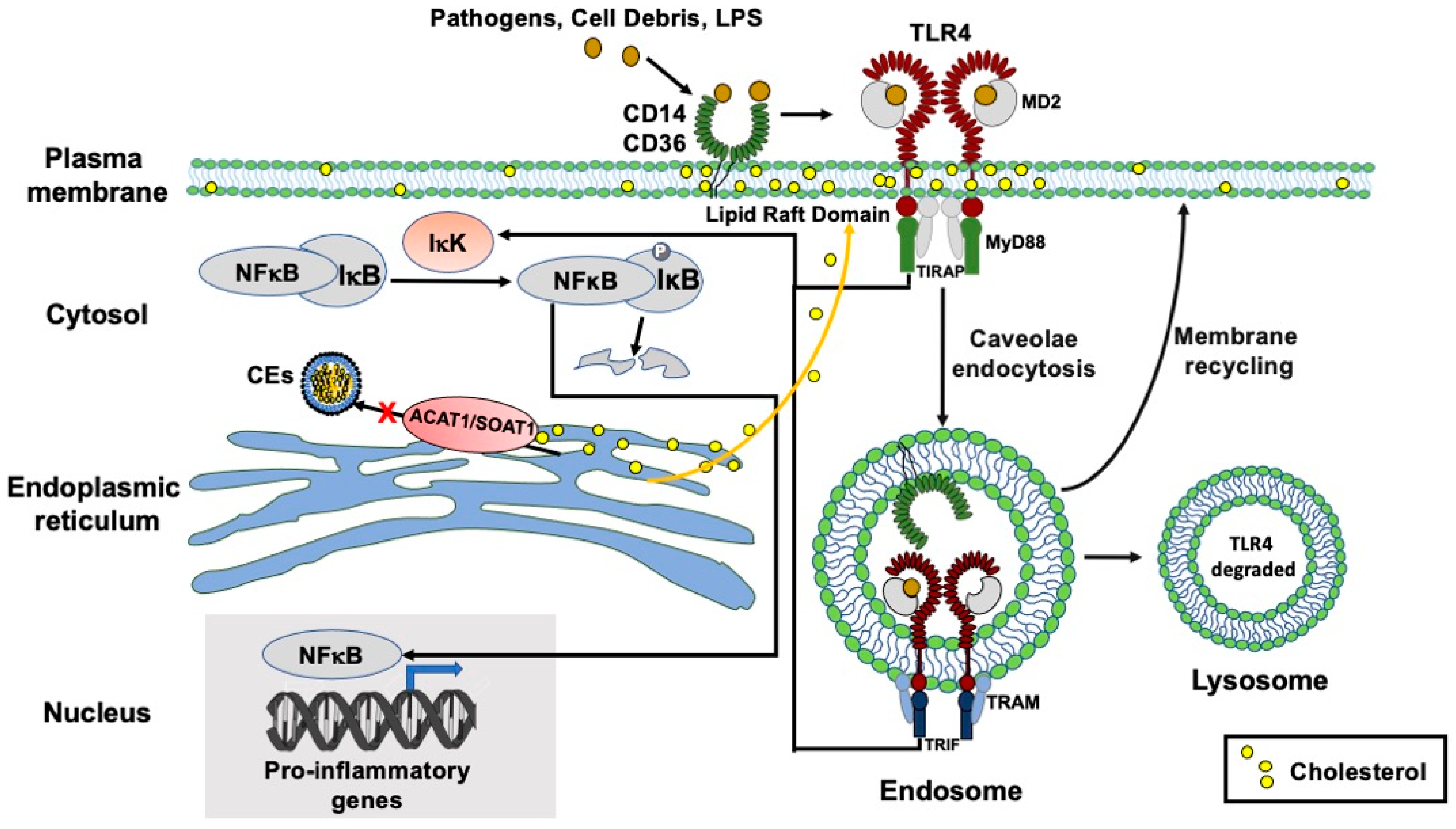
3. Discussion
4. Materials and Methods
4.1. Animals and Acute Neuroinflammation Induction
4.2. Cell Culture
4.3. RNA Isolation and qPCR
- β-actin forward: 5′-CAACGAGCGGTTCCGAT-3′, reverse: 5′-GCCACAGGATTCCATACCCA-3′;
- il-1β forward: 5′-ACAGAATATCAACCAACAAGTGATATT-3′, reverse: 5′-GATTCTTTCCTTTGAGGCCCA-3′;
- inos forward: 5′-ACATCGACCCGTCCACAGTAT-3′, reverse: 5′-CAGAGGGGTAGGCTTGTCTC-3′;
- mcp1 forward: 5′-CCCACTCACCTGCTGCTACT-3′, reverse: 5′-TCTGGACCCATTCCTTCTTG-3′;
- cxcl9 forward: 5′-GGAGTTCGAGGAACCCTAGTG-3′, reverse: 5′-GGGATTTGTAGTGGATCGTGC-3′;
- cxcl10 forward: 5′-CCAAGTGCTGCCGTCATTTTC-3′, reverse: 5′-GGCTCGCAGGGATGATTTCAA-3′;
- il6 forward: 5′-TCCAGTTGCCTTCTTGGGAC-3′, reverse: 5′-GTACTCCAGAAGACCAGAGG-3′;
- cox2 forward: 5′-AGCAACCCGGCCAGCAATCT-3′, reverse: 5′-CCTGCTGCCCGACACCTTCA-3′;
- arg1 forward: 5′-CTCCAAGCCAAAGTCCTTAGAG-3′, reverse: 5′-AGGAGCTGTCATTAGGGACATC-3′;
- erg2 forward: 5′-GCAAAGGACCTTGATGGAGC-3′, reverse: 5′-GGCCTAAGTTTTCGGAAGGC-3′;
- mrc1 forward: 5′-CTCTGTTCAGCTATTGGACGC-3′, reverse: 5′-CGGAATTTCTGGGATTCAGCTTC-3′;
- ym1 forward: 5′-CATGAGCAAGACTTGCGTGAC-3′, reverse: 5′-GGTCCAAACTTCCATCCTCCA-3′
4.4. OptiPrepTM Subcellular Fractionation
4.5. Whole Cell Protein Isolation and Western Blot Analyses
4.6. TLR4 Endocytosis Assay
4.7. Immunofluorescence Microscopy
4.8. K-604
4.9. LPS
4.10. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Luo, J.; Yang, H.; Song, B.L. Mechanisms and regulation of cholesterol homeostasis. Nat. Rev. Mol. Cell Biol. 2020, 21, 225–245. [Google Scholar] [CrossRef]
- Dietschy, J.M.; Turley, S.D. Thematic review series: Brain Lipids. Cholesterol metabolism in the central nervous system during early development and in the mature animal. J. Lipid Res. 2004, 45, 1375–1397. [Google Scholar] [CrossRef]
- Bjorkhem, I.; Meaney, S. Brain cholesterol: Long secret life behind a barrier. Arterioscler Thromb. Vasc. Biol. 2004, 24, 806–815. [Google Scholar] [CrossRef]
- Di Paolo, G.; Kim, T.W. Linking lipids to Alzheimer’s disease: Cholesterol and beyond. Nat. Rev. Neurosci. 2011, 12, 284–296. [Google Scholar] [CrossRef]
- Chang, T.Y.; Yamauchi, Y.; Hasan, M.T.; Chang, C. Cellular cholesterol homeostasis and Alzheimer’s disease. J. Lipid Res. 2017, 58, 2239–2254. [Google Scholar] [CrossRef] [PubMed]
- Chang, C.C.; Huh, H.Y.; Cadigan, K.M.; Chang, T.Y. Molecular cloning and functional expression of human acyl-coenzyme A:cholesterol acyltransferase cDNA in mutant Chinese hamster ovary cells. J. Biol. Chem. 1993, 268, 20747–20755. [Google Scholar] [CrossRef] [PubMed]
- Anderson, R.A.; Joyce, C.; Davis, M.; Reagan, J.W.; Clark, M.; Shelness, G.S.; Rudel, L.L. Identification of a form of acyl-CoA:cholesterol acyltransferase specific to liver and intestine in nonhuman primates. J. Biol. Chem. 1998, 273, 26747–26754. [Google Scholar] [CrossRef]
- Cases, S.; Smith, S.J.; Zheng, Y.W.; Myers, H.M.; Lear, S.R.; Sande, E.; Novak, S.; Collins, C.; Welch, C.B.; Lusis, A.J.; et al. Identification of a gene encoding an acyl CoA:diacylglycerol acyltransferase, a key enzyme in triacylglycerol synthesis. Proc. Natl. Acad. Sci. USA 1998, 95, 13018–13023. [Google Scholar] [CrossRef] [PubMed]
- Oelkers, P.; Behari, A.; Cromley, D.; Billheimer, J.T.; Sturley, S.L. Characterization of two human genes encoding acyl coenzyme A:cholesterol acyltransferase-related enzymes. J. Biol. Chem. 1998, 273, 26765–26771. [Google Scholar] [CrossRef]
- Liu, J.; Chang, C.C.; Westover, E.J.; Covey, D.F.; Chang, T.Y. Investigating the allosterism of Acyl Coenzyme A: Cholesterol acyltransferase (ACAT) by using various sterols: In vitro and intact cell studies. Biochem. J. 2005, 391, 389–397. [Google Scholar] [CrossRef] [PubMed]
- Chang, T.Y.; Li, B.L.; Chang, C.C.; Urano, Y. Acyl-coenzyme A:cholesterol acyltransferases. Am. J. Physiol. Endocrinol. Metab. 2009, 297, E1–E9. [Google Scholar] [CrossRef] [PubMed]
- Rogers, M.A.; Liu, J.; Song, B.L.; Li, B.L.; Chang, C.C.; Chang, T.Y. Acyl-CoA:cholesterol acyltransferases (ACATs/SOATs): Enzymes with multiple sterols as substrates and as activators. J. Steroid Biochem. Mol. Biol. 2015, 151, 102–107. [Google Scholar] [CrossRef]
- Chan, R.B.; Oliveira, T.G.; Cortes, E.P.; Honig, L.S.; Duff, K.E.; Small, S.A.; Wenk, M.R.; Shui, G.; Di Paolo, G. Comparative lipidomic analysis of mouse and human brain with Alzheimer disease. J. Biol. Chem. 2012, 287, 2678–2688. [Google Scholar] [CrossRef] [PubMed]
- Tajima, Y.; Ishikawa, M.; Maekawa, K.; Murayama, M.; Senoo, Y.; Nishimaki-Mogami, T.; Nakanishi, H.; Ikeda, K.; Arita, M.; Taguchi, R.; et al. Lipidomic analysis of brain tissues and plasma in a mouse model expressing mutated human amyloid precursor protein/tau for Alzheimer’s disease. Lipids Health Dis. 2013, 12, 68. [Google Scholar] [CrossRef] [PubMed]
- Puglielli, L.; Konopka, G.; Pack-Chung, E.; Ingano, L.A.; Berezovska, O.; Hyman, B.T.; Chang, T.Y.; Tanzi, R.E.; Kovacs, D.M. Acyl-coenzyme A: Cholesterol acyltransferase modulates the generation of the amyloid beta-peptide. Nat. Cell Biol. 2001, 3, 905–912. [Google Scholar] [CrossRef]
- Hutter-Paier, B.; Huttunen, H.J.; Puglielli, L.; Eckman, C.B.; Kim, D.Y.; Hofmeister, A.; Moir, R.D.; Domnitz, S.B.; Frosch, M.P.; Windisch, M.; et al. The ACAT inhibitor CP-113,818 markedly reduces amyloid pathology in a mouse model of Alzheimer’s disease. Neuron 2004, 44, 227–238. [Google Scholar] [CrossRef]
- Bryleva, E.Y.; Rogers, M.A.; Chang, C.C.; Buen, F.; Harris, B.T.; Rousselet, E.; Seidah, N.G.; Oddo, S.; LaFerla, F.M.; Spencer, T.A.; et al. ACAT1 gene ablation increases 24(S)-hydroxycholesterol content in the brain and ameliorates amyloid pathology in mice with AD. Proc. Natl. Acad. Sci. USA 2010, 107, 3081–3086. [Google Scholar] [CrossRef]
- Murphy, S.R.; Chang, C.C.; Dogbevia, G.; Bryleva, E.Y.; Bowen, Z.; Hasan, M.T.; Chang, T.Y. Acat1 knockdown gene therapy decreases amyloid-beta in a mouse model of Alzheimer’s disease. Mol. Ther. 2013, 21, 1497–1506. [Google Scholar] [CrossRef]
- Shibuya, Y.; Chang, C.C.; Huang, L.H.; Bryleva, E.Y.; Chang, T.Y. Inhibiting ACAT1/SOAT1 in microglia stimulates autophagy-mediated lysosomal proteolysis and increases Abeta1-42 clearance. J. Neurosci. 2014, 34, 14484–14501. [Google Scholar] [CrossRef]
- van der Kant, R.; Langness, V.F.; Herrera, C.M.; Williams, D.A.; Fong, L.K.; Leestemaker, Y.; Steenvoorden, E.; Rynearson, K.D.; Brouwers, J.F.; Helms, J.B.; et al. Cholesterol Metabolism Is a Druggable Axis that Independently Regulates Tau and Amyloid-beta in iPSC-Derived Alzheimer’s Disease Neurons. Cell Stem. Cell 2019, 24, 363–375.e9. [Google Scholar] [CrossRef]
- Nugent, A.A.; Lin, K.; van Lengerich, B.; Lianoglou, S.; Przybyla, L.; Davis, S.S.; Llapashtica, C.; Wang, J.; Kim, D.J.; Xia, D.; et al. TREM2 Regulates Microglial Cholesterol Metabolism upon Chronic Phagocytic Challenge. Neuron 2020, 105, 837–854.e9. [Google Scholar] [CrossRef] [PubMed]
- Shibuya, Y.; Niu, Z.; Bryleva, E.Y.; Harris, B.T.; Murphy, S.R.; Kheirollah, A.; Bowen, Z.D.; Chang, C.C.; Chang, T.Y. Acyl-coenzyme A:cholesterol acyltransferase 1 blockage enhances autophagy in the neurons of triple transgenic Alzheimer’s disease mouse and reduces human P301L-tau content at the presymptomatic stage. Neurobiol. Aging 2015, 36, 2248–2259. [Google Scholar] [CrossRef] [PubMed]
- Heneka, M.T.; Carson, M.J.; El Khoury, J.; Landreth, G.E.; Brosseron, F.; Feinstein, D.L.; Jacobs, A.H.; Wyss-Coray, T.; Vitorica, J.; Ransohoff, R.M.; et al. Neuroinflammation in Alzheimer’s disease. Lancet Neurol. 2015, 14, 388–405. [Google Scholar] [CrossRef] [PubMed]
- Huang, L.H.; Melton, E.M.; Li, H.; Sohn, P.; Jung, D.; Tsai, C.Y.; Ma, T.; Sano, H.; Ha, H.; Friedline, R.H.; et al. Myeloid-specific Acat1 ablation attenuates inflammatory responses in macrophages, improves insulin sensitivity, and suppresses diet-induced obesity. Am. J. Physiol. Endocrinol. Metab. 2018, 315, E340–E356. [Google Scholar] [CrossRef] [PubMed]
- Melton, E.M.; Li, H.; Benson, J.; Sohn, P.; Huang, L.H.; Song, B.L.; Li, B.L.; Chang, C.C.Y.; Chang, T.Y. Myeloid Acat1/Soat1 KO attenuates pro-inflammatory responses in macrophages and protects against atherosclerosis in a model of advanced lesions. J. Biol. Chem. 2019, 294, 15836–15849. [Google Scholar] [CrossRef]
- Cameron, B.; Landreth, G.E. Inflammation, microglia, and Alzheimer’s disease. Neurobiol. Dis. 2010, 37, 503–509. [Google Scholar] [CrossRef]
- Qin, L.; Wu, X.; Block, M.L.; Liu, Y.; Breese, G.R.; Hong, J.S.; Knapp, D.; Crews, F.T. Systemic LPS causes chronic neuroinflammation and progressive neurodegeneration. Glia 2007, 55, 453–462. [Google Scholar] [CrossRef]
- Zhao, J.; Bi, W.; Xiao, S.; Lan, X.; Cheng, X.; Zhang, J.; Lu, D.; Wei, W.; Wang, Y.; Li, H.; et al. Neuroinflammation induced by lipopolysaccharide causes cognitive impairment in mice. Sci. Rep. 2019, 9, 5790. [Google Scholar] [CrossRef]
- Zhang, Y.; Sloan, S.A.; Clarke, L.E.; Caneda, C.; Plaza, C.A.; Blumenthal, P.D.; Vogel, H.; Steinberg, G.K.; Edwards, M.S.; Li, G.; et al. Purification and Characterization of Progenitor and Mature Human Astrocytes Reveals Transcriptional and Functional Differences with Mouse. Neuron 2016, 89, 37–53. [Google Scholar] [CrossRef]
- Friedman, B.A.; Srinivasan, K.; Ayalon, G.; Meilandt, W.J.; Lin, H.; Huntley, M.A.; Cao, Y.; Lee, S.H.; Haddick, P.C.G.; Ngu, H.; et al. Diverse Brain Myeloid Expression Profiles Reveal Distinct Microglial Activation States and Aspects of Alzheimer’s Disease Not Evident in Mouse Models. Cell Rep. 2018, 22, 832–847. [Google Scholar] [CrossRef]
- Huang, L.H.; Melton, E.M.; Li, H.; Sohn, P.; Rogers, M.A.; Mulligan-Kehoe, M.J.; Fiering, S.N.; Hickey, W.F.; Chang, C.C.; Chang, T.Y. Myeloid Acyl-CoA:Cholesterol Acyltransferase 1 Deficiency Reduces Lesion Macrophage Content and Suppresses Atherosclerosis Progression. J. Biol. Chem. 2016, 291, 6232–6244. [Google Scholar] [CrossRef]
- Liu, H.-C.; Zheng, M.-H.; Du, Y.-L.; Wang, L.; Kuang, F.; Qin, H.-Y.; Zhang, B.-F.; Han, H. N9 microglial cells polarized by LPS and IL4 show differential responses to secondary environmental stimuli. Cell Immunol. 2012, 278, 84–90. [Google Scholar] [CrossRef] [PubMed]
- Zhou, S.; Guo, X.; Chen, S.; Xu, Z.; Duan, W.; Zeng, B. Apelin-13 regulates LPS-induced N9 microglia polarization involving STAT3 signaling pathway. Neuropeptides 2019, 76, 101938. [Google Scholar] [CrossRef] [PubMed]
- Ikenoya, M.; Yoshinaka, Y.; Kobayashi, H.; Kawamine, K.; Shibuya, K.; Sato, F.; Sawanobori, K.; Watanabe, T.; Miyazaki, A. A selective ACAT-1 inhibitor, K-604, suppresses fatty streak lesions in fat-fed hamsters without affecting plasma cholesterol levels. Atherosclerosis 2007, 191, 290–297. [Google Scholar] [CrossRef]
- Yin, Y.W.; Liao, S.Q.; Zhang, M.J.; Liu, Y.; Li, B.H.; Zhou, Y.; Chen, L.; Gao, C.Y.; Li, J.C.; Zhang, L.L. TLR4-mediated inflammation promotes foam cell formation of vascular smooth muscle cell by upregulating ACAT1 expression. Cell Death Dis. 2014, 5, e1574. [Google Scholar] [CrossRef] [PubMed]
- Ciesielska, A.; Matyjek, M.; Kwiatkowska, K. TLR4 and CD14 trafficking and its influence on LPS-induced pro-inflammatory signaling. Cell Mol. Life Sci. 2021, 78, 1233–1261. [Google Scholar] [CrossRef]
- Latz, E.; Visintin, A.; Lien, E.; Fitzgerald, K.A.; Monks, B.G.; Kurt-Jones, E.A.; Golenbock, D.T.; Espevik, T. Lipopolysaccharide rapidly traffics to and from the Golgi apparatus with the toll-like receptor 4-MD-2-CD14 complex in a process that is distinct from the initiation of signal transduction. J. Biol. Chem. 2002, 277, 47834–47843. [Google Scholar] [CrossRef] [PubMed]
- Kanarek, N.; Ben-Neriah, Y. Regulation of NF-kappaB by ubiquitination and degradation of the IkappaBs. Immunol. Rev. 2012, 246, 77–94. [Google Scholar] [CrossRef]
- Husebye, H.; Halaas, O.; Stenmark, H.; Tunheim, G.; Sandanger, O.; Bogen, B.; Brech, A.; Latz, E.; Espevik, T. Endocytic pathways regulate Toll-like receptor 4 signaling and link innate and adaptive immunity. EMBO J. 2006, 25, 683–692. [Google Scholar] [CrossRef]
- Triantafilou, M.; Morath, S.; Mackie, A.; Hartung, T.; Triantafilou, K. Lateral diffusion of Toll-like receptors reveals that they are transiently confined within lipid rafts on the plasma membrane. J. Cell Sci. 2004, 117 Pt 17, 4007–4014. [Google Scholar] [CrossRef] [PubMed]
- Rogers, M.A.; Chang, C.C.Y.; Maue, R.A.; Melton, E.M.; Peden, A.A.; Garver, W.S.; Lee, J.; Schroen, P.; Huang, M.; Chang, T.Y. Acat1/Soat1 knockout extends the mutant Npc1 mouse lifespan and ameliorates functional deficiencies in multiple organelles of mutant cells. Proc. Natl. Acad. Sci. USA 2022, 119, e2201646119. [Google Scholar] [CrossRef]
- Urano, Y.; Watanabe, H.; Murphy, S.R.; Shibuya, Y.; Geng, Y.; Peden, A.A.; Chang, C.C.; Chang, T.Y. Transport of LDL-derived cholesterol from the NPC1 compartment to the ER involves the trans-Golgi network and the SNARE protein complex. Proc. Natl. Acad. Sci. USA 2008, 105, 16513–16518. [Google Scholar] [CrossRef]
- Yin, J.; Liu, X.; He, Q.; Zhou, L.; Yuan, Z.; Zhao, S. Vps35-dependent recycling of Trem2 regulates microglial function. Traffic 2016, 17, 1286–1296. [Google Scholar] [CrossRef]
- Krause, B.R.; Bocan, T.M.A. ACAT inhibitors: Physiologic mechanisms for hypolipidemic and anti-atherosclerotic activities in experimental animals. In Inflammation: Mediators and Pathways; Ruffolo, R.R., Jr., Hollinger, M.A., Eds.; CRC Press Inc.: Boca Raton, FL, USA, 1995; pp. 173–198. [Google Scholar]
- Yang, W.; Bai, Y.; Xiong, Y.; Zhang, J.; Chen, S.; Zheng, X.; Meng, X.; Li, L.; Wang, J.; Xu, C.; et al. Potentiating the antitumour response of CD8(+) T cells by modulating cholesterol metabolism. Nature 2016, 531, 651–655. [Google Scholar] [CrossRef] [PubMed]
- Tarkowski, E.; Blennow, K.; Wallin, A.; Tarkowski, A. Intracerebral production of tumor necrosis factor-alpha, a local neuroprotective agent, in Alzheimer disease and vascular dementia. J. Clin. Immunol. 1999, 19, 223–230. [Google Scholar] [CrossRef] [PubMed]
- Akiyama, H.; Barger, S.; Barnum, S.; Bradt, B.; Bauer, J.; Cole, G.M.; Cooper, N.R.; Eikelenboom, P.; Emmerling, M.; Fiebich, B.L.; et al. Inflammation and Alzheimer’s disease. Neurobiol. Aging 2000, 21, 383–421. [Google Scholar] [CrossRef]
- Lei, L.; Xiong, Y.; Chen, J.; Yang, J.B.; Wang, Y.; Yang, X.Y.; Chang, C.C.; Song, B.L.; Chang, T.Y.; Li, B.L. TNF-alpha stimulates the ACAT1 expression in differentiating monocytes to promote the CE-laden cell formation. J. Lipid Res. 2009, 50, 1057–1067. [Google Scholar] [CrossRef] [PubMed]
- He, P.; Mei, C.; Cheng, B.; Liu, W.; Wang, Y.; Wan, J. Chlamydia pneumoniae induces macrophage-derived foam cell formation by up-regulating acyl-coenzyme A: Cholesterol acyltransferase 1. Microbes Infect. 2009, 11, 157–163. [Google Scholar] [CrossRef]
- Ruysschaert, J.M.; Lonez, C. Role of lipid microdomains in TLR-mediated signalling. Biochim. Biophys. Acta 2015, 1848, 1860–1867. [Google Scholar] [CrossRef]
- Zhou, Y.; Chen, Y.; Xu, C.; Zhang, H.; Lin, C. TLR4 Targeting as a Promising Therapeutic Strategy for Alzheimer Disease Treatment. Front. Neurosci. 2020, 14, 602508. [Google Scholar] [CrossRef] [PubMed]







Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Li, H.; Huynh, T.N.; Duong, M.T.; Gow, J.G.; Chang, C.C.Y.; Chang, T.Y. ACAT1/SOAT1 Blockade Suppresses LPS-Mediated Neuroinflammation by Modulating the Fate of Toll-like Receptor 4 in Microglia. Int. J. Mol. Sci. 2023, 24, 5616. https://doi.org/10.3390/ijms24065616
Li H, Huynh TN, Duong MT, Gow JG, Chang CCY, Chang TY. ACAT1/SOAT1 Blockade Suppresses LPS-Mediated Neuroinflammation by Modulating the Fate of Toll-like Receptor 4 in Microglia. International Journal of Molecular Sciences. 2023; 24(6):5616. https://doi.org/10.3390/ijms24065616
Chicago/Turabian StyleLi, Haibo, Thao N. Huynh, Michael Tran Duong, James G. Gow, Catherine C. Y. Chang, and Ta Yuan Chang. 2023. "ACAT1/SOAT1 Blockade Suppresses LPS-Mediated Neuroinflammation by Modulating the Fate of Toll-like Receptor 4 in Microglia" International Journal of Molecular Sciences 24, no. 6: 5616. https://doi.org/10.3390/ijms24065616
APA StyleLi, H., Huynh, T. N., Duong, M. T., Gow, J. G., Chang, C. C. Y., & Chang, T. Y. (2023). ACAT1/SOAT1 Blockade Suppresses LPS-Mediated Neuroinflammation by Modulating the Fate of Toll-like Receptor 4 in Microglia. International Journal of Molecular Sciences, 24(6), 5616. https://doi.org/10.3390/ijms24065616